
Abstract
Selenium is usually known as the ‘double-edged sword element’ for its dual toxic and beneficial character to health. Since the pioneer works by Schwarz and Foltz on the relationships between selenium deficiency and liver, muscle and heart diseases, many efforts have been undertaken to better understand the role of selenium in health. At the same time, an increasing number of publications have appeared during these last years on the selenium physico–chemical interactions within the environment. Both types of research represent ongoing efforts to correctly estimate the bioavailability of selenium species for health and the environment. Redox reactions, diffusion, adsorption and precipitation processes or interactions with organic matter and biota govern the speciation and mobility of selenium in the environment. This review intends to emphasize and collect the important advances made during these last years in the mechanistic understanding of processes which govern selenium cycling and bioavailability, like adsorption at the mineral/water interface, precipitation of elemental selenium, or bioavailability of nanoscaled precipitates. The advent of powerful spectroscopic techniques, like X-ray absorption spectroscopy, has allowed the structural description of adsorption and substitution processes that selenium undergoes in a variety of minerals. These and other structural details about selenium precipitates are reviewed here, together with their relationships to the bioavailability of the element in the environment.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Selenium (Se) was identified as a new substance in 1817 by Jöns Jacob Berzelius when studying a method to prepare sulfuric acid from sulfur-bearing rocks. A precipitate with a brownish/red color and tellurium (Tellus—Greek word meaning ‘Earth’) smell-like led him to name it selenium (from the Greek word ‘Selhum’, the ‘Moon’, (Berzelius 1817, 1818). The oldest histories related to selenium have their origin in the Middle Age. Marco Polo, during his travels in China in the 13th century, reported a hoof disease in horses that was later identified as selenium toxicosis (selenosis) (Oldfield 2006).
Selenium has been identified during long time as a dangerous substance because of its toxicity and it has been only in the recent past that its physiological importance as a trace element fundamental to health has been assessed. Schwarz and Foltz (1957), linked the existence of liver, muscle and heart diseases with selenium deficit. The Kashin–Beck disease, an articulation disease found in children from the north of China, north of Korea and Siberia, was shown to be related to selenium deficit in soils. A dietary selenium supplement helped to eradicate these health problems. Selenium is known to be a ‘double-edged sword’ element, having one of the narrowest ranges between dietary deficiency (<40 μg day−1) and toxic levels (>400 μg day−1) (Levander and Burk 2006). It is thus essential to understand the physico–chemical and biological processes that govern its bioavailability in the environment. Past research has demonstrated that it is an essential nutrient provided by plants as part of animal diets, even though it is not an essential element for plants. The evaluation of the limits between toxicity and deficiency is a problem with a difficult approach, as selenium toxicity and deficiency are influenced by bioavailability of selenium species in the environment, a factor entangled with many environmental variables, and by the bioaccumulation of selenium in some organisms like plants.
Bioavailability is a biogeochemical concept which relates the concentration of an element to its availability to organisms. It governs the uptake of selenium in animals and plants, influencing the biochemical functions that selenium fulfills in the organisms. Several studies over the past 30 years have reported detailed physico–chemical processes affecting bioavailability of selenium in different environments. Inorganic selenium oxyanions have been used as dietary supplements in fertilizers, and bioavailability of organic selenium compounds to different plants and animals has been largely studied. Advances in the field of drug delivery systems have recently focused some attention in the bioavailability of nanoparticles of elemental selenium, a chemical species that has been classically seen as biologically inert. Recent research on bacterial-mediated reduction of selenium oxyanions to elemental selenium has shown that different structures and nanoparticle sizes are bio-synthesized, enlarging the panorama of elemental selenium allotropes to a still uncertain number of new structures. In addition, the finding that solubility of certain elements increases when they are scaled down to nanometer sizes poses new questions about the mobility and bioavailability of elemental selenium nanoparticles and on their fate in the environment. This review intends to give a summary about the most important biogeochemical factors affecting selenium bioavailability in soils, waters and sediments. Special emphasis has been given to the structural characteristics of the sorption processes, and to review the new findings about elemental selenium nanoparticles of the recent years.
1.1 Structural and aqueous chemistry
Selenium is a nonmetal, of atomic number 34, atomic mass 78.96 and electronic configuration [Ar] 3d10 4s2 4p4. It is present in nature in five oxidation states: (−II), (−I), (0), (+IV) and (+VI). SeO4 2− and SeO3 2− are soluble oxyanions known respectively, as selenate and selenite.
-
The fully oxidized Se(VI) form, selenate, exists as a tetrahedral (point symmetry T d; see Fig. 1) oxyanion in solution as biselenate (HSeO4 −) or selenate (SeO4 2−) with a pK a2 = 1.8 ± 0.1 (Seby et al. 2001). The doubly protonated species do not exist under natural conditions, since pK a1 = − 2.01 ± 0.06 (Seby et al. 2001). The aqueous chemistry of selenate and sulfate are quite similar, and researchers have observed that similar surface complexes form with both oxyanions (Peak and Sparks 2002; Wijnja and Schulthess 2000b). Selenate is the predominant species at high redox potentials, e.g. in alkaline sub-desertic soils, and it is considered very soluble and with low adsorption and precipitation capacities (Fordyce 2007).
Fig. 1 
Selenite and selenate structures. Selenite has a C3v symmetry, with pyramidal-shape, which is reduced to C1 upon deprotonation. Selenate has a Td tetrahedral symmetry, reduced to C3v upon single protonation, and Cs upon double protonation
-
Se(IV) exists as the pyramidal oxyanion selenite (SeO3 2−), with point symmetry C3v (see Fig. 1) in its unprotonated state, and as the gas selenium dioxide, SeO2. Selenite is a weak acid that can exist as H2SeO3, HSeO3 −, or SeO3 2−, depending upon solution pH (pK a1 = 2.70 ± 0.06 and pK a2 = 8.54 ± 0.04, (Seby et al. 2001). In the moderate redox potential range, selenite is the major species and its mobility is mainly governed by sorption/desorption processes on various solid surfaces such as metal oxyhydroxides (Balistrieri and Chao 1987; Papelis et al. 1995; Saeki et al. 1995), clays (BarYosef and Meek 1987) and organic matter (Gustafsson and Johnsson 1994; Tam et al. 1995).
-
Se(−II) and Se(−I) are stable under strongly reducing conditions in a variety of metallic selenides and in a high number of organic compounds. Other compound, H2Se, is present in nature as a product of microbial processes (Oremland et al. 1994). Values for the acidic dissociation constants are pK a1 = 3.8 ± 0.1 and pK a2 = 14 ± 1. Comments on these large uncertainties are given in (Seby et al. 2001). Se(−II) is present in some metastable anions as selenosulfate SO3Se2− (Ball and Milne 1995), which is largely used as a source of selenium for the formation of metallic selenides in industrial processes (see e.g., Riveros et al. 2003).
-
Elemental selenium, Se(0), is present in nature as a non-soluble form, forming, at least, 11 different allotropes (Minaev et al. 2005): two amorphous phases (red, black, see Jovari et al. 2003), three different allotropes of crystalline monoclinic selenium (deep red), crystalline trigonal (or rhombohedral) selenium (metallic gray), orthorhombic selenium and three allotropes of crystalline cubic selenium formed under high-pressure conditions. Detailed structural information of some of these allotropes is given in Table 5.

A recent exhaustive compilation of published data of inorganic selenium–metal solubility constants, selenium acid-base equilibrium constants, standard potentials of selenium redox couples and some complex dissociation constants for the soluble species of selenium can be found in Seby et al. (2001) or in Olin et al. (2005).
Besides inorganic forms of selenium, non-volatile organic selenides such as seleno amino-acids and volatile methylated forms of selenides, principally dimethylselenide and dimethyldiselenide, can occur in surface water, groundwater and waste water (Cutter 1982; Lenz et al. 2006). Table 1 shows the main selenium-containing organic compounds found in nature.
1.2 Isotopes and radioactivity
There are five stable selenium isotopes (see selenium isotopes in Table 2). One of them, 82Se, occurs naturally and it has a decay half life of 1020 years. 79Se is of special interest nowadays due to the current growing interest in the nuclear waste disposal research, as it is a fission product of 235U (Shongsheng et al. 1997; ANDRA 2005). After long time storage, the main selenium radioactivity will be produced from this long-lived radionuclide, 79Se (half-life: 2.95 × 105 year). For this reason, the total cumulative dose of radioactivity could be affected by an eventual release of this radionuclide from waste repositories to the biosphere (ANDRA 2005). This has motivated the study of materials with high selenite retention capacities (the thermodynamically most stable chemical species in these environments), and the study of selenite interaction with clays (Breynaert et al. 2008; Bruggeman et al. 2007, 2005; Peak et al. 2006), cements (Bonhoure et al. 2006; Johnson et al. 2000b; Mace et al. 2007; Pointeau et al. 2006) or clays in the presence of Fe(II) (Charlet et al. 2007).
75Se is another isotope relevant in environmental studies. It has a half-life of 120 days. It is a gamma-emitter, which allows using it as a tracer, or a label material in a variety of studies due to the low detection limits achievable with radioactivity detectors (in the order of 10−9 mol selenium). 75Se–selenomethionine, 75Se–selenite and 75Se–selenate containing solutions are used as radioactive diagnostic materials in medical science applications (Irons et al. 2006), as tracers in hydrological studies (Li and Zheng 1990) or in biological investigations (Luoma et al. 1992).
2 Selenium cycling in the environment
2.1 Selenium in rocks, soils and waters
Selenium presents a complex chemical behaviour that allows it to combine with a variety of elements in nature. This makes selenium compounds widespread over all the Earth compartments: rocks, soils, waters and air. However, selenium is considered a trace element in the Earth crust, with average concentrations ranging from 0.05 to 0.09 mg kg−1 (Neal and Sposito 1989; Taylor 1985).
Rocks are the primary source of the element in the terrestrial system (Fleming 1980; Neal 1995; Rosenfeld and Beath 1964). The total amount of selenium in rocks comprises 40% of the total in the Earth’s crust, mainly in sandstone, quartzite and limestone (Wang and Gao 2001). Biogeochemical processes as weathering, rock–water interactions and biological activity control the transport of selenium from rocks to other compartments, distributing it over the planet and governing the uneven distribution of the element over the Earth. Sedimentary rocks present concentrations with a mean value of 0.0881 mg kg−1, as found by (Tamari et al. 1990). Because these major rock types account for most of the Earth’s surface it is straightforward to estimate that selenium-deficient environments are by far more widespread than selenium-excessive ones (Haygarth 1994). Average selenium concentrations in selected representative environmental compartments are given in Table 3. Contrary to sedimentary rocks, clay fractions in sediments, shales, phosphatic rocks, coals or organic-rich deposits contain usually high concentrations of selenium. Phosphatic rocks contain an average of 1 mg kg−1 (Charter et al. 1995), reflecting structural similarities between phosphate and selenate oxyanions. Selenium is often found in minerals substituting for sulfur, as in Chalcopyrite, Pyrite, or other common metallic sulfides (Masscheleyn et al. 1991; Neal and Sposito 1989).
Soil selenium concentration is mostly influenced by selenium concentration in the parent materials. The average selenium content in most soils ranges from 0.01 to 2 mg kg−1 (world mean 0.4 mg kg−1, Fordyce 2007). However, high concentrations have been reported in some seleniferous areas, reaching 1,200 mg kg−1 (Fleming 1980; Jacobs 1989; Neal 1995). On the other side, Dhillon and Dhillon (2003) proposed a value of 0.1 mg kg−1 for the limit in concentration under which soils are considered deficient in selenium. Soils containing more than 0.5 mg kg−1 of selenium are considered seleniferous soils, (Dhillon and Dhillon 2003), as the forages produced on such soils have more selenium than the maximum permissible level for animal consumption (2–5 mg kg−1 dry matter, regardless of the species (Mayland 1994). Changes in topographical features and leaching/erosion processes play an important role in the development of seleniferous soils in the different parts of the world. Areas of toxic, adequate and deficient selenium levels exist side by side. Soils with elevated levels of selenium are found in many countries including China (Wang and Gao 2001), India (Dhillon and Dhillon 2003), Ireland (Seby et al. 1997) and USA (Presser 1994). Usually, higher selenium concentrations are found in soils derived from Precambrian organic-rich carbonates, like in China (Ihnat 1989), or from Cretaceous shales, containing an average value of 4.5 mg kg−1 with maxima reaching up to 1,200 mg kg−1 (Lakin 1972). For instance, ~2 mg kg−1 occurs in the Cretaceous shale (Colorado, USA) and related shales, which are the parent materials for much of the seleniferous soils in the Northern Great Plains of the USA and the Prairie region of Canada (Ihnat 1989). Seleniferous marine sedimentary rocks from the coastal ranges in the San Joaquin Valley (California, USA) are the main source of selenium in waters at the Kesterson Reservoir, which is one of the most studied selenium-contaminated sites (Presser and Piper 1998). Weathering caused by infrequent storms and transport of alluvial deposits are key processes in the mobilization of selenium from parent rocks or sediments in this region, raising the selenium availability in the waters of the San Joaquin Valley (Presser and Swain 1990). Seleniferous rocks (like pyritic shales) in this area have mean values of 8.9 mg kg−1 of selenium. Soils concentrations are lower, ranging from 0.14 to 0.68 mg kg−1. The high presence of pyrite suggests that selenium is incorporated in its structure by replacing sulfur (Presser and Piper 1998). An extensive review on seleniferous soils has been compiled by Dhillon and Dhillon (2003), and a data compilation concerning selenium concentrations in rocks and seleniferous soils can be found in Rosenfeld and Beath (1964).
