
Abstract
Different substituents were introduced in positions 2 and 6 of 2,6 diaminopyridine in order to obtain new heterocyclic compounds. A new series of aza pyridine, imidazopyridine, benzodiazepine, indole, pyrimidine, and benzimidazole heterocyclic derivatives were synthesized in good yields. The anticancer activities of some of the new compounds were evaluated against liver cancer cell line HEPG2. Compounds 3, 4, 10, 11, 12, and 17 showed the highest activity when compared to 5-flurouracil (5-FU) and doxorubicin (DOX) chemotherapy.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The pyridine is among the most common heterocyclic compounds found in various therapeutic agents. The structure of 2,6-diaminopyridine (DAP) has the symmetrical feature that possesses three nitrogen lone pairs. N-acyl pyridine derivatives have also attracted considerable attention due to their hydrogen bonding motifs [1–3]. Poly functional pyridines are highly reactive reagents that have been used extensively in heterocyclic synthesis [4–7] and that possess biological as well as pharmacological activity [8–10]. In addition, triazolopyridines are also interesting compounds due to their pronounced biological activity, as they can be used as antidepressants [11–13]. Various pyridine derivatives are well known to possess an array of physiological activities, such as anticancer, muscle relaxant, hypnotic, anti-inflammatory, diuretic, and antihypertensive activities [14–17]. Imidazole derivatives are part of anticancer drugs like mercaptopurine. Imidazole is also a part of the theophylline molecule, found in tea leaves and coffee beans, which stimulate the central nervous system [18]. On the other hand, benzimidazole is a very important pharmacophore in drug discovery, and its derivatives are an important class of bioactive molecules in the field of drugs and pharmaceuticals [19]. Pyrimidines have been used as therapeutic agents possessing analgesics and anti-inflammatory activity [20–22]. Moreover, indoles and their derivatives are known to possess a wide variety of biological and pharmacological properties, including antibacterial and antifungal, cytotoxic, antioxidant and insecticidal activity [23, 24].
In this article, synthesis and characterization of some new heterocyclic compounds such as pyrido pyrimidine, diazepine, indole and imidazole derivatives and were performed to be evaluated against liver carcinoma cell line (HEPG2).
Experimental
Chemistry
All melting points are uncorrected and were recorded on an open glass capillary tube using an Electrothermal IA 9100 digital melting point apparatus. Elemental microanalyses were carried out at Micro analytical Unit, using Vario Elementar and were found within ±0.5% of the theoretical values. Infrared spectra were recorded on a Jasco FT/IR 300 E Fourier transform infrared spectrophotometer using the KBr disc technique. The spectra of 1H-NMR and C13-NMR were determined using a JEOL EX-270 and/or 500NMR spectrophotometers and were run in deuterated chloroform (CDCl3) or dimethyl sulphoxide (DMSO-d6). Chemical shifts were related to that of the solvent. The mass spectra were measured with a JEOL mass spectrometer and LC/MS-Finnigan at Central Services Laboratory, National Research Center, Cairo, Egypt. Follow-up of the reactions and checking purity of the compounds were made by thin-layer chromatograph technique on silica gel pre-coated aluminum sheets (Type 60, F 254, Merck, Darmstadt, Germany) and the spots were detected by exposure to UV lamp at 254 nm for a few seconds.
Synthesis of 6-amino-3,4-dihydro-2H-pyrido[1,2-a]pyrimidine-2-one (1)
A suspension of 2,6 diaminopyridine (0.1 mol), chloroacetic acid (10 g, 0.1 mol), anhydrous sodium acetate (1.5 g) in a mixture of acetic anhydride and acetic acid (1:3) was heated under reflux with stirring at 120 °C for 10 h. The reaction mixture was then poured onto ice-water (50 mL) and set aside in refrigerator at 0 °C. The solid formed was collected by filtration and recrystallization from benzene–petroleum ether (60–80 °C).
Yield 72%, mp. 140–142 °C. 1H-NMR (270 MHz, chloroform) δ: 3.60 (2H, t, CH2), 3.90 (2H, t, CH2), 6.50 (2H, br, J = 11.0 Hz, NH2 D2O-exchangeable), 7.20–7.60 (3H, m, Ar’H pyridine), 13C-NMR (270 MHz, chloroform) δ: 39.80, 40.20 (CH2), 120.50, 122.70, 131.90 (pyridine carbon), 158.90 (C=N), 160.90 (C=N), 170.90 (C=O). IR (KBr) cm−1: 3350 (NH2), 1720(C=O), 1640–1650 (C=N). Anal. Calcd. for C8H9N3O: C, 58.88; H, 5.56; N, 25.75. Found: C, 58.80; H, 5.50; N; 25.70.
Synthesis of 6-amino-2H-pyrido[1,2-a]pyrimidine-2,4(3H)-dione (2) and 1-aminobenzo[e]pyrido[1,2-a][1,3]diazepine-6,11-dione (2-3): general procedure
A mixture of 2,6 diaminopyridine (0.02 mol) and (0.02 mol) malonic acid and/or phthalic acid anhydride in ethanol (25 mL) in presence of few drops of TEA was refluxed for 8–10 h. Cooling the mixture followed by filtration and recrystallization from ethanol afforded the target molecules 2 and 3.
6-amino-2H-pyrido[1,2-a]pyrimidine-2,4(3H)-dione (2)
Yield 77%, mp. 130–133 °C. 1H-NMR (270 MHz, chloroform) δ: 3.95 (2H, s, J = 7.0 Hz, CH2), 6.50 (2H, br, J = 11.0 Hz, NH2 D2O-exchangeable), 7.20–7.60 (3H, m, Ar’H pyridine). 13C-NMR (270 MHz, chloroform) δ: 40.20 (CH2), 120.50, 122.70, 131.90 (pyridine carbon), 158.90 (C=N), 160.90 (C=N), 168.90 (C=O), 169.90 (C=O). IR (KBr) cm−1: 3350 (NH2), 1720–1740 (C=O), 1640–1650 (C=N). Anal. Calcd. for C8H8N3O2: C, 53.93; H, 4.53; N, 23.58. Found: C, 53.90; H, 4.55; N, 23.68, MS: m/z = 178 [M+].