The concentrations and chemical forms of selenium in soils or drainage water are governed by various physical and chemical factors, including pH, chemical and mineralogical composition, adsorbing surface, and oxidation–reduction status (Dhillon and Dhillon 1999). These processes include as well weathering of elemental selenium or metallic selenides (selenium-bearing FeS2, FeSe2), and oxidation of Se(−II) to selenite (under acidic conditions) or selenate (under alkaline conditions) (Presser and Piper 1998). The general trend for the occurrence of the different species of selenium in soils is deduced directly from their physico–chemical properties: the oxyanions selenite and selenate exist predominantly in the aqueous solutions of aerated alkaline soils, while insoluble selenides and elemental forms of selenium are mostly present in poorly aerated, acid, organic-rich soils of strong reducing conditions (Fordyce 2007; Neal 1995). However, this is a very general rule that is not always fulfilled.
Because selenium escapes as high-temperature gases during volcanic activity, selenium concentrations left in volcanic rocks such as basalts and rhyolites are usually very low (Fleming 1980; Jacobs 1989; Neal 1995; Nriagu 1989). A mean value of 0.0086 mg kg−1 has been reported for igneous rocks in Japan (Tamari et al. 1990). On the other side, volcanic-ashes produced simultaneously to volcanic gases and soils and sediments developed from these ashes are usually rich in selenium (Byers et al. 1938; Davidson and Powers 1959; Ihnat 1989; Lakin 1972). However, volcanic soils have been usually reported as having low soil contents of selenium (Rayman 2000). The origin in this apparent discrepancy is two folded: on one hand, some soils in volcanic regions present specific mineralogical characteristics which lead to the immobilization of large quantities of organic matter (Wang and Chen 2003); selenium associates with organic matter being immobilized and thus becomes unavailable to the biota (Wang and Gao 2001). Clear difference should be made then between low bioavailability and deficiency, terms that have been used in a confusing way by some authors (Rayman 2000; Reilly 1997; Sirichakwal et al. 2005). On the other hand, little is known about the specific interactions of selenium oxyanions (predominant species of inorganic selenium in volcanic soils) and the mineralogical components specific to these soils (imogolite and allophane), which could be responsible of the low selenium bioavailability. More research is needed to better understand these interactions at a mechanistic level.
Volcanic soils are usually very porous acidic soils. Reducing conditions prevail only in some paddy fields, where selenite can be transformed into insoluble forms, such as elemental selenium (Yamada et al. 1999). Otherwise, in these acidic environments, selenate is unstable and leaches easily (Neal et al. 1987a, b), and selenite is the major species (Nakamaru et al. 2006). Selenium bioavailability in these soils is thus controlled by the adsorption of selenite on mineral surfaces and organic matter. Andosols are the soils with higher selenium distribution coefficients (Nakamaru et al. 2005). The particle/solution distribution coefficient (K d) of selenium in Japanese agricultural andosols are as high as 600–800 L kg−1. Sequential extractions performed on andosol samples show high correlation coefficients between occurrence of short-range-ordered aluminosilicates such as allophane, imogolite and active aluminum, and selenium K d values (Nakamaru et al. 2005). The high specific surface areas and positive charges of these volcanic soil components may be the responsible for this high affinity of selenium. Other soil components with high affinity for selenium are iron–humus complexes (Ihnat 1989) and poorly crystalline iron oxides such as ferrihydrite, as e.g. for New Zealand soils (John et al. 1975). In Hawaiian volcanic soils, the selenium content varies from 1 to 20 mg kg−1, but this selenium is unavailable for vegetation, due to its complexation with iron and aluminum bearing minerals (Davidson and Powers 1959). Volcanic soils are thus an example of soils with high total selenium content but low bioavailability. This contrast requires further structural studies to be fully understood.
Hydrological systems are one of the most significant transport processes for selenium (Cutter and Bruland 1984). Nriagu (1989, 1991) has undertaken a detailed study of these processes estimating that fluxes of about 14,000 tons of selenium per year are transferred along aquatic pathways from the continents to the oceans, via either selenium-bearing sediments, responsible of roughly 85% of this transport (Cutter and Bruland 1984), or via soluble species. Leaching of selenium species from soil to groundwater is another component of selenium hydrological transport. However, processes governing this leaching are poorly understood, due to the heterogeneity of the water/rocks, soils and sediment interfaces (Dhillon et al. 2008). Mean residence time of selenium in the deep ocean is about 1,100 years, i.e., is of the order of water residence time (Ihnat 1989). This is typical of highly soluble species, poorly adsorbed on sedimentary particles.
2.2 Selenium associations with organic matter
Organic matter (OM) plays a fundamental role among the soil biogeochemical variables (pH, Eh, surface charge, aggregation, cation exchange capacity…) governing the mobility of trace elements by complexation processes (Filella and Town 2003). There is a general agreement on the fact that selenium is associated generally with OM in aquatic and terrestrial environments (Abrams et al. 1990; Zhang and Moore 1996). This is reflected in the distribution coefficient for organic matter used for selenium by the international atomic energy agency (IAEA 1994). The (K d) is often used to predict the sorption behavior of an element in soil. The (K d) for organic matter associated selenium is twice that for soil clays, with a value K d SeOM = 1,800 L kg−1. However, mechanistic processes of the interactions governing selenium/OM associations are still poorly understood. The organic compounds found in soils and waters are highly heterogeneous (Sutton and Sposito 2005), which makes more complicated the study of OM properties, like its ion complexation ability, or the biogenic processes underlying their formation. Selenium is associated with OM through two different kinds of processes.
2.2.1 Selenium complexation to organic matter
Selenium can be immobilized through the formation of selenium metal–humic ternary complexes, as suggested by Gustafsson and Johnsson (1994) for Fe(III)–OM complexes, althought little is known about the ions involved in this process. Associations of selenite with natural polysaccharides of different molecular weight has been studied by Ferri and Sangiorgio (2001). They showed that formation constants were independent of the charge of the organic ligands, the strength of the complexes stability being more dependent of their structure. Abiotic selenium fractionation between humic and fulvic substances has been studied by some authors who related the effects of these associations to the bioavailability of selenium to plants (Kang et al. 1991; Zhang and Moore 1996). Wang et al. (1996) reported a decrease in the selenium uptake by plant roots due to interactions between selenite and fulvic acid. The same effects were found to affect selenium bioavailability to animals (Wang et al. 1996). Wang and Gao (2001), showed that selenium is immobilized in soils in the northeastern regions of China, where selenium deficiency diseases (Keshan and Kaschin–Beck diseases) are widespread. Selenium content in these soils is not low (it ranges from 0.1 to 0.3 mg kg−1), but these soils are very rich in OM, which appears to be the responsible of the selenium immobilization and of its low bioavailability. The retention of negatively charged selenium oxyanions by OM highlights the importance of ternary selenium-cation–OM complexes. These complexes seem to be important mechanisms of selenium immobilization by OM, although little is know about their structure and stability.
Other environments where selenium immobilization by OM is a relevant factor are the geological environments dedicated to nuclear waste disposal (see Sect. 1.2). Bruggeman et al. (2007, 2005) have recently studied the interaction between selenium oxyanions and dissolved humic substances derived from such environments. Thermodynamically, and due to the presence of a reducing phase (FeS2), selenium oxyanions are expected to be reduced to elemental selenium or Se(−II), and to be removed from solution by precipitation (Bruggeman et al. 2005). However, in the case of selenate, no speciation change was observed in the experiments of Bruggeman et al. (2005, 2007). This implies that selenate is kinetically inert and that it is meta-stable in groundwaters rich in humic substances in equilibrium with reducing solid phases, and that it may only be reduced by micro-organisms. In the case of selenite equilibrated in the presence of humic substances, the selenium solution speciation changed drastically, accompanied by a decrease of the total selenium dissolved concentration after centrifugation. In addition, a newly-formed selenium species was detected, but could not be identified. The authors propose that this new species was associated with the dissolved humic substances (Bruggeman et al. 2007). Thus an abiotic route for the transformation of selenite to organically-bound selenium was observed to exist, although the exact nature of this species is not known. OM–metal complexes are thus relevant factors that may play an important role in the mobility and bioavailability of selenium in anoxic environments. There is no information in the literature about the degree of association between reduced selenium species (elemental selenium and selenides) with stable humic substances. Transport of Se(0) bound to bacterial walls has been suggested by Fevrier et al. (2007) and similar colloidal-mediated transport could be imagined for reduced species of selenium adsorbed onto OM colloids.
The adsorption of adsorption of selenium oxyanions on aluminum oxides is affected by competitive effects of the oxyanions with some organic acids. While oxalic and citric acids compete with selenite and selenate for adsorption sites, formic and acetic acids do not modify the yield of adsorption of selenite, and even promote the adsorption of selenate (Dynes and Huang 1997; Wijnja and Schulthess 2000a, b). Formic and acetic acids form outer-sphere complexes at the aluminum oxide/water interface, as opposed to oxalic and citric acids that are adsorbed by an inner-sphere mechanism (Dynes and Huang 1997; Wijnja and Schulthess 2000a, b).
Fulvic acids have been shown not to have any effect on sorption of selenite on aluminum oxides (Zuyi et al. 2000), but to increase the total adsorption of selenite on amorphous iron oxides (Tam et al. 1995). These effects can be explained by the high affinity of selenium for aluminium oxides and for some iron oxides, as will be discussed later on, or by selenium–Fe–humic substance ternary complexes bound to the iron oxides.
A recent study by Ogaard et al. (2006) shows the effect of manure on retention of selenium in soil. Addition of cattle manure in combination with selenite and selenate reduced the adsorption of both anions to a loam soil in batch experiments. The results were explained by the content of low-molecular-weight organic acids in the manure (as acetic, formic and oxalic acids) which compete with selenium for the sorption sites (Ogaard 1996).
There is a lack in the literature of mechanistic descriptions of these abiotic processes. This is probably due to the major impact of biotic processes on the cycling of selenium (Dowdle and Oremland 1998). For this reason, correct estimations on the degree of selenium associations with OM cannot be made presently, which suggest that more research should be done in the future in this direction.
2.2.2 Biotic transformations
Selenium speciation, mobility and bioavailability are highly affected by the presence of microorganisms in the environment. Their main influence on the bioavailability is through the control of selenium oxidation state, which directly relates to the solubility of different selenium compounds (see Sect. 3.3). Different biotic pathways have been identified nowadays by which a selenium oxyanion can be reduced to Se(0) or Se(−II). Bacteria can use selenate and selenite as terminal electron acceptors in energy metabolism (dissimilatory reduction). They can also reduce and incorporate selenium into organic compounds (assimilatory reduction). Other relevant processes are alkylation, dealkylation and oxidation (see Sect. 3.3). Many authors have studied the influence of these microbial processes on the selenium cycling in soils. One example is the study on the distribution of organic selenium in selected California soils (Abrams et al. 1990). Extracted organic selenium compounds were fractionated into humates and hydrophobic and hydrophilic fulvates. Part of the selenium associated with the OM was identified as selenomethionine, which suggests that micro-organisms are involved in the selenium cycling in this area. Gustafsson and Johnsson (1994) added 75Se-labelled and unlabelled selenite to two slightly acidic forest floors and to an organic-rich brown lake water. It was observed that the forest floors fixed most of the added selenite by means of microbial reductive incorporation, preferentially into low-molecular-weight fractions of humic substances. Inorganic complexation of selenite to metal–humic complexes seems to be an important process which can control the kinetics of biotic selenium transformations. Zhang and Moore (1996) added selenate to a wetland sediment and observed that formation of organically-bound selenium (i.e., formation of both organic selenium compounds and selenium adsorbed to OM) was one of the most important processes removing selenate from water, together with selenium reduction to the elemental form. Both processes were interpreted as being the result of uptake or transformation by micro-organisms. Resuming, it can be stated that most experiments dealing with the interaction or incorporation of different selenium forms with soil OM rely on microbial action. Only few authors (Ferri and Sangiorgio 2001; Gustafsson and Johnsson 1994) have specifically focussed on the abiotic route discussed above.
2.3 Atmospheric selenium
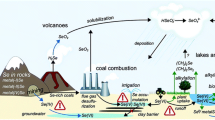
Volcanism contributes significantly to the different geochemical cycles of selenium, being one of the natural sources of selenium to the atmosphere and hydrosphere. The high chemical similarity of selenium and sulfur implies that selenium accounts for up to 5% of the amount of sulfur emitted by volcanoes in the form of sulfur dioxide (SO2), forming a gaseous flux of SeO2(g) instead (Seiler 1998). This atmospheric source of selenium accounts for 300–1,000 tons of selenium emitted to the atmosphere per year, which is in the same order of magnitude of the total selenium released by all the natural sources in Earth (4,500 tons per year) (Nriagu 1991). Different authors report different values for the amount of naturally emitted gaseous selenium to the atmosphere, all of them in the order of magnitude of the 4,500 tons per year estimated by Nriagu (1991). A review containing absolute values by different authors is provided by Wen and Carignan (2007).
Haygarth et al. (1994) have reported on the importance of atmospheric deposition processes as a mean of soil and plant fertilization in The UK. The author noted that wet deposition led to a 15% increase in soil selenium concentration in the last century, while it contributed to 33–82% of plant leaf selenium uptake. Deposition is heavily influenced by geographical proximity to an emission source, with highest levels associated with industrial and coastal zones, being considerably higher during the winter months. Industrial processes and, in particular, coal combustion, are important sources as well of atmospheric particulate selenium. Selenium containing dusts derived from volcanoes and wind erosion of the Earth’s surface contribute to the transfer from land-to-atmosphere selenium with 180 tons per year, while suspended sea salts contribute to an additional amount of 550 tons per year more (Nriagu 1989).
Terrestrial biogenic sources are one of the main atmospheric selenium sources together with marine biota (Nriagu 1989). Most of this gaseous biogenic selenium is thought to be in dimethylselenide form, which is the biogenic species derived from microbial activity. However, these compounds have short lifetimes in the atmosphere, as they suffer decomposition reactions with either ozone or the nitrate and hydroxyl radicals (Atkinson et al. 1990). Several factors affect the selenium cycling in the atmosphere. Soil moisture content may influence heavily the volatilization of selenium. As a result of dimethylselenide’s high solubility in water, soil moisture decreases the rate of gaseous diffusion in soil (Zhang and Frankenberger 1999). Other soil factors known to affect the rate of dimethylselenide volatilization are temperature, pH, and redox status (Frankenberger and Karlson 1994). These soil factors, as well as soil moisture content, directly control microbial activity and, hence, the selenium biomethylation process. The addition of organic amendments to selenium-contaminated soil and sediment increases the selenium volatilization rate by stimulating indigenous microorganisms to methylate selenium (Doran and Alexander 1977a; Karlson and Frankenberger 1989). Organic carbon is often a limiting factor to soil microbial methylation of selenium. Microorganisms capable of demethylating volatile selenium compounds have also been isolated (Doran and Alexander 1977a; Oremland and Zehr 1986). Thus, demethylation of dimethylselenide may be an additional significant rate-controlling factor of selenium volatilization. Other authors noticed the unstability of dimethylselenide in the atmosphere as a result of its reactions with atmospheric OH, NO3 and O3 free radicals (Atkinson et al. 1990) and oxidation catalyzed by suspended soil particles (Martens and Suarez 1999).
Micro-organisms are capable of performing a process called bio-methylation, which has been referred by some authors as a selenium remediation process for soils from seleniferous areas or soils that were amended with selenium (Abu-Erreish et al. 1968; Banuelos and Lin 2007; Chasteen and Bentley 2003; Doran and Alexander 1977a; Francis et al. 1974; Frankenberger and Karlson 1994; Ganje and Whitehead 1958; Zieve and Peterson 1981). This is a complex process consisting in a series of redox reactions driving selenium from oxidation states +IV or +VI to −II followed by a methylation reaction by which different gaseous or aqueous selenide species are formed, with 1 or 2 methyl groups (Chasteen and Bentley 2003; Eisler 1985). This mechanism is considered to be an enzymatically driven phenomenon (Ranjard et al. 2002), although abiotic methylation pathways have shown to be also possible (Guo et al. 2003). Thayer (2002) proved that abiotic methylation of selenium may occur from inorganic selenide–metal cation complexes in the presence of strong carbo-cation donors such as halomethane or sulphonium compounds, and Amouroux et al. (2000) have shown that stable volatile species can be formed from bio-organic selenium (seleno-methionine and seleno-cystine) in the marine photic zone.
Microbial methylation is responsible for selenium volatilization from the Earth to the atmosphere. This process is affected by bioavailability of selenium, carbon sources, oxygen availability and temperature, as shown by Haygarth (1994), and it is supposed to be an important pathway for land-to-atmosphere selenium flux. However, little attention has been paid to the fate of the organic selenium species produced by methylation (Dungan et al. 2002).
An extensive review on selenium biomethylation by microorganisms and plants has been published by Chasteen and Bentley (2003).
Methylated selenium is also to a large extent produced in the ocean. Biogenic volatile flux from the ocean is considerably greater than those coming from the Earth’s surface (Wen and Carignan 2007). Methylated selenides related to the algae’s biomass have been found to be expelled to the atmosphere in three major European estuaries, the Gironde (F), the Rhine (NL) and the Scheldt (B/NL) estuaries (Tessier et al. 2002). Some microalgae have been studied to be used as possible detoxification mechanisms, as in the case of the single-celled freshwater microalgae Chlorella sp. (Neumann et al. 2003). This has been identified as a high rate metabolic transformer of selenate to volatile dimethylselenide, showing selenium-volatilization rates orders of magnitude higher than those similarly measured for wetland macroalgae and higher plants (Neumann et al. 2003).
An important input to atmospheric selenium is the transport of selenium associated with wind-eroded particules, an input estimated to be equal to up to 180 tons per year, with particles containing up to 0.5 mg selenium kg−1 (Haygarth 1994). Mobilization of sea salts from marine systems is thought to be responsible of an input 550 tons of selenium per year to the atmosphere. The reader is referred to an excellent review on selenium cycling in atmosphere written by Wen and Carignan (2007).
2.4 Anthropogenic sources
Selenium is released to the atmosphere through coal and petroleum fuel combustion, and during the extraction and processing of a variety of elements, such as copper, zinc, uranium, phosphate or lead. Coal combustion is thought to be the main selenium releasing anthropogenic process, being responsible of about 7,300 tons of selenium per year (Yudovich and Ketris 2006). Selenium is one of the most volatile trace elements in coal, and is largely released in vapor phase to the atmosphere, mainly as the SeO2(g), SeO(g) and solid selenium species (Yan et al. 2001). Haygarth et al. (1994), demonstrated from selenium herbage historical data a positive correlation between the highest concentrations found in 1940 and 1970 with the high coal combustion activity observed in these years. Nowadays, Asiatic countries, especially China, account for about half the worldwide increase in global coal production, which may result in a new environmental problem. These concerns have stimulated some research on the treatment of wastes derived from flue gas desulphurization systems. These systems are used to decrease sulfur dioxide emissions, but result in wastewaters containing high concentrations of arsenic, selenium and mercury, in addition to sulfur (Sundberg-Jones and Hassan 2007). Anthropogenic emissions of selenium to the environment account for 50–65% of the total emissions at the global scale (Mosher and Duce 1987), a number that has been increasing ever since the onset of industrialization (Wen and Carignan 2007). Around 88,000 selenium tons per year are released globally to the environment from anthropogenic activities (Fordyce 2005).
On the other hand, anthropogenic activities in the mid-1970s such as irrigation, increased the release of selenium to the environment, which had a strong impact in the selenium cycling in the biosphere and hence in the occurrence of selenium-rich regions. The use of phosphate pesticides mobilizes a big amount of selenium to the environment: Kabata-Pendias and Pendias (1984), reported selenium concentrations in phosphate fertilizers ranging from 0.5 to 25 mg kg−1 (some phosphatic rocks contain high concentrations of selenium, 1–178 mg selenium kg−1; Robbins and Carter 1970). These are high values, considering that limestones have a selenium background concentration of only 1 mg kg−1 (Ihnat 1989). An example of selenium application to soils is the case of low-selenium soils in Finland, described by Hartikainen (2005). Finnish soils are not exceptionally low in selenium, but they are young acidic and weakly weathered, and high in adsorptive oxides. All these soil characteristics contribute to the low bioavailability of this element. Low levels were detected in human tissues and correlated with coronary heart diseases (Virtamo et al. 1985). Selenium uptake by plants was increased by using fertilizers rich in selenium. This raised the level of selenium uptake by humans to acceptable values for human health. However, while the use of fertilizers raises the total concentration of selenium in soils, it is the bioavailability of selenium what is relevant for health (Rayman 2004). Repeated use of metal-enriched chemicals, fertilizers, and organic amendments such as sewage sludge as well as wastewater is thought to be a possible cause of contamination at a large scale (He et al. 2005). A recent paper by Haug et al. (2007) discusses on the most effective treatments to soils and food in order to increase the selenium intake by humans. These treatments rely on the present knowledge about the undergoing physico–chemical processes governing the solubility and uptake behavior of the selenium species, which have to be understood for each particular soil.
3 Selenium bio-availability
3.1 Bioavailability
The bioavailability of a trace element is related to the factors that make it available to an organism, that is, in a form that can be transported across the organism’s biological membrane (Reeder et al. 2006). However, this concept is not very precise, as a substance can be adsorbed on a colloidal particle small enough to pass through the membrane. This has motivated the use of the term “bioaccessibility”, representing “the fraction of a substance that becomes soluble within the gut or lungs and therefore available for absorption through a membrane” (Fig. 2) (Reeder et al. 2006; Ruby et al. 1996, 1999). Bioavailability and bioaccessibility rely then on a variety of entangled physico–chemical factors affecting mainly solubility of the substances, like speciation, ionic strength, pH or redox potential. An example that emphasizes the relevance of bioavailability against the high selenium concentrations is provided by Amweg et al. (2003). The authors have studied the effects of an algae-bacterial selenium-reduction system applied to irrigation waters of the San Joaquin Valley (California, USA). The system helped to reduce 80% of the influent selenium in waters, but the concentration of selenite and organic selenium suffered an 8-fold increase. These two forms of selenium, which are more bioavailable to biota than the original species, made that selenium concentration in organisms increased 2–4 times as a result of the water treatment. Speciation is thus a key factor in the fate of selenium species in the environment and in their availability to organisms. Usually, Se(0) is considered to have little toxicological significance to most organisms (Combs et al. 1996; Schlekat et al. 2000), although biological activity has been reported for elemental selenium nanoparticles (Zhang et al. 2005). Selenite and selenate are both water soluble inorganic species typically found in aerobic water sources. Selenite is both more bioavailable and ~5–10 times more toxic than selenate (Lemly 1993). Organic selenium, in the form of selenide, Se(−II), is the most bioavailable form, and it is taken up by algae 1,000 times more readily than inorganic forms (Lemly 1993; Maier et al. 1993). Another example showing the importance of selenium speciation in soils on bioavailability is the case of selenium deficiency in the Zhangjiakou District of the Hebei province in China (Johnson et al. 2000a). Soils from the Zhangjiakou district present an average of 0.15 mg of selenium kg−1, which can be considered a low concentration, but not a critically low level. In this area, the Keshan disease, a heart disease, affects some part of the population. This disease is usually attributed to a lack of selenium in diet. Johnson et al. (2000a) demonstrated that the prevalence of the disease is not correlated with a lack of selenium in the soil as might be expected. The cause for the selenium deficiency is rather a result of the fact that the soil-bound selenium is not in a form available for plants. These soils are rich in organic matter, which can be the responsible of the selenium immobilization, either through direct adsorption or through redox processes: reduction of selenate to selenite would favor the adsorption of the latter onto iron and aluminum oxyhydroxides.

Difference between bioavailability and bioaccessibility of selenium. The left part (red pathway) represent the pathway followed by selenium oxyanions adsorbed in mineral particules, which enter the cellular membrane (bioavailable) but are not metabolized by the organism (not bioaccessible). At the left, a selenium aqua-oxyanion enters the cellular membrane and is bioaccessible to the organism. Adapted from Reeder et al. (2006)
Selenium bioavailability is thus influenced by different factors that will be reviewed herein: adsorption properties of soils, sediments and aquifer substrates; speciation in aqueous solution; mobility of the different species and solubility with respect to solid phases. A review with data on selenium acidity constants influencing speciation in aqueous solutions and solubility constants of different selenium-containing solid phases can be found in Seby et al. (2001) or Olin et al. (2005).
3.2 Selenium mineralogy and sorption processes
3.2.1 Selenium retention at the mineral/water interface
The study of selenium mineralogy is being restricted in this subsection to the description of selenium sorption processes at the mineral/water interface. Readers interested in the mineralogy of selenium-rich minerals are referred to the reviews by Seby et al. (2001) and Olin et al. (2005), in which solubility constants are given and associations with different minerals are described, and to the inorganic crystal structure database (ICSD) (http://icsd.ill.fr), where the crystallographic details for each phase can be found.
Sorption processes are responsible for the immobilization of selenium species at the mineral/water interface. Detailed studies of these processes are thus required to make accurate estimations of the bioavailability of selenium species in soils, sediments and waters. In a general way, selenium adsorbed onto mineral phases accounts only for about a 20% of the total selenium present in soils and sediments, although this number is strongly dependent on mineralogical characteristics and geochemical conditions (Selim and Sparks 2001). Another fraction of the soil, organic matter, is considered to be responsible 50% of the selenium retention in soils and sediments, as reported by Abrams et al. (1990) and Gustafsson and Johnsson (1994), in Californian Tirs soils and forest soils, respectively. However, these numbers are estimations mainly done from chemical extraction procedures, which usually are precision-limited due to the non-specifity of much of the extractants used (Gruebel et al. 1988; Lenz et al. 2008b). In anoxic conditions, elemental selenium can account for a 20–60% of the total selenium in soils and sediments (Zawislanski et al. 2003; Zhang and Moore 1996).