1-aminobenzo[e]pyrido[1,2-a][1,3]diazepine-6,11-dione (3)
Yield 74%, mp. 154–156 °C. 1H-NMR (270 MHz, chloroform) δ: 6.50 (2H, br, NH2 D2O-exchangeable), 7.20–7.50 (3H, m, J = 9.5 Hz, Ar’H pyridine), 7.60–7.80 (4H, m, Ar’H).13C-NMR (270 MHz, chloroform) δ: 120.50, 122.70, 131.90 (pyridine carbon), 124.90, 125.40, 129.90, 130.70, 132.90, 135.60 (aromatic carbon), 158.90 (C=N), 160.50 (C=N), 166.30 (C=O), 168.90 (C=O). IR (KBr) cm−1: 3350 (NH2), 1710–1720 (C=O), 1640–1650 (C=N). Anal. Calcd. for C13H9N3O2: C, 65.27; H, 3.79; N, 17.56. Found: C, 65.20; H, 3.75; N; 17.50.
General procedure for the synthesis of compounds (4–6)
A mixture of compound 1–3 (0.001 mol) and the appropriate aldehyde, namely, 5-methylfuran-2-carbaldehyde (0.0015 mol) in ethanol (20 mL) and glacial acetic acid (1 mL) was refluxed for 6–8 h. The reaction mixture was poured into ice-cold water and the solid product was filtered off, washed with petroleum ether and recrystallization from methanol to form compounds 4–6, respectively.
-6-((4-methylfuran-2-yl)methyleneamino)-3,4-dihydro-2H-pyrido[1,2-a]pyrimidin-2-one (4)
Yield 75%, mp. 162–164 °C. 1H-NMR (270 MHz, DMSO) δ: 1.90 (3H, s, CH3), 3.60 (2H, t, CH2), 3.90 (2H, t, CH2), 6.65 (1H, d, furan), 6.75 (1H, d, furan), 7.20–7.50 (3H, m, J = 9.5 Hz, Ar’H pyridine), 7.65 (1H, s, J = 12.0 Hz, N=CH). IR (KBr) cm−1: 1710 (C=O), 1640–1650 (C=N). Anal. Calcd. for C14H13N3O2: C, 65.87; H, 5.13; N, 16.46. Found: C, 65.80; H, 5.10; N, 16.40, MS: m/z = 253[M+].
6-((4-methylfuran-2-yl)methyleneamino)-2H-pyrido[1,2-a]pyrimidine-2,4(3H)-dione (5)
Yield 72%, mp. 170–172 °C. 1H-NMR (270 MHz, DMSO) δ: 1.90 (3H, s, CH3), 3.95 (2H, s, CH2), 6.65 (1H, d, furan), 6.75 (1H, d, furan), 7.20–7.50 (3H, m, Ar’H pyridine), 7.75 (1H, s, J = 12.0 Hz, N=CH), IR (KBr) cm−1: 1710–1720 (C=O), 1640–1650 (C=N). Anal. Calcd. for C14H11N3O3: C, 62.45; H, 4.12; N, 15.61. Found: C, 62.50; H, 4.18; N, 15.66.
(4a)-1-((4-methylfuran-2-yl)methyleneamino)benzo[e]pyrido[1,2-a][1,3]diazepine-6,11-dione (6)
Yield 75%, mp. 180–183 °C. 1H-NMR (270 MHz, DMSO) δ: 1.90(3H, s, J = 5.0 Hz, CH3), 6.65 (1H, d, furan), 6.75 (1H, d, furan), 7.20–7.50 (3H, m, Ar’H pyridine), 7.60–7.80 (4H, m, Ar’H), 7.90 (1H, s, J = 12.0 Hz, N=CH). IR (KBr) cm−1: 1710–1720 (C=O), 1640–1650 (C=N). Anal. Calcd. for C19H13N3O3: C, 68.88; H, 3.95; N, 12.68. Found: C, 68.85; H, 3.89; N, 12.60, MS: m/z = 331[M+].
Synthesis of N2,N6-bis ((1H-indol-3-yl)methylene)pyridine-2,6-diamine (7)
A mixture of starting compound 2,6 diaminopyridine (0.001 mol) and indole 3-carboxaldehyde (0.001 mol) in ethanol and few drops of glacial acetic acid was refluxed for 8 h. The reaction mixture was poured into ice-cold water and the solid product was filtered off, washed with petroleum ether, and recrystallization from ethanol to give compound 7.
Yield 68%, mp. 182–184 °C. 1H-NMR (270 MHz, DMSO) δ: 7.20–7.50 (3H, m, Ar’H pyridine), 7.60–7.80(5H, m, J = 13.00 Hz, Ar’H), 7.90 (1H, s, J = 11.0 Hz, N=CH), 10.75 (1H, br, NH benzimidazole). IR (KBr) cm−1: 3290 (NH), 1640 (C=N). Anal. Calcd. for C23H17N5: C, 67.01; H, 4.71; N, 19.27. Found: C, 67.08; H, 4.76; N, 19.20. MS: m/z = 253 [M+].
Reaction of compound (7) with oxalyl chloride: preparation of -2-(3-((6-((1-(2-chloro-2-oxoacetyl)-1H-indol-3-yl)methyleneamino)pyridin-2-yl)methyleneamino)-1H-indol-1-yl)-2-oxoacetyl chloride (8)
A mixture of compound 7 (0.01 m mol) and oxalyl chloride (0.01 mmol) in diethyl ether (30 mL) with stirring for 12 h. The reaction mixture was poured into ice water and the solid product was filtered off, washed with petroleum ether, and recrystallization from ethanol to obtain compound 8.
Yield 65%, mp. 192–195 °C. 1H-NMR (270 MHz, DMSO) δ: 7.20–7.50 (3H, m, Ar’H pyridine), 7.60–7.80 (5H, m, J = 13.00 Hz, Ar’H), 7.90 (1H, s, J = 11.0 Hz, N=CH). IR (KBr) cm−1: 1640–1660 (C=N), 1680–1695 (C=O), 1250–1260 (C–Cl). Anal. Calcd. for C29H21N5O4: C, 60.64; H, 3.68; Cl, 12.34; N, 12.19. Found: C, 60.68; H, 3.62; Cl, 12.39; N, 12.15, MS: m/z = 545 [M+ +1].