3.2.2 Selenium adsorption
Different processes at the mineral/water interface can be considered responsible for the selenium associations with mineralogical soil components: adsorption, co-precipitation and surface precipitation processes, the two latter depending on solubility of the target solid phases. Adsorption is defined as ‘the process through which a chemical substance accumulates at the common boundary of two contiguous phases’ (Sposito 2004). It highly depends on factors like ionic strength of the medium, which can reduce the adsorption properties of some minerals through the reduction of the size of Stern layer of adsorption (Sposito 2004). Competition effects are also an important factor which may reduce selenium adsorption, e.g., when fertilizers are applied on soils, like phosphates or nitrates. For instance, selenite sorption in Indian soils have been studied by Dhillon and Dhillon (2000), showing that selenite adsorption is reduced by a 50% when phosphate anions are present as competitors. Selenium species can be adsorbed through two different mechanisms at the mineral/water interface (Balistrieri and Chao 1987; Hansmann and Anderson 1985; Neal et al. 1987a, b): outer-sphere and inner-sphere complexation. Formation of outer-sphere complexes is an electrostatic-driven sorption mechanism, strongly dependent on surface charge and thus on solution ionic strength (Sposito 2004). Inner-sphere complexes form when an ion is adsorbed “specifically” on a “crystallographic site”, i.e., when covalent or ionic bonds are created with functional sites present on the mineral faces. These bonds have a stronger degree of covalence and are more stable than outer-sphere complex formation (Sposito 2004). They are responsible in much cases of the long-term immobilization of ions at the mineral/water interface (Duc et al. 2003). Regarding selenium sorption kinetics, very few studies have been published so far. Most of them report very fast selenium adsorption processes, where selenite and selenate sorption reaches equilibrium typically in periods shorter than 2 h, independently of the soil physic–chemical conditions (Dhillon and Dhillon 1999, 2000; Goh and Lim, 2004; Neal et al. 1987a). Of special interest from a structural point of view is the article by Zhang and Sparks (1990), in which the two-steps mechanism originating an inner-sphere complex is described.
Traditional studies determine the adsorption capacities of the sorbent phase by using sorption isotherms in which either the pH, the ionic strength or the selenium concentration are used as a variable while the two other magnitudes are kept constant (Balistrieri and Chao 1987; Neal et al. 1987a). Other studies have pointed out the importance of parameters like particle aggregation (Hansmann and Anderson 1985) or humidity (Manceau and Charlet 1994) on the adsorption behavior.
Selenium associations with iron, aluminum and manganese oxides and hydroxides, carbonates and organic matter have been widely reported (Selim and Sparks 2001). Iron and aluminum oxides have surfaces with a variable charge in relation to pH, which implies a higher sorption process of selenite and selenate at low pH, where the positive charge is developed in the mineral surface (Yu 1997). Both selenite and selenate species adsorb onto ferric oxy-hydroxides, but the affinity of these solids for selenate is generally smaller than for selenite (Balistrieri and Chao 1987; Hayes et al. 1987; Lo and Chen 1997; Su and Suarez 2000; Zhang and Sparks 1990). This behavior may be related to differences in the nature of respective surface complexes, and to geometrical factors that may affect the extent of inner-sphere complexation. In general, selenite is sorbed by inner-sphere complexation, although the exact coordination (mono- or bi-dentate) and species protonated or deprotonated depends on the structure and mineral surface charge. Ferric oxy-hydroxides whose surface reactivity toward selenium species has been studied include: goethite (Balistrieri and Chao 1987; Hayes et al. 1987; Hiemstra and Van Riemsdijk 1999; Lo and Chen 1997; Manceau and Charlet 1994; Parida et al. 1997; Peak and Sparks 2002; Su and Suarez 2000; Zhang and Sparks 1990) HFO (hydrous ferric oxide) (Davis and Leckie 1980; Hayes et al. 1987; Lo and Chen 1997; Manceau and Charlet 1994; Peak and Sparks 2002; Su and Suarez 2000), other iron oxy-hydroxide polymorphs (Parida et al. 1997) and hematite (Peak and Sparks 2002). A list of the different complexes formed on the surfaces of these solids is given in Table 4.
In the case of selenate oxyanions sorption on goethite and HFO, the use of modern spectroscopic techniques like EXAFS has provided a new view of the adsorption process. Some authors had pointed out that the sorption of selenate showed an ionic strength dependence, which was traditionally linked with an outer-sphere (electrostatic) adsorption (Hayes et al. 1987). Manceau and Charlet (1994), demonstrated that selenate binds onto the goethite and HFO structures by forming inner-sphere complexes. The influence of humidity and drying processes in the sample conditioning may be responsible for these different observations. A recent paper by Fukushi and Sverjensky (2007), in which an extended triple layer model taking into account the electrostatics of water dipole desorption during ligand exchange reactions has predicted these differences in behavior. The lower ionic strength and higher surface coverage used by Hayes et al. (1988) is predicted to favor an outer-sphere selenate species, whereas the higher ionic strength used by Manceau and Charlet, (1994), is predicted to favor an inner-sphere selenate species, as suggested also by Peak and Sparks (2002). This implies a high effect of the electrostatic potential of the mineral surface on the surface complexation mechanisms, as confirmed by Hiemstra and Van Riemsdijk (1999). These authors were able to fit the data of Hayes et al. (1988) assuming the formation of only a bidentate inner-sphere complex (Hiemstra and Van Riemsdijk 1999). Peak and Sparks (2002) employed EXAFS and attenuated total reflectance–fourier transform infrared (ATR–FTIR) spectroscopies to determine selenite bonding mechanisms on hematite, goethite, and hydrous ferric oxide (HFO). They also showed that selenate forms only inner-sphere surface complexes on hematite, but it forms a mixture of outer- and inner-sphere surface complexes on goethite and HFO. This continuum of adsorption mechanisms is strongly affected by both pH and ionic strength.
Selenite sorption on apatites and iron oxides has been studied by Duc et al. (2003). Both types of solids were shown to retain selenite ions from aqueous solution with a high efficiency, iron oxides giving higher distribution coefficient values than apatites. These solids were suggested to be used as additives to the engineered barriers built around radioactive wastes and to compensate the low sorption ability of clay minerals for anionic species (Duc et al. 2003). In natural (geologic) barriers, frequently occurring minerals such as apatites and iron oxides could play an important role in selenite immobilization (ANDRA 2005).
Aluminum oxide reactions to selenite and selenate have been studied to a lower extent than their iron oxides counterparts. The sorption properties and mechanisms are however, very similar. Concerning selenite, Papelis et al. (1995), showed by EXAFS inner-sphere complexes to be formed on gibbsite. This was corroborated by the ionic strength independent behavior of the adsorption, although this is not a conclusive experiment. Same results were found by Schulthess and Hu (2001). Peak (2006), showed that selenite forms a mixture of outer-sphere and inner-sphere bidentate-binuclear (corner-sharing) surface complexes on hydrous aluminum oxides (HAO) and selenate forms primarily outer-sphere surface complexes on HAO. An interesting result shows that selenate forms outer-sphere surface complexes on corundum at pH 3.5 but inner-sphere monodentate surface complexes at pH 4.5 and above, on the same surface (Fig. 3). This difference in behavior may be related to proton-promoted structural changes in the surface of corundum or to the co-existence of outer-sphere and inner-sphere complexes at low pH, which would be affecting the intensity of the EXAFS signal. Changes in the protonation state of the selenate molecule are not expected due to the low pK a2 of the selenate molecule (pK a2 = 1.8) (Peak 2006).
Clay–selenium interactions are nowadays a research field of increasing interest due to its relevance to nuclear waste barrier efficiency. Clays are indeed an important part of these barriers (ANDRA 2005). However, up to date selenium sorption on clays has not been extensively studied. Some works by Frost and Griffin (1977), and BarYosef and Meek (1987), showed that kaolinite and montmorillonite do not show high selenite adsorption capacities, the latter being a slightly better sorbent. According to Peak et al. (2006), selenite forms inner-sphere bidentate binuclear complexes on Montmorillonite edges. Charlet et al. (2007), showed that selenite forms an outer sphere complex on the edges of Montmorillonite (Fig. 3).

Atomistic view of the two more common adsorption mechanisms of selenite at the mineral/water interface. Only two Fe(III) octahedra have been recreated here to represent the mineral surface. Inner-sphere adsorption is characterized by a ligand exchange mechanism, by which selenite shares one (monodentate) or two (bidentate) oxygen atoms with the mineral surface. In the outer-sphere mechanism, selenite conserves its hydration shell, and it is only retained by electrostatic interactions. violet selenium, red oxygen, white hydrogen, blue iron
The geochemistry of selenium seems to play an important role in the geochemical mobility of fluoride ions. Jinadasa and Dissanayake (1992), described a competition effect between selenium oxyanions and fluoride for adsorption sites on kaolinite. Although the concentrations used in their studies (>0.1 mM selenium) are not relevant for the environment, the formation of an inner-sphere selenite–kaolinite complex is reported, which represent useful structural information that can be extended to other kaolinitic soils.
3.2.3 Selenium substitutions
The ability of some minerals to host selenium oxyanions or Se(−II) in their crystalline structure through substitution processes can be a relevant mechanism controlling the concentration of selenium in some environments (Reeder et al. 1994), or as an immobilization mechanism for detoxification of contaminated waters. For instance, apatites are potential candidates to control the mobility of selenate in contaminated soils due to the similar symmetry between selenate and phosphate anions (Duc et al. 2003). In addition to their stability and the high affinity that they exhibit toward numerous possible cationic pollutants, it was shown that they could also be the basis of anionic substitutions (Monteil-Rivera et al. 2000). The substitution of phosphate sites was implied by the occurrence of natural or synthetic modified apatites containing carbonate, sulfate, vanadate, and arsenate groups (Boechat et al. 2000; Fleet and Liu 2008; McConnell and Foreman 1978; Sneddon et al. 2005). Substitution of selenite oxyanions by phosphate in hydroxyapatites has been reported (Duc et al. 2003; Monteil-Rivera et al. 2000). Calcite can also be the host phase controlling the activity of selenite ions in carbonate rich-environments, as demonstrated by (Reeder et al. 1994). Structural similarities between carbonate and selenite oxyanions can facilitate the incorporation of significant amounts of selenite to the bulk of calcite crystals, as previously demonstrated for arsenic oxyanions (Alexandratos et al. 2007; Roman-Ross et al. 2006). Other substitution processes have not been reported yet but may be present in nature and should be quantified for a correct estimation of the fate of selenium oxyanions in the environment. Possible substitutions should be studied in view of the molecular structure similarities between the anions involved in the process. The chemical affinity between sulfur and selenium has been cited already in this article and is the responsible of the high concentrations of Se(−II) present in minerals as pyrite or chalcopyrite (Ihnat 1989).
Materials presenting high retention properties towards selenium in aqueous media are required in engineered and natural barriers used to prevent the dispersion of radioactivity from waste repositories. The most frequently proposed materials for engineered barriers are essentially made of clays or cements, but these materials have low retention properties towards anionic species such as selenite and selenate. Therefore, there is a need for materials able to sorb anionic species, and which can be used as additives in engineered barriers. Apatites are also frequently present in soils and geologic media and can also contribute to the control of the migration of radionuclides in natural media through substitutions in their crystallographic lattice. Iron oxy-hydroxides are present in practically every natural media in contact with water: soils, sedimentary rocks or cracks in igneous rocks. They constitute also corrosion products of steel containers, playing thus an important role in the prediction of the sorption and migration of radionuclides in underground water (ANDRA 2005). Further studies in this direction will help to the evaluation of the more appropriate mineral phases for the retention of selenium radionuclides emerging from these wastes.
3.3 Selenium oxido-reduction processes
The solubility of selenium is largely controlled by its oxidation state. While oxidized form of selenium like selenite and selenate are highly soluble under a wide range of geochemical conditions, elemental selenium and selenides formed with Se(−II) have very low solubilities (Seby et al. 2001). Biotic-mediated processes of selenium reduction have been already presented in the Sect. 2.2, and will be explained here in detail. The name organic selenium stands for organic Se(−II) compounds formed mainly by biotic processes (see Table 1). The chemical similarities between sulfur and selenium make possible that selenium displaces sulfur in some amino-acids (e.g., selenomethionine, selenocysteine) through assimilatory processes (Fordyce 2007). These seleno amino-acids are usually found later in the humic fraction of soil OM (Kang et al. 1991; Lemly 1997). Some micro-organisms can then metabolize these seleno amino-acids to produce H2Se, which is finally oxidized to elemental selenium (Doran and Alexander 1977b); (Herbel et al. 2003; Wessjohann et al. 2007). These biological reactions are very similar to the reactions sulfur undergoes. For this reason, Shrift (1964) proposed a microbial selenium cycle in nature, which is similar to the existing sulfur cycle.
In the late 1980s, other pathways for the selenium cycling in the environment were reported. After the discovery by Presser and Ohlendorf (1987) of low selenium concentrations in surface waters of the Kesterson National Wildlife Refuge salt marsh environments in California, Zehr and Oremland (1987) reported that certain sulfato-respiring bacteria have the ability to reduce selenate to selenide, providing thus a pathway for depletion of selenium in surface waters. Later, dissimilatory sulfate-independent bacterial respiration was discovered (Oremland et al. 1989) (this sulfate-independent respiration has been recently proved by Lenz et al. (2008b), who fed bacteria with sulfate in 2,600 molar excess to selenate in a continuous manner, showing that selenate still was completely reduced). Since then, many bacterial cultures capable of selenate and selenite respiratory growth have been observed (Basaglia et al. 2007; Blum et al. 1998; Ghosh et al. 2008; Hunter 2007; Hunter et al. 2007; Macy et al. 1989; Narasingarao and Haggblom 2007; see review by Stolz et al. 2006; Stolz and Oremland 1999). To date, about 16 diverse species of bacteria and archaea have been described that grow anaerobically by linking the oxidation of organic substrates or H2 to the dissimilatory reduction of selenium oxyanions (Oremland and Stolz 2000; Stolz and Oremland 1999). The decoupling of this mechanism from bacterial sulfate-respiration is an important discovery, since it explains the reduction of selenium to elemental selenium or metallic selenides in a variety of environments, independently of their salinity (Herbel et al. 2003). Selenite has been found to be an intermediate product in some of these reductive-precipitation processes, revealing thus a parallel pathway for the fate of selenite in sedimentary environments: selenite produced by reduction of selenate by bacteria would adsorb specifically onto mineral surfaces. This would explain the high concentrations of selenite reported in oxygenated seawater (Stolz and Oremland 1999). Other pioneer studies in this field reported the redox potentials for these processes to occur in different types of sediments (Steinberg and Oremland 1990), or reaction rates for the reduction of selenate in sediments (Oremland et al. 1990).
Within these microbe-mediated processes, dissimilatory reduction has been widely reported as an active process by which selenate or selenite are reduced to elemental selenium by utilizing the oxyanions as terminal electron acceptors during the respiration of organic carbon. For instance, in agricultural drainage water of the San Joaquin Valley, California, selenate was effectively reduced to elemental selenium by Enterobacter taylorae (Zhang et al. 2003) and by Thauera selenatis (Macy et al. 1993) using this pathway. Some bacteria, like Enterobacter cloacae SLD1a-1, have known pathways for the detoxification of selenate which include a series of reductive transformations of the anion and in the presence of an electron donor (Watts et al. 2003). Several bacteria strains have been identified to reduce toxic selenium oxyanions using lactate as electron donor, and producing elemental selenium nanospheres that accumulated inside and outside the cell wall (Oremland et al. 2004). This process has been observed even at very low selenium concentrations (μM) (Lenz et al. 2008a). Other researchers have reported the reduction of selenite to intracellular granules of elemental monoclinic selenium, Se(0), by Ralstonia metallidurans CH34 (Sarret et al. 2005). This same bacteria was shown to reduce selenate following an assimilatory pathway with alkyl selenide as a final product (Sarret et al. 2005).
Some biotic routes of selenium reduction are being studied nowadays as a potential methodology to remediate selenium contamination of wastewaters (Cantafio et al. 1996; Fujita et al. 2002; Lenz et al. 2008c). Recently, some authors (Astratinei, et al., 2006; Lenz et al. 2006) have demonstrated the ability of some bacteria strains present in anaerobic granular sludge biofilms to reduce selenate to amorphous elemental selenium and to some Se(−II) bearing crystalline phases (Lenz et al. 2008b). Both species of selenium are non-soluble, which gives these processes an interesting potential applicability as removal methods for selenium in wastewaters.
Reduction of selenate oxyanions to elemental selenium in the absence of external metabolic electron donors has been found to occur on the cellular walls of Shewanella putrefaciens 200R. Kenward et al. (2006) showed using XANES spectroscopy that selenate forms outer-sphere complexes on the cell walls, and that elemental selenium was formed. The authors claim that given the number of potential electron donors found within the cell wall, it is possible that the cell wall is sufficient to reduce selenium, even in the absence of active metabolism. This process would imply that bacterial cellular walls could be an important sink of selenium in natural subsurface environments, where bacteria experience nutrient poor conditions and are often found as non-metabolizing cells and cell fragments. Therefore, the impact of bacteria on the toxicity, mobility, and cycling of selenium may extend beyond that exerted by selenium-reducing bacteria that employ selenium as terminal electron acceptors. Other biotic mechanism involving a pathway for selenium reduction in the environment is the reduction of selenium oxyanions by sulfides produced by dissimilatory mechanisms in sulfate-reducing bacteria (Hockin and Gadd 2003). Some strains of bacteria may further reduce elemental selenium to selenide or oxidize selenide to form elemental selenium (Doran and Alexander 1977b; Herbel et al. 2003).
On the other hand, some microorganisms have the ability of oxidize elemental selenium or some selenides to selenite or selenate (Dowdle and Oremland 1998; Sarathchandra and Watkinson 1981; Zhang et al. 2004). However, oxidation of selenium is usually considered as a slow phenomenon, with kinetics in the order of 103 times slower than those of bio-reduction processes (Dowdle and Oremland 1998).
Abiotic reduction of selenium oxyanions by Fe(II)-containing mineral surfaces has been found to occur under reducing environments, which may be significant for natural environments as sediments, groundwater or nuclear waste disposal geological media (Scheinost et al. 2008). Selenite is the stable valence state under mildly reducing or anoxic conditions (0.26 V < Eh <0.55 V at pH 7 and 1 μmol l−1 total concentration; (White et al. 1991). The slow homogeneous kinetic reactions between the different redox states (Gruebel et al. 1995) make that selenite is found in some oxic environments, as an intermediate product of biotic processes (Oremland and Stolz 2000). Selenite and selenate are reduced abiotically by Fe(II)-bearing minerals like green rust (Myneni et al. 1997; Scheidegger et al. 2003), magnetite, mackinawite and siderite (Scheinost and Charlet 2008), pyrite (Bruggeman et al. 2005), troilite (Breynaert et al. 2008), and by Fe(0) (Loyo et al. 2008). In the case of selenium reduction by Fe(II)-bearing minerals, elemental selenium seems to be the product in most of the laboratory experiments performed (precipitation of Fe selenides has been only shown by Scheinost and Charlet 2008 and by Breynaert et al. 2008). This is in clear contrast with the prevalent existence of Fe–selenides in soils, sediments and ore-deposits. Scheinost et al. (2008) have shown that fast reduction kinetics favor the precipitation of Fe(II)–selenides, while elemental is being precipitated in slow redox processes. The concentration of HSe- and the low solubility of Fe(II)–selenides seem to be the controlling factors of these redox reactions.
The Fe(II) hexaqua ion, which is ubiquitous in anoxic soils and aquifers, does not seem to reduce significant amounts of selenite. However, Charlet et al. (2007) have shown that Fe(II) adsorbed specifically on clay edges reduces selenite in a slow reaction, which continues for several weeks. The reduction is kinetically decoupled from the rapid oxidation of Fe(II) to Fe(III), most likely by formation and storage of an hydrogen intermediate at the clay mineral surface (Géhin et al. 2007). Regarding reduction kinetics, Bruggeman et al. (2005) reported an increase in the kinetics of selenite reduction by pyrite (FeS2) probably due to the effect of Fe(II) associated with humic substances.
3.4 Elemental selenium
Selenium is among the various geogenic contaminants which can be reductively precipitated, with biotic processes governing most of the redox reactions. The low solubility of Se(0) and of the metal selenides (Seby et al. 2001; Olin et al. 2005) and the long-held concept that Se(0) is biologically inert, makes possible that some of these redox processes are considered as effective bioremediation techniques to remove selenium from agricultural drainage water (Ehrlich 1997). However, recent advances in the understanding of microbiological processes leading to the formation of elemental selenium nano-precipitates have raised some questions about the structure of these precipitates (Oremland et al. 2004). It is known than some thermodynamic properties are different for particles in the nano-scale than for their bulk counterparts (Hochella et al. 2008). The reactivity of these Se(0) nanoparticles is thus an unexplored field of study with strong importance in studies on nuclear waste disposal (Charlet et al. 2007), where these nanoparticles have been reported to exist, or in the medical community (Sieber 2003), where they are starting to be used as dietary supplements.
In addition, the structure and dynamics of the non-crystalline phases of bulk selenium (glassy, supercooled liquid, normal liquid and amorphous films) are still not perfectly understood, which makes more difficult to understand the properties of the nanometric phases. Amorphous selenium precipitates (nano or bulk) are thought to be formed by the same atomic units present in the crystalline phases of selenium: linear chains, Se6 rings or Se8 rings. The published structures of crystalline ‘bulk’ elemental selenium (ICSD 2008) are composed by selenium atoms with the following atomic arrangements (see Table 5):
-
The three allotropes of monoclinic elemental selenium (deep red) have Se8 rings with D4d symmetry as structural units within their unit cell.
-
The trigonal allotropes of elemental selenium (metallic grey) are composed by polymeric helical chains of selenium atoms.
-
Amorphous selenium (red or black) is thought to contain two kinds of molecules: polymeric chains (Sen) and monomeric rings (Se8 or Se6) (Popov 1976). However, different experimental studies over the last century have reported contradictory conclusive statements on the nature of these elemental chains or sub-structures (Jovari et al. 2003), a puzzling question still under debate today.
-
The cubic phases of selenium have been synthesized at high pressure, and up to date, there is no report or evidence of their occurrence in the environment.
A vast number of experimental and theoretical investigations about the amorphous selenium structure have been published using a variety of techniques. In brief, structural, dynamical, and photo-induced properties have been studied by inelastic neutron scattering, Raman and infrarred spectroscopy, X-ray absorption fine structure, viscosimetric techniques, X-ray and neutron diffraction, inelastic X-ray scattering, electron spin resonance, small-angle neutron scattering, ultraviolet photoemission spectroscopy, nuclear magnetic resonance, reverse Monte Carlo, and numerous classical molecular dynamics and ab initio molecular dynamics simulations (Yannopoulos and Andrikopoulos 2004, and references therein).
Studies reporting bacterial redox-mediated formation of nanoscaled elemental selenium precipitates usually describe the final precipitate of red color, which can be ascribed to the formation of an amorphous phase or to one of the monoclinic allotropes in which Se(0) crystallizes (Sarret et al. 2005). Only very recently, some authors have tried to characterize the structure of these nanoscaled precipitates by Raman and UV-vis studies. Oremland et al. (2004), revealed differences in the local structure of the end products formed by three different bacteria. Although the same crystallographic system was found for the three precipitates (monoclinic selenium), the optical properties differed, which was attributed to different atomic arrangements: two of the precipitates were described to have Se6 structural units, while the third one was assigned Se8 units with D4d point symmetry. However, the use of Raman spectroscopy in the study of these nanometric precipitates is not straightforward, due to the difficulties associated to the assignment of the mode geometry to the different wavenumbers. Different assignments can be found in the literature, leading to different predictions regarding the structural units of elemental selenium. A recent study by Pearce et al. (2008), has used in situ EXAFS spectroscopy to follow the formation of elemental selenium nanoparticles and metal selenides by Vellonella atypica, bacteria capable of reducing selenite. The experiment has shown that the bacteria reduce selenite to Se(−II), with Se(0) as an intermediate stage. When a divalent metal as Zn(II) or Cd(II) is added after the reduction of Se(0) to Se(−II), a nano-precipitate of ZnSe or CdSe is formed. This is the only article in the literature where a structural description of the redox process from selenite to Se(−II) is presented. However, the authors do not describe the structure of the intermediate Se(0) precipitate. A recent paper by Lenz et al. (2008b) describe by XANES spectroscopy a Se(0) precipitate with neither monoclinic nor trigonal structure. More insight is needed in this field to extract values of the solubility of elemental selenium nanoparticles.
Regarding the fate of these biotic elemental selenium precipitates in the environment, (Zhang et al. 2004), have shown that Se(0) is removed from water columns following two pathways: (1) flocculation and sedimentation and (2) oxidation to selenite or selenate. However, other pathways have been reported. Some researchers have investigated the uptake of elemental selenium by some bivalves as Macoma Balthica or Potamocorbula amurensis (Luoma et al. 1992; Schlekat et al. 2000). M. Balthica ingests sediments with >1.5 μg selenium g−1, a concentration that could achieve steady-state tissue burdens approaching the level toxic to fish. Luoma et al. (1992) thus suggest that this pathway should be taken into account in remediation efforts that attempt to reduce hazardous exposures to selenium at highly contaminated sites by stimulation of microbial dissimilatory reduction of selenium oxyanions. New lines of research are opening in the study of colloidal transport of elemental selenium. Fontes et al. (1991), showed how bacteria can be transported through porous media. Se(0) nanoparticles adsorbed onto bacteria cell walls could then be mobilized by means of colloidal transport (Losi and Frankenberger 1997), with the whole cell acting as a carrier.