Synthesis of 2,2′-(3,3′-(pyridine-2,6-diylbis(aza-1-yl-1-ylidene))bis(methan-1-yl-1-ylidene)bis(1H-indole-3,1-diyl))bis(1-(piperazin-1-yl)ethane-1,2-dione) (9)
A mixture of the appropriate compound 8 (0.005 mol) and piperazine (0.005 mol) was dissolved in hot absolute ethanol (20 mL) and anhydrous potassium carbonate (0.5 g) was added with stirring. The mixture was heated under reflux with stirring for 8 h. Then, an excess of solvent was evaporated under vacuum, poured onto ice and then neutralized with diluted HCl. The residue formed was treated with petroleum ether, collected and recrystallization from ethanol to form compound 9.
Yield 62%, mp. 206–208 °C. 1H-NMR (270 MHz, DMSO) δ: 2.80–3.30 (8H, m, 4CH2 piprazine), 7.20–7.50 (3H, m, Ar’H pyridine), 7.60–7.80 (5H, m, Ar’H), 7.90 (1H, s, J = 11.0 Hz, N=CH), 9.95 (1H, s, NH piprazine). IR (KBr) cm−1: 3225 (NH), 1640–1660 (C=N), 1680–1695 (C=O). Anal. Calcd. for C36H35N9O4: C, 65.74; H, 5.36; N, 19.17. Found: C, 65.70; H, 5.31; N, 19.10.
Synthesis of 3,3′-(pyridine-2,6-diyl)bis (aza-1-yl-1-ylidene)) diindolin-2-one (10)
2,6 diaminopyridine (0.02 mol) was added to 2,3 indolinedione (0.02 mol, 2.94 g) in (50 mL) absolute ethanol containing few drops of glacial acetic acid. The reaction mixture was heated under reflux for 6 h and then the reaction mixture was filtered off. Recrystallization of the product from ethanol then drying afforded product 10.
Yield 78%, mp. 225–227 °C. 1H-NMR (500 MHz, DMSO) δ: 7.20–7.50 (3H, m, Ar’H pyridine), 7.60–7.90 (4H, m, J = 13.0 Hz, Ar’H), 10.19 (1H, s, NH). 13C-NMR (500 MHz, DMSO) δ: 122.3, 127.98, 132.70 (pyridine carbon), 111.80, 118.90, 124.80, 125.70, 133.60, 135.20 (aromatic carbon), 165.77 (C=N), 167.70 (C=N), 170.90 (C=O). IR (KBr) cm−1: 3340 (NH), 1645–1660 (C=N), 1690 (C=O) Anal. Calcd. for C21H13N5O2: C, 68.66; H, 3.57; N, 19.06. Found: C, 68.62; H, 3.52; N, 19.10. MS: m/z = 367 [M+].
Synthesis of -1-(2-hydroperoxyethyl)-2-oxoindolin-3-ylideneamino) pyridin-2-ylimino)-2-oxoindolin-1-yl) acetic acid derivative (11)
A mixture of potassium hydroxide (0.01 mol) and compound 10 (0.01 mol) was dissolved in (25 mL) absolute ethanol. Then chloroacetic acid (0.01 mol, 1.22 g) was added drop wisely with stirring and then the reaction mixture was refluxed for 8 h. After cooling, the solvent was evaporated under vacuum and the formed solid was recrystallization from ethanol.
Yield 74%, mp. 285–287 °C. 1H-NMR (500 MHz, DMSO) δ: 4.20 (2H, s, J = 7.5 Hz, CH2), 7.20–7.50 (3H, m, Ar’H pyridine), 7.60–7.95 (4H, m, J = 9.0 Hz, Ar’H), 11.20 (1H, s, OH). 13C-NMR (500 MHz, DMSO) δ: 40.50 (CH2), 122.3, 127.98, 132.70 (pyridine carbon), 115.90, 117.30, 124.50, 125.98, 129.47, 131.77 (aromatic carbon), 155.30 (C=N), 163.22 (C=N), 170.90 (C=O), 173.90 (COOH). IR (KBr) cm−1: 3370 (OH), 1645–1660 (C=N), 1690 (C=O), 1750 (COOH). Anal. Calcd. for C25H17N5O6: C, 62.11; H, 3.54; N, 14.49. Found: C, 62.18; H, 3.54; N, 14.55. MS: m/z = 483 [M+].
Synthesis of 3,3′-(pyridine-2,6-diyl)bis(aza-1-yl-1-ylidene))bis(2-oxoindoline-1-yl-3-ylidene)bis(methylene)bis(1H-benzo[d]imidazole-6-carboxylic acid) (12) and 3,3′-(pyridine-2,6-diylbis(aza-1-yl-1-ylidene))bis(1-((3H-imidazo[4,5-b]pyridin-2-yl)methyl)indolin-2-one) (13) derivatives: general procedure
Compound 11 (0.01 mol) was dissolved in ethanol (10 mL) and was added to 2,3 diaminopyridine and/or o-phenylendiamine-5-carboxylic acid (0.01 mol) in ethanol (10 mL) in presence of few drops of TEA. The reaction mixture was heated under reflux with stirring for 10–12 h. The solvent was evaporated under vacuum and then crystallization from methanol to afford the products 12–13.
3,3′-(pyridine-2,6-diyl)bis(aza-1-yl-1-ylidene))bis(2-oxoindoline-1-yl-3-ylidene)bis(methylene)bis(1H-benzo[d]imidazole-6-carboxylic acid) derivative (12)
Yield 75%, mp. 196–198 °C. 1H-NMR (500 MHz, DMSO) δ: 4.20 (2H, s, J = 7.0 Hz, CH2), 6.90–7.10 (4H, m, J = 12.0 Hz, Ar’H), 7.20–7.50 (4H, m, Ar’H pyridine), 7.60–7.90 (4H, m, Ar’H), 10.90 (1H, s, NH), 11.20 (1H, s, OH). 13C-NMR (500 MHz, DMSO) δ: 39.96 (CH2), 122.3, 127.98, 132.70 (pyridine carbon), 114.20, 115.90, 116.50, 117.80, 118.50, 119.20, 121.90, 124.50, 125.50, 126.80, 128.60, 131.70 (aromatic carbon), 163.20 (C=N), 165.90 (C=N), 167.22 (C=N), 170.90 (C=O), 174.90 (COOH), IR (KBr) cm−1: 3340 (OH), 1640–1665 (C=N), 1690 (C=O), 1750 (COOH). Anal. Calcd. for C39H25N9O6: C, 65.45; H, 3.52; N, 17.61. Found: C, 65.52; H, 3.46; N, 17.55.