Several applications of Se(0) ‘subnanoparticles’ (with 0.4 nm–1 nm diameter sizes) have been reported in medical applications as manifold as anti-tumor effects (Miyagi et al. 2003), anti-leukemia/lymphoma agents (Sieber 2003; Sieber et al. 2007) or as anti-oxidants, which delete free radicals in vitro and improve the activity of the seleno-enzyme, glutathione peroxidase, preventing free radicals from damaging cells and tissues in vivo (Gao et al. 2002). The use of modifiers to stabilize these nanoparticles in the aqueous phase has been studied as well (Bai et al. 2006; Gao et al. 2002). These stabilizers are essential to increase the solubility of the nanoparticles. In their absence, the nanoparticles aggregate, forming bigger precipitates which crystallize into black or grey-colored phases (Zhang et al. 2005), which decreases their biological activity. In fact, the biological activity of these nanoparticles seems to be directly related to their surface area. Huang et al. (2003) recently demonstrated that selenium sub-nanoparticles in different sizes had marked differences in directly scavenging a battery of free radicals in vitro, i.e. the smaller the particles, the higher the efficacy. Zhang et al. (2001) found that red elemental selenium nanoparticles of 20–60 nm had equal bioavailability compared to selenite in terms of induction of seleno-enzymes in both cultured cells and selenium-deficient rats. In addition they showed that elemental selenium nanoparticles had notably low acute toxicity (1/7 of selenite). However, little information is given about the structure of these nanoparticles. Most of the authors use the color of the precipitates to assign the amorphous or monoclinic structures, or measure X-ray diffraction patterns which give broad peaks, characteristic of poorly crystalline or nanocrystalline materials.
3.5 Plant uptake
Even though selenium uptake by organisms as plants is determined by its bioavailability selenium concentration in plants generally reflects the levels of selenium in the environment where they have grown (with the exception of selenium hyperaccumulators). Usually, low selenium soils have developed on selenium-defficient parent materials with a very low flux of selenium between soil and plants (Wang and Gao 2001). Selenium has never been shown to be essential for higher plants, but is taken up and assimilated readily because of its chemical similarity to sulfur (Lauchli 1993). In general, it can be stated that selenate is taken up at higher rates than selenite, and it has been suggested that selenate may be taken up actively by plants, whereas selenite may be absorbed passively and in lesser concentrations (Sors et al. 2005).
Plants vary considerably in their physiological response to selenium. Most plant species contain <5 μg selenium g−1 dry weight and cannot tolerate high levels of selenium in the environment (White et al. 2004). Other plant species of the genera Astragalus, Neptunia and Stanleya, among others, are able to hyperaccumulate selenium at concentrations up to 10–15 mg selenium g−1 dry weight in their shoots while growing on naturally-occurring soils containing only 2–10 μg selenium g−1 dry weight (Virupaksha and Shrift 1965). The reviews by Terry et al. (2000) and Ellis and Salt (2003) summarize the knowledge about the physiology and biochemistry of both types of plants.
Molecular approaches are providing new insights into the role of sulfate transporters and sulfur assimilation enzymes in selenate uptake and metabolism, as well as the question of selenium essentiality in plants (Terry et al. 2000). In fact, the non-specific integration of the seleno amino acids seleno cysteine and seleno methionine into proteins is believed to be the major contributor of selenium toxicity in plants (Brown and Shrift 1981). The ability of selenium hyper-accumulator plants to accumulate and tolerate high concentrations of selenium is thought to be associated with a distinct metabolic capacity that enables them to divert selenium away from incorporation into proteins (Brown and Shrift 1981).
Banuelos et al. (2005), described the formation of the enzyme adenosine triphosphate sulfurylase by the Indian mustard plants. This enzyme is a key element in the plant’s ability to convert selenate into a non-toxic form of selenium, allowing the plant to accumulate more selenate without incurring harm. Aquatic plants were shown to remove selenium from agricultural or industrial wastewater through selenium accumulation and volatilization, i.e., through the conversion of selenate or selenite to volatile forms of selenium as selenium-alkyls (Pilon-Smits et al. 1999). This ability (phytovolatilization) has led to their use in phytoremediation processes, a technology for the cleanup of selenium and other toxic trace elements (Banuelos et al. 2005). However, these techniques are currently limited by the slow rate of the biological processes involved.
3.6 Selenium and health
Selenium is an essential element for nutrition of a capital importance in the human biology. This relevance to health was highlighted in the 1950s. Nutrition scientists proclaim the year 1957 as the date when selenium started to be considered as an essential nutrient (Schwarz and Foltz 1957). After its discovery by Berzelius, several episodes have underlined the importance of selenium in the human body. In 1935, 57 people from Keshan county (Heilongjiang province, China) died after a degenerative cardiac illness, related to the ‘white muscle disease’ suffered by animals (Gu and Cheng 1987). These illnesses were known to be related to a selenium deficit. Another sickness, the Kashin–Beck disease, responsible for a disorder of the bones and joints of the hands and fingers, elbows, knees, and ankles of children and adolescents mainly from the regions of North China, North Korea and Siberia, has been related to a deficit in selenium (Moreno-Reyes et al. 1998). Soil selenium content analyses have shown an important deficit in these regions, which implies a daily dietary consumption <10 μg day−1 (Kohrle et al. 2000; Moreno-Reyes et al. 1998).
Selenium, when incorporated in some mammalian enzymes, acts as a redox active center: for instance, it reduces nucleotides during DNA synthesis in thioredoxin reductase, controlling as well the intracellular redox state. The best known example of this redox function is the reduction of H2O2 or of lipidic or phospholipidic hydro-peroxides into non-dangerous substances as water or alcohol by the family of glutathione-peroxidases (Kohrle et al. 2000). This function allows to keep the integrity of the membranes, decrease the production of prostacyclines and to decrease the danger associated with oxidative stresses to some bio-molecules as lipids, lipoproteins or DNA. This stresses are associated with the atherosclerosis and carcinogenesis.
The number of identified seleno proteins is increasing every day, although the role of some of them has not being identified yet. In the last years, the development of new methods of analysis in proteomics, like 2D gel electophoresis or MALDI TOF MS has allowed the detection of new seleno-proteins (Encinar et al. 2003; Tastet et al. 2008). Selenocysteine is considered the 21st amino acid (Johansson et al. 2005).
In our alimentation, more than 80% of total selenium is present in the form of organic selenium, associated to proteins: the l(+)-selenomethionine (from vegetal and animal sources) and the selenocysteine (from animal sources) (Cesarini 2004). Other organic forms as S-methylselenocysteine or selenocystathionine are present in some vegetables grown on selenium-rich soils. Sodium selenate is an inorganic form than is also found in alimentation, but in lower proportions. Regarding selenium metabolism, the human intestine absorbs better selenomethionine and selenate than selenite or selenocysteine (Schrauzer 2000). The reasons are not clearly known, but they could be related to the formation of ion-pairs or complexes with other elements, like mercury (Mykkanen and Metsaniitty 1987), zinc, cadmium (Feroci et al. 2005) or lead (Mykkanen and Humaloja 1984). For instance, Mykkanen and Metsaniitty (1987) have proposed the formation of a soluble Se–Hg complex not capable of permeating through the intestinal wall. This complex have been identified by other workers in the blood and other tissues of animals injected with mercury chloride and sodium selenite (Burk et al. 1974).
It is worth to note that, although black and grey elemental selenium has been reported to not to be absorbed through the intestine walls (Spallholz 1994), new studies have highlighted the biological activity of red elemental selenium nanoparticles of reduced size down to the nanometer (Jia et al. 2005). However, nothing is known about the precise structure of these red nanoparticles or about their surface chemistry. Another non-solved issue is which chemical form of bioavailable selenium has to be used as dietary supplement in cases of deficiency. Inorganic forms (selenite and selenate) have been largely used in the past. Today, the use of organic forms (as present in plants) as selenomethionine is preferred, being selenium-enriched yeast the more popular supplement (Schrauzer 2000). On the other hand, methionine and selenomethionine are competitive molecules for their incorporation in proteins. Due to the non-specific substitution of methionine by selenomethionine, the latter compound is less bioavailable than its sulfur analogue. New studies on the biological interactions of selenium chemical forms within the organism will lead the decision-taking procedure for the design of selenium dietary supplements. Some effort has been devoted in the recent years to the design of drug delivery systems with reduced toxicity. A study by Sarin et al. (2007), reports the potential use of carbon nanoparticles as a vehicle for cell delivery of Se(0) nanoparticles. (Montes-Hernandez 2008), have studied the use of a portlandite carbonation technique at supercritical conditions in the presence of selenocysthine to synthesize Se(0) nanoparticles sorbed onto calcite nanocrystals.
Haug et al. (2007) have reviewed the current use of selenium resources, and concluded that supplementation should be made directly in the food production chain, by enriching seeds and adding resulting sprouts to food products, and not through the use of fertilizers. In this way, the impact of selenium environmental cycling in food would be reduced and problems like, e.g., the low selenium levels found in some areas where phosphate fertilizers have been applied (Dhillon and Dhillon 2000), would be no longer an issue. However, from a physic–chemical point of view, the interactions within the selenium–soil–water–plant system have to be deeply understood in order to correctly assess the problems of selenium deficiency and/or toxicity in organisms (Schrauzer 2000).
4 Conclusions and perspectives
Selenium is a toxic element when high concentrations of bioaccesible forms are uptaken by the organism, but it is an essential element as demonstrated by the dramatic effects affecting health of people with selenium deficit. Its variety of chemical forms and its associations with organic and inorganic natural phases gives it a very complex behavior, with different species being present at the same time in one environment. It is thus a complicated issue to determine, which forms are to be supplemented (referring to soils amendments or health care products) in cases of deficiency, and what are the best remediation procedures for contaminated sites. The advent of nanotechnology has promoted the development of new tools to study the molecular processes underlying these phenomena. For instance, spectroscopic tools have allowed studying selenium adsorption processes at the nanoscale, increasing our knowledge about the selenium retention mechanisms at the mineral/water interface. A better understanding of the environmental cycling of selenium is required, and many efforts have to be devoted to the comprehension of the basic processes governing it. Interactions with mineral phases and organic matter are far from being well understood, mostly due to the complex geochemistry of selenium. Other complex mechanisms, as the formation of OM–metal–selenium complexes, have been reported recently, but little is known about their stability constants and their structure. The same holds for the interactions between selenium and other elements which have been reported to have an effect on the absorption of selenium through the intestine walls. Special environments have attracted the attention of selenium researchers last years: (1) nuclear waste disposal engineering involves the study of radionuclide transport, like in the case of 79Se. The anoxic environment formed by compacted clays surrounding the nuclear waste canister opens a challenge to researchers studying the complex reactions taking place in it; (2) naturally occurring contaminated sites, as the Kesterson reservoir (USA) still wait for the development of new remediation techniques; (3) environments such as volcanic soils, where low bioavailability of selenium has been reported, require further investigations about the processes responsibles of their retention mechanisms; (4) recent research on elemental selenium nanoscaled precipitates and on the design of selenium cell delivery systems is being benefited from spectroscopic techniques developed over the last 15 years, with techniques focusing on the structure of selenium nanoparticles; (5) biotic reactions in selenium contaminated environments and remediation systems produce selenium nano-precipitates whose reactivity and surface chemistry are not known. Research in this direction will enlarge our knowledge about the reactivity of selenium nanoparticles. All these different subjects are attracting renovated interest by researchers from different areas nowadays, which makes the study of selenium bioavailability an exciting field of research.
References
Abrams MM, Burau RG, Zasoski RJ (1990) Organic Selenium Distribution in Selected California Soils. Soil Sci Soc Am J 54:979–982
Abu-Erreish GM, Whitehead EI, Olson OE (1968) Evolution of volatile selenium from soils. Soil Sci 106:415–420
Alexandratos VG, Elzinga EJ, Reeder RJ (2007) Arsenate uptake by calcite: macroscopic and spectroscopic characterization of adsorption and incorporation mechanisms. Geochim Cosmochim Acta 71:4172–4187
Amouroux D, Pecheyran C, Donard OFX (2000) Formation of volatile selenium species in synthetic seawater under light and dark experimental conditions. Appl Organomet Chem 14:236–244
Amweg EL, Stuart DL, Weston DP (2003) Comparative bioavailability of selenium to aquatic organisms after biological treatment of agricultural drainage water. Aquat Toxicol 63:13–25
ANDRA (2005) Dossier 2005 Argile. Evaluation de surete du stockage geologique. Agence National pour la gestion des Dechets Radioactifs, Paris
Astratinei V, van Hullebusch E, Lens P (2006) Bioconversion of selenate in methanogenic anaerobic granular sludge. J Environ Qual 35:1873–1883
Atkinson R, Aschmann SM, Hasegawa D, Thompsoneagle ET, Frankenberger WT (1990) Kinetics of the atmospherically important reactions of dimethyl selenide. Environ Sci Technol 24:1326–1332
Bai SH, Thomas C, Rawat A, Ahsan F (2006) Recent progress in dendrimer-based nanocarriers. Crit Rev Ther Drug Carrier Syst 23:437–495
Balistrieri LS, Chao TT (1987) Selenium adsorption by goethite. Soil Sci Soc Am J 51:1145–1151
Ball S, Milne J (1995) Studies on the interaction of selenite and selenium withy sulfur donors 3 Sulfite. Can J Chem-Revue Canadienne De Chimie 73:716–724
Banuelos GS, Lin ZQ (2007) Acceleration of selenium volatilization in seleniferous agricultural drainage sediments amended with methionine and casein. Environ Pollut 150:306–312
Banuelos GS, Lin ZQ, Arroyo I, Terry N (2005) Selenium volatilization in vegetated agricultural drainage sediment from the San Luis Drain, Central California. Chemosphere 60:1203–1213
BarYosef B, Meek D (1987) Selenium sorption by kaolinite and montmorillonite. Soil Sci 144:11–19
Basaglia M, Toffanin A, Baldan E, Bottegal M, Shapleigh JP, Casella S (2007) Selenite-reducing capacity of the copper-containing nitrite reductase of Rhizobium sullae. FEMS Microbiol Lett 269:124–130
Berzelius JJ (1817) Lettre de M. Berzelius a M. Berthollet sur deux metaux nouveaux. Annales de Chimie et de Physique 7:199–207
Berzelius JJ (1818) Recherches sur un nouveau corps mineral trouve dans le souffre fabrique a Fahlun. Annales de Chimie et de Physique 9:160–180 (225–267, 337–365)
Blum JS, Bindi AB, Buzzelli J, Stolz JF, Oremland RS (1998) Bacillus arsenicoselenatis, sp nov, and Bacillus selenitireducens, sp nov: two Haloalkaliphiles from Mono Lake, California that respire oxyanions of selenium and arsenic. Arch Microbiol 171:19–30
Boechat CB, Eon JG, Rossi AM, Perez CAD, San Gil RAD (2000) Structure of vanadate in calcium phosphate and vanadate apatite solid solutions. Phys Chem Chem Phys 2:4225–4230
Bonhoure I, Baur I, Wieland E, Johnson CA, Scheidegger AM (2006) Uptake of Se(IV/VI) oxyanions by hardened cement paste and cement minerals: an X-ray absorption spectroscopy study. Cem Concr Res 36:91–98
Boyle-Wight EJ, Katz LE, Hayes KF (2002) Spectroscopic studies of the effects of selenate and selenite on cobalt sorption to gamma-Al2O3. Environ Sci Technol 36:1219−1225
Breynaert E, Bruggeman C, Maes A (2008) XANES–EXAFS analysis of se solid-phase reaction products formed upon contacting Se(IV) with FeS2 and FeS. Environ Sci Technol 42:3595–3601
Brown TA, Shrift A (1981) Exclusion of selenium from proteins of selenium-tolerant astragalus species. Plant Physiol 67:1051–1053
Bruggeman C, Maes A, Vancluysen J, Vandenmussele P (2005) Selenite reduction in boom clay: effect of FeS2, clay minerals and dissolved organic matter. Environ Pollut 137:209–221
Bruggeman C, Maes A, Vancluysen J (2007) The interaction of dissolved boom clay and gorleben humic substances with selenium oxyanions (selenite and selenate). Appl Geochem 22:1371–1379
Burk RF, Foster KA, Greenfie PM, Kiker KW (1974) Binding of simultaneously administered inorganic selenium and mercury to a rat plasma-protein (37894). Proc Soc Exp Biol Med 145:782–785
Byers HG, Miller JT, Williams KT, Lakin HW (1938) Selenium occurrence in certain soils in the United States with a discussion of related topics. Third Report U.S.D.A. Tech Bul 601., US
Cantafio AW, Hagen KD, Lewis GE, Bledsoe TL, Nunan KM, Macy JM (1996) Pilot-scale selenium bioremediation of San Joaquin drainage water with Thauera selenatis. Appl Environ Microbiol 62:3298–3303
Catalano JG, Zhang Z, Fenter P, Bedzyk MJ (2006) Inner-sphere adsorption geometry of Se(IV) at the hematite (100)—water interface. J Colloid Interface Sci 297:665−671
Cesarini JP (2004) Le selenium: actualites. John Libbey Eurotext, Paris
Chabroullet C (2007) Etude de la remobilisation d’elements traces a partir d’un sol de surface contamine: influence du vieillissement des composes organiques du sol sur la remobilisation du selenium. Thesis of the University of Grenoble p. 234 Grenoble
Charlet L, Scheinost AC, Tournassat C, Greneche JM, Gehin A, Fernandez-Martinez A, Coudert S, Tisserand D, Brendle J (2007) Electron transfer at the mineral/water interface: selenium reduction by ferrous iron sorbed on clay. Geochim Cosmochim Acta 71:5731–5749
Charter RA, Tabatabai MA, Schafer JW (1995) Arsenic, molybdenum, selenium, and tungsten contents of fertilizers and phosphate rocks. Commun Soil Sci Plant Anal 26:3051–3062
Chasteen TG, Bentley R (2003) Biomethylation of selenium and tellurium: microorganisms and plants. Chem Rev 103:1–25
Combs GF, Garbisu C, Yee BC, Yee A, Carlson DE, Smith NR, Magyarosy AC, Leighton T, Buchanan BB (1996) Bioavailability of selenium accumulated by selenite-reducing bacteria. Biol Trace Elem Res 52:209–225
Cutter GA (1982) Selenium in reducing waters. Science 217:829–831
Cutter GA, Bruland KW (1984) The marine biogeochemistry of selenium—a re-evaluation. Limnol Oceanogr 29:1179–1192
Davidson DF, Powers HA (1959) Selenium content of some volcanic rocks from Western United States and Hawaiian Islands. Bull Geol Surv US 1084(C):69–81
Davis JA, Leckie JO (1980) Surface-ionization and complexation at the oxide–water interface 3 adsorption of anions. J Colloid Interf Sci 74:32–43
Dhillon KS, Dhillon SK (1999) Adsorption–desorption reactions of selenium in some soils of India. Geoderma 93:19–31
Dhillon SK, Dhillon KS (2000) Selenium adsorption in soils as influenced by different anions. J Plant Nutr Soil Sci-Zeitschrift Fur Pflanzenernahrung Und Bodenkunde 163:577–582
Dhillon KS, Dhillon SK (2003) Distribution and management of seleniferous soils. In: Sparks D (ed) Advances in agronomy, 1st edn. Elsevier, Amsterdam, pp 119–184
Dhillon SK, Dhillon KS, Kohli A, Khera KL (2008) Evaluation of leaching and runoff losses of selenium from seleniferous soils through simulated rainfall. J Plant Nutr Soil Sci-Zeitschrift Fur Pflanzenernahrung Und Bodenkunde 171:187–192
Doran JW, Alexander M (1977a) Microbial formation of volatile selenium-compounds in soil. Soil Sci Soc Am J 41:70–73
Doran JW, Alexander M (1977b) Microbial transformations of selenium. Appl Environ Microbiol 33:31–37
Dowdle PR, Oremland RS (1998) Microbial oxidation of elemental selenium in soil slurries and bacterial cultures. Environ Sci Technol 32:3749–3755
Duc M, Lefevre G, Fedoroff M, Jeanjean J, Rouchaud JC, Monteil-Rivera F, Dumonceau J, Milonjic S (2003) Sorption of selenium anionic species on apatites and iron oxides from aqueous solutions. J Environ Radioactiv 70:61–72
Dungan RS, Yates SR, Frankenberger WT (2002) Volatilization and degradation of soil-applied dimethylselenide. J Environ Qual 31:2045–2050
Dynes JJ, Huang PM (1997) Influence of organic acids on selenite sorption by poorly ordered aluminum hydroxides. Soil Sci Soc Am J 61:772–783
Ehrlich HL (1997) Microbes and metals. Appl Microbiol Biotechnol 48:687–692
Eisler R (1985) Selenium hazards to fish, wildlife and invertebrates: a synoptic review. US Fish and Wildlife Service Biological Report, Maryland
Ellis DR, Salt DE (2003) Plants, selenium and human health. Curr Opin Plant Biol 6:273–279
Encinar JR, Ruzik R, Buchmann W, Tortajada J, Lobinski R, Szpunar J (2003) Detection of selenocompounds in a tryptic digest of yeast selenoprotein by MALDI time-of-flight MS prior to their structural analysis by electrospray ionization triple quadrupole MS. Analyst 128:220–224
Feroci G, Badiello R, Fini A (2005) Interactions between different selenium compounds and zinc, cadmium and mercury. J Trace Elem Med Biol 18:227–234
Ferri T, Sangiorgio P (2001) Selenium speciation in waters: role of dissolved polysaccharides on the mobilization process. Ann Chim 91:229–238
Fevrier L, Martin-Garin A, Leclerc E (2007) Variation of the distribution coefficient (K d) of selenium in soils under various microbial states. J Environ Radioactiv 97:189–205
Filella M, Town RM (2003) Implications of natural organic matter binding heterogeneity on understanding trace metal complexation in aquatic systems. J Phys IV France 107:479–482
Fleet ME, Liu X (2008) Accommodation of the carbonate ion in fluorapatite synthesized at high pressure. Am Mineral 93:1460–1469
Fleming GA (1980) Essential micronutrients II: iodine and selenium. In: Davis BE (ed) Applied soils trace elements, 1st edn. John Wiley & Sons, New York, pp 199–234
Fontes DE, Mills AL, Hornberger GM, Herman JS (1991) Physical and chemical factors influencing transport of microorganisms through porous-media. Appl Environ Microbiol 57:2473–2481
Fordyce F (2005) Selenium deficiency and toxicity in the environment. In: Selinus O (ed) Essentials of medical geology, 1st edn. Elsevier, Uppsala, pp 373–415
Fordyce F (2007) Selenium geochemistry and health. Ambio 36:94–97
Foster AL, Brown GE, Parks GA (2003) X-ray absorption fine structure study of As(V) and Se(IV) sorption complexes on hydrous Mn oxides. Geochim Cosmochim Acta 67:1937–1953
Francis AJ, Duxbury JM, Alexander M (1974) Evolution of dimethylselenide from soils. Appl Microbiol 28:248–250
Frankenberger WT, Karlson U (1994) Soil-management factors affecting volatilization of selenium from dewatered sediments. Geomicrobiol J 12:265–278
Frost RR, Griffin RA (1977) Effect of pH on adsorption of arsenic and selenium from landfill leachate by clay–minerals. Soil Sci Soc Am J 41:53–57
Fujita M, Ike M, Kashiwa M, Hashimoto R, Soda S (2002) Laboratory-scale continuous reactor for soluble selenium removal using selenate-reducing bacterium, Bacillus sp SF-1. Biotechnol Bioeng 80:755–761
Fukushi K, Sverjensky DA (2007) A surface complexation model for sulfate and selenate on iron oxides consistent with spectroscopic and theoretical molecular evidence. Geochim Cosmochim Acta 71:1–24
Ganje TJ, Whitehead EI (1958) Evolution of volatile selenium from a pierre shale supplied with selenium-75 as selenite or selenate. Proc S D Acad Sci 37:81–84
Gao XY, Zhang JS, Zhang L (2002) Hollow sphere selenium nanoparticles: their in vitro anti hydroxyl radical effect. Adv Mater 14:290–294
Géhin A, Greneche J-M, Tournassat C, Brendle J, Rancourt DG, Charlet L (2007) Reversible surface-sorption-induced electron-transfer oxidation of Fe(II) at reactive sites on a synthetic clay mineral. Geochim Cosmochim Acta 71:863–876
Ghosh A, Mohod AM, Paknikar KM, Jain RK (2008) Isolation and characterization of selenite- and selenate-tolerant microorganisms from selenium-contaminated sites. World J Microbiol Biotechnol 24:1607–1611
Goh KH, Lim TT (2004) Geochemistry of inorganic arsenic and selenium in a tropical soil: effect of reaction time, pH, and competitive anions on arsenic and selenium adsorption. Chemosphere 55:849–859
Gruebel KA, Davis JA, Leckie JO (1988) The feasibility of using sequential extraction techniques for arsenic and selenium in soils and sediments. Soil Sci Soc Am J 52:390–397
Gruebel KA, Davis JA, Leckie JO (1995) Kinetics of oxidation of selenite to selenate in the presence of oxygen, titania, and light. Environ Sci Technol 29:586–594
Gu BQ, Cheng TO (1987) The international textbook of cardiology. Pergamon, New York
Guo XM, Sturgeon RE, Mester Z, Gardener GK (2003) Photochemical alkylation of inorganic selenium in the presence of low molecular weight organic acids. Environ Sci Technol 37:5645–5650