3,3′-(pyridine-2,6-diyl)bis(aza-1-yl-1-ylidene))bis(1-((3H-imidazo[4,5-b]pyridin-2-yl)methyl)indolin-2-one) derivative (13)
Yield 78%, mp. 175–177 °C. 1H-NMR (500 MHz, DMSO) δ: 4.23 (2H, s, CH2), 6.90–7.10 (3H, m, Ar’H pyridine), 7.20–7.50 (3H, m,, Ar’H pyridine), 7.60–7.90 (3H, m, J = 11.0 Hz, Ar’H), 10.50 (1H, s, NH). 13C-NMR (500 MHz, DMSO) δ: 40.90 (CH2), 122.89, 127.80, 132.50 (pyridine carbon), 115.70, 117.90, 123.80, 126.90, 130.90 (pyridine carbon), 115.90, 117.80, 119.90, 124.50, 126.98, 131.91 (aromatic carbon), 158.40 (C=N), 161.90 (C=N), 163.22 (C=N), 165.24 (C=N), 170.90 (C=O). IR (KBr) cm−1: 3350 (NH),1640–1660 (C=N), 1695 (C=O) Anal. Calcd. for C35H23N11O2: C, 66.77; H, 3.68; N, 24.47. Found: C, 66.70; H, 3.61; N, 24.40.
Synthesis of 1,1′-(pyridine-2,6-diyl) bis (3-isopropylthiourea) (14), 1,1′-(pyridine-2,6-diyl) bis (3-butylurea) (15) and N,N′-(pyridine-2,6-diyl)bis (aza-diyl)) bis (thioxomethylene) dibenzamid (16) derivatives: general procedure
To a solution of 2,6 diaminopyridine (0.01 mol) in ethanol (20 mL) containing isocyanate or isothiocyanate derivatives; namely, isopropyl isothiocyanate, butylisocyanate, and benzoyl isothiocyanate (0.02 mol) added dropwisely with stirring. The reaction mixture was heated under reflux for 6–8 h. After cooling, the reaction mixture washed with water. The formed solids were collected by filtration, dried, and crystallization from methanol to produce compounds 14–16.
1,1′-(pyridine-2,6-diyl) bis (3-isopropylthiourea) derivative (14)
Yield 70%, mp. 103–105 °C. 1H-NMR (500 MHz, DMSO) δ:1.20–1.34 (6H, m, 2CH3), 4.55–4.70 (1H, m, CH), 7.60–7.90 (3H, m, Ar’H pyridine), 8.90 (1H, s, NH),11.50 (1H, s, NH). IR (KBr) cm−1: 3280–3290 (NH), 1640 (C=N), 1260 (C=S). Anal. Calcd. for C13H21N5S2: C, 50.13; H, 6.80; N, 22.48; S, 20.59 Found: C, 50.20; H, 6.70; N, 20.60; S, 20.55. MS: m/z = 311 [M+].
1,1′-(pyridine-2,6-diyl) bis (3-butylurea) derivative (15)
Yield 67%, mp. 130–132 °C. 1H-NMR (500 MHz, DMSO) δ: 1.18–1.30 (3H, m, CH3), 4.25–4.50 (6H, m, 3CH2), 7.20–7.50 (3H, m, Ar’H pyridine), 8.90 (1H, s, NH), 9.60 (1H, s, NH). IR (KBr) cm−1: 3250–3270 (NH), 1690 (C=O), 1640 (C=N). Anal. Calcd. for C15H25N5O2: C, 58.61; H, 8.20; N, 22.78. Found: C, 58.70; H, 4.28; N, 22.86. MS: m/z = 307 [M+].
N,N′-(pyridine-2,6-diyl)bis(aza-diyl))bis(thioxomethylene)dibenzamide derivative (16)
Yield 70%, mp. 120–122 °C. 1H-NMR (500 MHz, DMSO) δ: 6.90–7.50 (5H, m, Ar’H), 7.60–7.90 (3H, m, Ar’H pyridine), 10.50 (1H, s, NH), 11.40 (1H, s, NH). IR (KBr) cm−1: 3260–3280 (NH), 1700 (C=O), 1640 (C=N), 1265 (C=S). Anal. Calcd. for C21H17N5O2S2: C, 57.91; H, 3.93; N, 16.08; S, 14.72. Found: C, 57.82; H, 4.10; N, 16.02; S, 14.67.
Synthesis of 3,3′-(pyridine-2,6-diyl)bis(dihydropyrimidine-4,6(1H,5H)-dione) derivatives (17–19): general procedure
A mixture of compounds 14–16 (0.001 mol) and (0.002 mol) of malonic acid in presence of sodium methoxide (20 mL) added dropwisely and heated under reflux for 5–6 h. The mixture was cooled, followed by filtration and recrystallization from methanol afforded the target molecules 17–19.
3,3′-(pyridine-2,6-diyl)bis(1-isopropyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione) derivative (17)
Yield 70%, mp. 156–158 °C. 1H-NMR (500 MHz, DMSO) δ: 1.20–1.40 (6H, m, 2CH3), 4.10 (2H, s, CH2), 4.50–4.70 (1H, m, CH), 7.60–7.90 (3H, m, Ar’H pyridine). 13C-NMR (500 MHz, DMSO) δ: 19.90 (CH3), 29.70 (CH3), 39.20 (CH3), 50.90 (CH), 124.77, 129.66, 135.32 (pyridine carbon), 166.33 (C=N), 168.92 (C=O), 170.40 (C=O), 179.50 (C=S). IR (KBr) cm−1: 1680–1695 (C=O), 1645 (C=N), 1265 (C=S). Anal. Calcd. for C19H21N5O4S2: C, 51.06; H, 4.69; N, 15.65, S, 14.33 Found: C, 51.02; H, 4.74; N, 15.70; S, 14.30. MS: m/z = 447 [M+].
3,3′-(pyridine-2,6-diyl)bis(1-butyl-2-dihydropyrimidine-4,6(1H,5H)-dione) derivative (18)
Yield 67%, mp. 178–180 °C. 1H-NMR (500 MHz, DMSO) δ: 1.20–1.30 (3H, m, CH3), 4.10 (2H, s, CH2), 4.25–4.50 (6H, m, 3CH2), 7.60–7.90 (3H, m, Ar’H pyridine). 13C-NMR (500 MHz, DMSO) δ: 20.90(CH3), 27.90 (CH2), 29.20 (CH2), 30.99 (CH2), 39.70 (CH2), 124.50, 135.74, 137.70 (pyridine carbon), 149.80 (C=O), 165.70 (C=O), 169.70 (C=O). IR (KBr) cm−1: 1680–1698 (C=O), 1645 (C=N). Anal. Calcd. for C21H25N5O6: C, 56.88; H, 5.64; N, 15.80. Found: C, 56.80; H, 5.60; N, 15.86. MS: m/z = 443 [M+].
3,3′-(pyridine-2,6-diyl)bis(1-benzoyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione) derivative (19)
Yield 72%, mp. 146–148 °C. 1H-NMR (500 MHz, DMSO) δ: 4.20 (2H, s, J = 5.0 CH2), 6.90–7.50 (5H, m, J = 10.2, Ar’H), 7.60–7.90 (3H, m, Ar’H pyridine). IR (KBr) cm−1: 1680–1690 (C=O), 1645 (C=N), 1265 (C=S). Anal. Calcd. for C27H17N5O6S2: C, 56.74; H, 3.00; N, 12.25; S, 11.22. Found: C, 56.70; H, 3.10; N, 12.28; S, 11.26.
Results and discussion
In the present work, we report on the synthesis and preliminary biological activity screening of several heterocycles compounds based on 2,6 diaminopyridine (DAP) could be considered as a starting material was reacted with chloroacetic acid as well as malonic acid and phthalic acid in refluxing ethanol containing TEA afforded a single product identified as 6-amino-3,4-dihydro-2H-pyrido[1,2-a]pyrimidin-2-one (1), 6-amino-2H-pyrido[1,2-a]pyrimidine-2,4(3H)-dione (2) and 1-aminobenzo[e]pyrido[1,2-a][1,3]diazepine-6,11-dione (3), respectively (Scheme 1). The structures of compounds 1–3 were identified by elemental analyses and spectroscopic data. The structure of compounds 1, 2, and 3 were confirmed by their IR, 1H-NMR and 13C-NMR spectra. The IR spectra of compounds 1–3 appearance of bands at 3,350 cm−1 attributed to NH2 stretching frequency are good evidence for the structure given to those compounds, 1710–1740 cm−1 due to the presence of two C=O groups for compound 2 and 3 in addition to C=N groups at 1640–1650 cm−1. All the synthesized compounds 1–3 were confirmed by thin-layer chromatograph.
On the other hand, compounds 1–3 were reacted similarly with 5-methylfuran-2-carbaldehyde in refluxing ethanol containing a few drops of acetic acid to yield the corresponding 6-((4-methylfuran-2-yl)methyleneamino)-3,4-dihydro-2H-pyrido[1,2-a]pyrimidin-2-one (4), 6-((4-methylfuran-2-yl)methyleneamino)-2H-pyrido[1,2-a]pyrimidine-2,4(3H)-dione (5) and -1-((4-methylfuran-2-yl)methyleneamino)benzo[e]pyrido[1,2-a][1,3]diazepine-6,11-dione (6), respectively (Scheme 1). The identities of the isolated products were assigned by their elemental analysis and spectral data. The 1H-NMR analysis of compounds (4–6) showed characteristic resonance at δ 7.20–7.50 ppm due to the presence of (Ar’H pyridine) function and 7.65–7.90 ppm due to the presence of (N=CH) group of compounds 4–6 and 1.90 ppm for (CH3) group of compounds 4–6, respectively. All synthesized compounds were confirmed by thin-layer chromatography.

Synthesis of 2H-pyrido[1,2-a]pyrimidine derivatives 4–5 and pyrido[1,2-a][1,3]diazepine derivative 6 from 2,6 diaminopyridine (DAP). Reagents and conditions: (i) chloro acetyl chloride/anhydrous sodium acetate/reflux, (ii) malonic acid/ethanol/TEA/reflux, (iii) phthalic acid/ethanol/TEA/reflux, (iv) 5-methylfuran-2-carboxaldehyde/ethanol/acetic acid/reflux
In addition, condensation reaction of 2,6 diaminopyridine with indole 3-carboxaldehyde afforded the corresponding of N2,N6-bis ((1H-indol-3-yl)methylene)pyridine-2,6-diamine (7), which was reacted with oxalyl chloride to give -2-(3-((6-((1-(2-chloro-2-oxoacetyl)-1H-indol-3-yl)methyleneamino)pyridin-2-yl)methyleneamino)-1H-indol-1-yl)-2-oxoacetyl chloride (8). The condensation reaction of compound 8 with piperazine led to the formation of 2,2′-(3,3′-(pyridine-2,6-diylbis(aza-1-yl-1-ylidene))bis(methan-1-yl-1-ylidene)bis(1H-indole-3,1-diyl))bis(1-(piperazin-1-yl)ethane-1,2-dione) (9) (Scheme 2). All the synthesized compounds 7–9 were characterized by their physical, chemical, and spectral data. Thus, the IR spectra of compound 7 shows the presence of characteristic absorption peaks around 3,290 cm−1 (NH stretching), C=N at 1,640 cm−1 and 1H-NMR spectra of compound 7 showed broad band’s at 10.75 ppm that revealed the presence of NH absorption peak in addition to MS at m/z = 253 [M+].

Synthesis of 2,2′-(3,3′-(pyridine-2,6-diylbis(aza-1-yl-1-ylidene))bis(methan-1-yl-1-ylidene)bis(1H-indole-3,1-diyl))bis(1-(piperazin-1-yl)ethane-1,2-dione) 9 from 2,6 DAP. Reagents and conditions: (i) indole 3-carboxaldehyde/ethanol/glacial acetic acid/reflux, (ii) oxally chloride/diethylether/rt, (iii) piprazine/ethanol/anhydrous potassium carbonate/reflux
In a similar manner, treatment of compound 2,6 diaminopyridine with 2,3 indolinedione upon refluxing in ethanol and few drops of acetic acid gave the corresponding Schiff’s base 3,3′-(pyridine-2,6-diylbis(aza-1-yl-1-ylidene))diindolin-2-one (10), which reacted with chloroacetic acid yielded the corresponding 1-(-2-hydroperoxyethyl)-2-oxoindolin-3ylideneamino)pyridin-2-ylimino)-2-oxoindolin-1-yl) acetic acid derivative (11), which continue to react with diamines, namely, o-phenylendiamine-5-carboxylic acid and 2,3 diaminopyridine yielded the corresponding 3,3′-(pyridine-2,6-diyl) bis (aza-1-yl-1-ylidene))bis(2-oxoindoline-1-yl-3-ylidene)bis(methylene)bis(1H-benzo[d] imidazole-6-carboxylic acid) derivative (12) and 3,3′-(pyridine-2,6-diylbis(aza-1-yl-1-ylidene)) bis (1-((3H-imidazo [4,5-b] pyridin-2-yl) methyl) indolin-2-one) derivative (13), respectively (Scheme 3). The 1H -NMR spectra of compounds 10 showed broad band at 10.19 ppm revealed the presence of the NH peak. The structure of compound 10 was further supported by its MS spectra, indicating the molecular ion peak was at m/z 367. The structures of compounds 10, 11, 12, and 13 were confirmed on the basis of elemental analyses as well as spectral data. The synthesized compounds were confirmed by thin-layer chromatography.

Synthesis of 3,3′-(pyridine-2,6-diyl)bis(aza-1-yl-1-ylidene))bis(2-oxoindoline-1-yl-3-ylidene)bis(methylene)bis(1H-benzo[d]imidazole-6-carboxylic acid) 12 and 3,3′-(pyridine-2,6-diylbis(aza-1-yl-1-ylidene))bis(1-((3H-imidazo[4,5-b]pyridin-2-yl)methyl)indolin-2-one) 13 from 2,6 diaminopyridine (DAP). Reagents and conditions: (i) 2,3 indolinedione/ethanol/acetic acid/reflux, (ii) chloroacetic acid/ethanol/reflux, (iii) 2,3 diaminopyridine and/or o-phenylendiamine-5-carboxylic acid/ethanol/TEA/reflux
Moreover, 2,6-diaaminopyridine, which upon treatment with appropriate isocyanate and/or isothiocyanate derivatives, namely, isopropyl isothiocyanate, butyl isocyanate and benzoyl isothiocyanate, afforded the corresponding urea and thiourea derivatives 14–16, respectively, which was followed by cyclization when treated with malonic acid in presence of sodium methoxide to gave the corresponding pyrimidine derivatives; 3,3′-(pyridine-2,6-diyl)bis(1-isopropyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione) (17), 3,3′-(pyridine-2,6-diyl)bis(1-butyl-2-dihydropyrimidine-4,6(1H,5H)-dione) (18) and 3,3′-(pyridine-2,6-diyl)bis(1-benzoyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione) (19) derivatives. The spectral data of 14–19 are in agreement with the assigned structures (Scheme 4). All the synthesized compounds 14–19 were confirmed by thin-layer chromatography.

Synthesis of 3,3′-(pyridine-2,6-diyl)bis(1-isopropyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione) derivative 17, 3,3′-(pyridine-2,6-diyl)bis(1-butyl-2-dihydropyrimidine-4,6(1H,5H)-dione) derivative 18 and 3,3′-(pyridine-2,6-diyl)bis(1-benzoyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione) derivative 19 from 2,6 diaminopyridine. Reagents and conditions: (i) (CH3)2CHNCS/ethanol/reflux, (ii) CH3 (CH2)3NCO/ethanol/reflux, (iii) C6H5CONCS/ethanol/reflux, (iv) HCOOCH2COOH/sodium methoxide/reflux
Pharmacology
Antitumor activity
Cytotoxic and biological effects of tested compounds against liver cancer cell line
Compounds 3, 4, 5, 6, 7, 8, 10, 11, 12, 13, 17, and 18 were soluble in DMSO at concentrations high enough to allow cell experiments; the in vitro biological activity of these compounds was evaluated by their growth-inhibitory potency in liver HEPG2 cancer cell lines. The cytotoxic potency of compounds 3, 4, 5, 6, 7, 8, 10, 11, 12, 13, 17, and 18 were studied in comparison to the known anticancer drugs 5-flurouracil (5-FU) and doxorubicin (DOX). Moreover, the biochemical effects of the tested compounds on some enzymes such as aspartate and alanine aminotransferases (AST and ALT) and alkaline phosphates (ALP), in addition to albumin, globulins, creatinine, total lipids, cholesterol, triglycerides, and bilirubin in serum of mice were investigated.
Measurement of potential cytotoxicity by SRB assay
Compounds (3, 4, 5, 6, 7, 8, 10, 11, 12, 13, 17, and 18) were evaluated for their antitumor activity against liver HEPG2 cancer cell lines in comparison to the known anticancer drugs: 5-FU and DOX. Potential cytotoxicity of the tested derivatives was tested according the method of Skehan et al. [25] as follows:
Cells were plated in 96-multiwell plates (104 cells/well) for 24 h before treatment with the tested compounds to allow attachment of cells to the wall of the plate. Different concentrations of the compound under test (0, 1, 2.5, 5, 10 μg/mL) were added to the cell monolayer. Triplicate wells were prepared for each individual dose. Monolayer cells were incubated with the compounds for 48 h at 37 °C and in an atmosphere of 5% CO2. Cultures were then fixed with trichloroacetic acid and stained for 30 min with 0.4% (wt/vol) sulforhodamine B (SRB) dissolved in 1% acetic acid. Unbound dye was removed by four washes with 1% acetic acid, and protein-bound dye was extracted with 10 mM un-buffered Tris base [tris (hydroxymethyl) amino methane] for determination of optical density in a computer-interfaced, 96-well micro titer plate reader. The SRB assay results were linear with the number of cells and with values for cellular protein measured by both the Lowry and Bradford assays modified by Zor and Selinger [26] at densities ranging from sparse sub-confluence to multilayered supra-confluence. The signal-to-noise ratio at 564 nm was approximately 1.5 with 1,000 cells per well. The relation between surviving fraction and drug concentration is plotted to get the survival curve of both cancer cell lines after the specified compounds.
Biochemical analysis
Animals
Male albino mice weighing 18–20 g were used in the present study. Mice were divided into three main groups as follows:
-
1-
Group (1): untreated or control group (five mice).
-
2-
Group (2): divided into two subgroups (five mice for each subgroup) and treated with 5-FU or DOX as reference anticancer drugs.
-
3-
Group (3): divided into eight subgroups (five mice for each subgroup) and treated with the tested compounds.
-
4-
Treatment.
Group (1): each mouse was given a single intraperitoneal injection of 0.1 mL DMSO.
Group (2): each mouse was given a single intraperitoneal injection of 0.1 mL containing 12 mg/kg body weight 5-FU or DOX dissolved in sterile water.
Group (3): each mouse was given a single intraperitoneal injection of 0.1 mL containing 12 mg/kg body weight of the tested compounds (3, 4, 5, 6, 7, 8, 10, 11, 12, 13, 17 and 18, respectively) dissolved in DMSO. Blood was collected after 7 days from all mice groups.
The biochemical effects of the tested compounds, on some liver enzymes such as aspartate and alanine, aminotransferases (AST and ALT) [27] and alkaline phosphatase (ALP) [28], were done using blood auto analyzer (Olympus AV 400, Japan). Moreover, albumin [29], globulins [30] and creatinine [31], total lipids [32], cholesterol [33], triglycerides [34] and bilirubin [35] in serum of mice were evaluated in comparison to 5-FU and DOX.
Statistical analysis of the results was performed using Chi-square values (SPSS computer program).
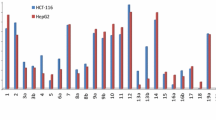
Preliminary screening of the tested compounds showed that all compounds exhibited a moderate to strong growth inhibition activity on the tested cell line between 1 and 10 μg/mL concentrations in comparison to the known anticancer drugs: 5-flurouracil and doxorubicin. Table 1 indicated the cytotoxic activity of the tested compounds (3, 4, 5, 6, 7, 8, 10, 11, 12, 13, 17, and 18) against liver HEPG2 cancer cell lines in comparison to the traditional anticancer drugs: 5-FU and DOX. It can be deduced from the results that compounds (3, 4, 10, 11, 12 and 17) were the most active and induced a reasonable growth inhibition, in a dose-dependent manner against HEPG2 when compared to 5-FU and DOX (IC50 equals 2.08, 2.89, 3.02, 3.09, 3.15, and 3.36 μg/mL, while 5-FU and DOX were 5 and 3.56 μg/mL).
Effect of antitumor compounds on the biochemical parameters
Data obtained in Table 2 represented the liver enzymatic activities (ALT, AST, and ALP) in serum of control and treated groups of mice. The results showed that the values recorded for AST and ALT were significantly higher (p < 0.001) with 5-Fu and DOX treated groups of mice than the control. On the other hand, treatment of compounds, caused inverse effects, where some values recorded for AST and ALT were non-significant (n.s.) or slightly higher (p < 0.01) in comparison to control. Moreover, the recorded data showed that ALP activities were significantly increased (p < 0.001) with the treatment of 5-Fu and DOX, while there were no significant changes in ALP activities upon treatment with some of the new compounds.
Data listed in Table 3 demonstrated the comparison between the levels of total lipids, cholesterol, triglycerides, and bilirubin in serum of treated mice and the control group. It can be deduced from the present data that 5-FU and DOX caused a significant increase in the level of these parameters while treatment with the selected compounds showed moderate or no significant changes.
Table 4 represented a comparison between the levels of albumin, globulins, and creatinine in serum of control and treated groups of mice. It is clear from the results in the table that there was a slight increase in the level of albumin and creatinine and globulins in the 5-FU and DOX treated groups of mice while there were moderate or non-significant changes in the other treated groups.
Cytotoxic drugs are being administered with novel ways of therapy such as inhibitors of signals [36]. It is therefore important to discover novel cytotoxic agents with spectra of activity and toxicity that differ from those current agents. The antitumor activities of compounds were assessed against HEPG2 cancer cell line in comparison to the traditional anticancer drugs: 5-Fu and DOX. Regarding the antitumor activity study, some of the compounds showed reasonable antitumor activity in comparison to 5-FU and DOX. Moreover, study of the induced biochemical parameters of the tested compounds in mice showed insignificant differences relative to the control group, which indicates a moderate margin of safety for the tested compounds. Comparable to 5-FU and DOX, a dose augmentation of compounds 11 and 4 may be higher potency. Furthermore, the tested compounds have important potential advantages over 5-FU and DOX because of their lower toxicity and their ability to induce lower biochemical parameters. These results are in agreement with Espinosa et al. [37] and Kamalakannan and Venkappayya [38].
On the basis of monitoring the inhibition of the growth of human cancer cells, a series of synthesized compounds possessing a broader spectrum of antitumor activity and fewer toxic side-effects than traditional anticancer drugs have been studied. Twenty tested compounds (3, 4, 5, 6, 7, 8, 10, 11, 12, 13, 17, and 18) were subjected to a screening system for investigation of their antitumor potency against liver (HEPG2) cell line. Moreover, the biochemical effects of the tested compounds on some enzymes such as aspartate and alanine aminotransferases (AST and ALT) and alkaline phosphatase (ALP), in addition to albumin, globulins, creatinine, total lipids, cholesterol, triglycerides and bilirubin in serum of mice were studied in comparison to 5-flurouracil and doxorubicin.
The antitumor activity results indicated that most of the compounds showed good growth inhibition activity against the tested cell line but with varying intensities extents in comparison to the known anticancer drugs: 5-flurouracil and doxorubicin. Moreover, compounds (11, 4, 10, 17, 12 and 3) showed cytotoxic activity (IC50 equals 2.08, 2.89, 3.02, 3.09, 3.15, and 3.36 μg/mL, respectively). Results of the biochemical investigations indicated that 5-flurouracil and doxorubicin caused significant changes in the level of all parameters tested while treatment with the tested compounds showed slight, moderate, or no significant changes.
Conclusions
In the present investigation, we report the design and synthesis of a new series of 2, 6 disubstituted pyridine, which include in their structures various heterocyclic moieties directly attached to the pyridine nucleus at positions 2 and 6 aiming to potentiate the biological activity as anticancer. In addition, our study was extended to the synthesis of additional derivatives containing a substituted pyridine moiety directly attached to diazepine, indole, benzimidazole, pyrimidine, and imidazole nucleus at positions 2 and 6. The anticancer activity data indicate that compounds 11, 4, 10, 17, 12, and 3 were the most active compounds against HEPG2.
References
M. Ma zik, D. Bläser, R. Boese, Tetrahetron 55, 12771 (1999)
S. Chang, D.A. Hamilton, J. Am. Chem. Soc. 110, 1318 (1988)
S. Chang, D. Van Engen, E. Fan, D.A. Hamilton, J. Am. Chem. Soc. 113, 7640 (1991)
H.M. Elnagdi, A.S. Ghozlan, M.F. Abdel-Razik, S.A. Maghraby, Chem. Synop. 5, 116 (1991)
A.F. Attaby, M.S. Eldin, F.M. Abdel-Razik, Phosphorus Sulfur Silicon Relat. Elem. 21, 106 (1995)
A.K. Asadov, R.N. Burangulova, F.H. Guseninov, R.Z. Gilmanov, I.P. Phaljachov, Chem. Heterocycl. Compd. 39, 392 (2003)
M. Miletin, J. Hartl, M. Dolezal, Z. Odlerova, K. Kralova, M. Machacek, Molecules 5, 208 (2000)
A. Abdel-Rahman, E.A. Bakhite, E.A. Al-Laifi, J. Chin. Chem. Soc. 49, 223 (2002)
C.S. Rao, V. Venkaleswarlu, G. Achaiah, Bioorg. Med. Chem. Lett. 16, 2134 (2006)
A.H. Todd, Brit 203, 149 (1970)
G.A. Younghale, U.S.P 288, 440 (1980)
G.A. Younghale, Chem. Abstr. 96, 6596c (1982)
F. Li, Y. Feng, Q. Meng, W. Li, Q. Wang, F. Tao, ARKIVOC 1, 40 (2007)
M.A. Abdel-Rahman, M.J. Morsy, S. El-Edfawy, A.H. Ameneand, Pharmazie 54, 347 (1999)
M.R. Abdel-Rahman, M.J. Morsy, F. Hanafy, A.H. Abdel-Salam, Pharmazie 54, 3 (1999)
Z. El-Gendy, M.R. Abdel-Rahman, Indian J. Heterocycl. Chem. 4, 295 (1995)
M.R. Abdel-Rahman, Trends Heterocycl. Chem. (India) 8, 187 (2002)
T. Mosmann, J. Immunol. Method 65, 55 (1983)
J. Valdez, R. Cedillo, A. Hernández-Campos, L. Yépez, F. Hernández-Luis, G. Navarrete-Vázquez, A. Tapia, R. Cortés, M. Hernández, R. Castillo, Bioorg. Med. Chem. Lett. 12, 2221 (2002)
G.H. Garg, C. Prakash, J. Med. Chem. 4, 175 (1971)
J.C. Shishoo, S.U. Pathak, S.I. Rathod, S.K. Jain, Indian J. Chem. 38B, 684 (1999)
M. Mostafa, I.H. Heiba, A.H. Amina, B.A. Amany, G. Marwa, J. Am. Sci. 7(1), 1063 (2011)
M. Lounasmaa, A. Tolvanen, Nat. Prod. Rep. 17, 175 (2000)
S. Hibino, T. Chozi, Nat. Prod. Rep. 18, 66 (2001)
P. Skehan, R. Storeng, D. Scudiero, A. Anne Monks, J. McMahon, D. Vistica, J. Warren, H. Bokesch, S. Kenney, M. Boyd, J. Natl. Cancer Inst. 82, 1107 (1990)
T. Zor, Z. Selinger, Anal. Biochem. 236, 302 (1996)
M.H. Abo-Ghalia, A.M. Soliman, Acta Pol. Pharm. 57, 53 (2000)
K. Spencer, C.P. Price, Clin. Chim. Acta 95, 263 (1979)
A. Mays, Lab. Anim. Sci. 19, 838 (1969)
V.V. Joseph, Clin. North Am. Exot. Anim. Pract. 2, 689 (1999)
A.M. Soliman, L.M. Faddah, Egypt. J. Bilh. 16, 8 (1994)
A.H. Garde, A.M. Hansen, L.T. Skovgaard, J.M. Christensen, Clin. Chem. 47, 1877 (2001)
E.B. Rietz, G.C. Guilbault, Clin. Chem. 23, 286 (1977)
D.C. Turnell, Ann. Clin. Biochem. 22, 217 (1985)
N.L. Guilbaud, F. Kraus-Berthier, V. Meyer-Losic, C. Malivet, M. Chacun, F. Jan, S. Tillequin, M. Michel, B. Koch, Clin. Cancer Res. 7, 2573 (2001)
N.K. Sathish, V.S. Rajendra Prasad, N.M. Raghavendra, S.M. Shanta Kumar, Y.C. Mayur, Pharm 77, 19 (2009)
A. Espinosa, J. Marchal, A. Aránega, M. Gallo, S. Aiello, J. Campos, Farmaco 60, 91 (2005)
P. Kamalakannan, D. Venkappayya, J. Inorg. Biochem. 90, 22 (2002)
Acknowledgments
The authors are grateful to the National Research Center for financial support of this work (Project No: 8040204).
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s11164-017-2877-8.
Rights and permissions
About this article
Cite this article
Bassyouni, F.A., Tawfik, H.A., Soliman, A.M. et al. Synthesis and anticancer activity of some new pyridine derivatives. Res Chem Intermed 38, 1291–1310 (2012). https://doi.org/10.1007/s11164-011-0413-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-011-0413-9