Gustafsson JP, Johnsson L (1994) The association between selenium and humic substances in forested ecosystems–laboratory evidence. Appl Organomet Chem 8:141–147
Hansmann DD, Anderson MA (1985) Using electrophoresis in modeling sulfate, selenite, and phosphate adsorption onto goethite. Environ Sci Technol 19:544–551
Hartikainen H (2005) Biogeochemistry of selenium and its impact on food chain quality and human health. J Trace Elem Med Biol 18:309–318
Haug A, Graham RD, Christiphersen OA, Lyons GH (2007) How to use the world’s scarce selenium resources effectively to increase the selenium concentration in food. Microb Ecol Health Dis 19:209–228
Hayes KF, Roe AL, Brown GE, Hodgson KO, Leckie JO, Parks GA (1987) In situ X-ray absorption study of surface complexes–selenium oxyanions on α-FeOOH. Science 238:783–786
Hayes KF, Papelis C, Leckie JO (1988) Modeling ionic-strength effects on anion adsorption at hydrous oxide solution interfaces. J Colloid Interf Sci 125:717–726
Haygarth PM (1994) Global importance and global cycling of selenium. In: Frankenberger WT Jr, Benson S (eds) Selenium in the environment. Marcel Dekker, Inc., Hong Kong, pp 1–28
Haygarth PM, Fowler D, Sturup S, Davison BM, Jones KC (1994) Determination of gaseous and particulate selenium over a rural grassland in the UK. Atmos Environ 28:3655–3663
He ZLL, Yang XE, Stoffella PJ (2005) Trace elements in agroecosystems and impacts on the environment. J Trace Elem Med Biol 19:125–140
Herbel MJ, Blum JS, Oremland RS, Borglin SE (2003) Reduction of elemental selenium to selenide: experiments with anoxic sediments and bacteria that respire Se-oxyanions. Geomicrobiol J 20:587–602
Hiemstra T, Van Riemsdijk WH (1999) Surface structural ion adsorption modeling of competitive binding of oxyanions by metal (hydr)oxides. J Colloid Interf Sci 210:182–193
Hochella MF, Lower SK, Maurice PA, Penn RL, Sahai N, Sparks DL, Twining BS (2008) Nanominerals, mineral nanoparticles, and Earth systems. Science 319:1631–1635
Hockin SL, Gadd GM (2003) Linked redox precipitation of sulfur and selenium under anaerobic conditions by sulfate-reducing bacterial biofilms. Appl Environ Microbiol 69:7063–7072
Huang B, Zhang JS, Hou JW, Chen C (2003) Free radical scavenging efficiency of Nano-Se in vitro. Free Radic Biol Med 35:805–813
Hunter WJ (2007) An Azospira oryzae (syn Dechlorosoma suillum) strain that reduces selenate and selenite to elemental red selenium. Curr Microbiol 54:376–381
Hunter WJ, Kuykendall LD, Manter DK (2007) Rhizobium selenireducens sp Nov.: a selenite-reducing alpha-Proteobacteria isolated from a bioreactor. Curr Microbiol 55:455–460
IAEA (1994) Handbook of parameter values for the prediction of radionuclides transfer in temperate environments. International Atomic Energy Agency, Vienne
Ihnat M (1989) Occurence and distribution of Selenium. CRC Press, Florida
International Crystal Structure Database (2008). Institut Laue-Langevin. http://icsd.ill.fr
Irons R, Carlson BA, Hatfield DL, Davis CD (2006) Both selenoproteins and low molecular weight selenocompounds reduce colon cancer risk in mice with genetically impaired selenoprotein expression. J Nutr 136:1311–1317
Jacobs LW (1989) Selenium in agriculture and the environment. Soil Science of America Special Publication, Wisconsin
Jia X, Li N, Chen J (2005) A subchronic toxicity study of elemental Nano-Se in Sprague-Dawley rats. Life Sci 76:1989–2003
Jinadasa K, Dissanayake CB (1992) The effect of selenium on fluoride–clay interactions—possible environmental–health implications. Environ Geochem Health 14:3–7
Johansson L, Gafvelin G, Arner ESJ (2005) Selenocysteine in proteins—properties and biotechnological use. Biochim Biophys Acta-Gen Subj 1726:1–13
John MK, Saunders WMH, Watkinson JH (1975) Selenium adsorption by New Zealand soils I. Relative adsorption of selenite by representative soils and their relationship to soil properties. NZ J Agric Res 19:143–151
Johnson CC, Ge X, Green KA, Liu X (2000a) Selenium distribution in the local environment of selected villages of the Keshan disease belt, Zhangjiakou district, Hebei province, People’s Republic of China. Appl Geochem 15:385–401
Johnson EA, Rudin MJ, Steinberg SM, Johnson WH (2000b) The sorption of selenite on various cement formulations. Waste Manage 20:509–516
Jovari P, Delaplane RG, Pusztai L (2003). Structural models of amorphous selenium. Physical Review B 67
Kabata-Pendias A, Pendias H (1984) Trace elements in soils and plants. CRC Press, Florida
Kang Y, Yamada H, Kyuma K, Hattori T, Kigasawa S (1991) Selenium in soil humi-acid. Soil Sci Plant Nutr 37:241–248
Karlson U, Frankenberger WT (1989) Accelerated rates of selenium volatilization from California soils. Soil Sci Soc Am J 53:749–753
Kenward PA, Fowle DA, Yee N (2006) Microbial selenate sorption and reduction in nutrient limited systems. Environ Sci Technol 40:3782–3786
Kohrle J, Brigelius-Flohe R, Bock A, Gartner R, Meyer O, Flohe L (2000) Selenium in biology: facts and medical perspectives. Biol Chem 381:849–864
Lakin HW (1972) Selenium accumulation in soils and its absorption by plants and animals. Geol Soc Am Bull 83:181–189
Lauchli A (1993) Selenium in plants—uptake, functions, and environmental toxicity. Botanica Acta 106:455–468
Lemly AD (1993) Guidelines for evaluating selenium data from aquatic monitoring and assessment studies. Environ Monit Assess 28:83–100
Lemly AD (1997) A teratogenic deformity index for evaluating impacts of selenium on fish populations. Ecotoxicol Environ Saf 37:259–266
Lenz M, Gmerek A, Lens PNL (2006) Selenium speciation in anaerobic granular sludge. Inter J Environ Anal Chem 86:615–627
Lenz M, Smit M, Binder P, van Aelst AC, Lens PNL (2008a) Biological alkylation and colloid formation of selenium in methanogenic UASB reactors. J Environ Qual 37:1691–1700
Lenz M, Van Hullebusch ED, Farges F, Nikitenko S, Borca CN, Grolimund D, Lens PNL (2008b) Selenium speciation assessed by X-ray absorption spectroscopy of sequentially extracted anaerobic biofilms. Environ Sci Technol 42:7587–7593
Lenz M, Van Hullebusch ED, Hommes G, Corvini PFX, Lens PNL (2008c) Selenate removal in methanogenic and sulfate-reducing upflow anaerobic sludge bed reactors. Water Res 42:2184–2194
Levander OA, Burk RF (2006) Update of human dietary standards for selenium. In: Hatfield DL, Berry MJ, Gladyshev VN (eds) Selenium its molecular biology and role in human health, 2nd edn. Springer, New York, pp 399–410
Li J, Zheng CJ (1990) The handbook of the environmental background values. China Environmental Science Press, Beijing
Lo SL, Chen TY (1997) Adsorption of Se(IV) and Se(VI) on an iron-coated sand from water. Chemosphere 35:919–930
Losi ME, Frankenberger WT (1997) Reduction of selenium oxyanions by Enterobacter cloacae SLD1a-1: isolation and growth of the bacterium and its expulsion of selenium particles. Appl Environ Microbiol 63:3079–3084
Loyo RLD, Nikitenko SI, Scheinost AC, Simonoff M (2008) Immobilization of selenite on Fe3O4 and Fe/Fe3C ultrasmall particles. Environ Sci Technol 42:2451–2456
Luoma SN, Johns C, Fisher NS, Steinberg NA, Oremland RS, Reinfelder JR (1992) Determination of selenium bioavailability to a benthic bivalve from particulate and solute pathways. Environ Sci Technol 26:485–491
Mace N, Landesman C, Pointeau I, Grambow B, Giffaut E (2007) Characterisation of thermally altered cement pastes influence on selenite sorption. Adv Cem Res 19:157–165
Macy JM, Michel TA, Kirsch DG (1989) Selenate reduction by a Pseudomonas species—a new mode of anaerobic respiration. FEMS Microbiol Lett 61:195–198
Macy JM, Lawson S, Demolldecker H (1993) Bioremediation of selenium oxyanions in San Joaquin drainage water using Thauera selenatis in a biological reactor system. Appl Microbiol Biotechnol 40:588–594
Maier KJ, Foe CG, Knight AW (1993) Comparative toxicity of selenate, selenite, seleno-dl-methionine and seleno-dl-cystine to Daphnia–Magna. Environ Toxicol Chem 12:755–763
Manceau A, Charlet L (1994) The mechanism of selenate adsorption on goethite and hydrous ferric-oxide. J Colloid Interf Sci 168:87–93
Martens DA, Suarez DL (1999) Transformations of volatile methylated selenium in soil. Soil Biol Biochem 31:1355–1361
Masscheleyn PH, Delaune RD, Patrick WH (1991) Biogeochemical behavior of selenium in anoxic soils and sediments—an equilibrium thermodynamics approach. J Environ Sci Health A Environ Sci Eng Toxic Hazard Subst Control 26:555–573
Mayland HF (1994) Selenium in plant and animal nutrition. In: Frankenberger WT, Benson S (eds) Selenium in the environment, 1st edn. Marcel Dekker, New York, pp 29–46
McConnell D, Foreman DW (1978) Model for carbonate apatite. Inorg Chem 17:2039–2040
Minaev VS, Timoshenkov SP, Kalugin VV (2005) Structural and phase transformations in condensed selenium. J Optoelectron Adv Mater 7:1717–1741
Miyagi K, Sampson RW, Tsujino I, Gunther WHH, Krieg M, Sieber F (2003) Proteinated subnanoparticles of elemental selenium potentiate the anti-tumor effect of ionizing radiation and selected chemotherapeutic agents. Proc Am Assoc Cancer Res 44:385
Montes-Hernandez G, Fernandez-Martinez A, Charlet L, Renard F, Scheinost AC, Bueno M (2008). Synthesis of a Se(0)/calcite composite using hydrothermal carbonation of Ca(OH)2 coupled to a complex selenocystine fragmentation. Crystal Growth Des 8:2497–2504
Monteil-Rivera F, Fedoroff M, Jeanjean J, Minel L, Barthes MG, Dumonceau J (2000) Sorption of selenite (SeO3 2−) on hydroxyapatite: an exchange process. J Colloid Interf Sci 221:291–300
Moreno-Reyes R, Suetens C, Mathieu F, Begaux F, Zhu D, Rivera MT, Boelaert M, Neve J, Perlmutter N, Vanderpas J (1998) Kashin–Beck osteoarthropathy in rural Tibet in relation to selenium and iodine status. N Engl J Med 339:1112–1120
Mosher BW, Duce RA (1987) A global atmospheric selenium budget. J Geophys Res-Atmospheres 92:13289–13298
Mykkanen H, Humaloja T (1984) Effect of lead on the intestinal-absorption of sodium selenite and selenomethionine (Se-75) in chicks. Biol Trace Elem Res 6:11–17
Mykkanen HM, Metsaniitty L (1987) Selenium–mercury interaction during intestinal-absorption of Se-75 compounds in chicks. J Nutr 117:1453–1458
Myneni SCB, Tokunaga TK, Brown GE (1997) Abiotic selenium redox transformations in the presence of Fe(II, III) oxides. Science 278:1106–1109
Nakamaru Y, Tagami K, Uchida S (2005) Distribution coefficient of selenium in Japanese agricultural soils. Chemosphere 58:1347–1354
Nakamaru Y, Tagami K, Uchida S (2006) Effect of phosphate addition on the sorption-desorption reaction of selenium in Japanese agricultural soils. Chemosphere 63:109–115
Narasingarao P, Haggblom MM (2007) Pelobacter seleniigenes sp nov., a selenate respiring bacterium. Int J Syst Evol Microbiol 57:1937–1942
Neal RH (1995) Selenium. Blackie Academic & Professional, London
Neal RH, Sposito G (1989) Selenate adsorption on alluvial soils. Soil Sci Soc Am J 53:70–74
Neal RH, Sposito G, Holtzclaw KM, Traina SJ (1987a) Selenite adsorption on alluvial soils. 1. Soil composition and ph effects. Soil Sci Soc Am J 51:1161–1165
Neal RH, Sposito G, Holtzclaw KM, Traina SJ (1987b) Selenite adsorption on alluvial soils. 2. Solution composition effects. Soil Sci Soc Am J 51:1165–1169
Neumann PM, De Souza MP, Pickering IJ, Terry N (2003) Rapid microalgal metabolism of selenate to volatile dimethylselenide. Plant Cell Environ 26:897–905
Nriagu JO (1989) Occurrence and distribution of selenium. CRC Press, Florida
Nriagu JO (1991) Heavy metals in the environment. CEP Consultants, Edinburgh
Ogaard AF (1996) Effect of fresh and composted cattle manure on phosphate retention in soil. Acta Agric Scand B Soil Plant Sci 46:98–105
Ogaard AF, Sogn TA, Eich-Greatorex S (2006) Effect of cattle manure on selenate and selenite retention in soil. Nutr Cycl Agroecosyst 76:39–48
Oldfield JE (2006) Selenium: a historical perspective. Springer, US
Olin A, Nolang G, Osadchii E, Ohman LO, Rosen E (2005) Chemical thermodynamics of selenium. North Holland Elsevier Science Publishers B. V, The Netherlands
Oremland RS, Stolz JF (2000) Dissimilatory reduction of selenate and arsenate in nature. In: Lovley DR (ed) Environmental metal–microbe interaction, 1st edn. ASM Press, Washington DC, pp 199–224
Oremland RS, Zehr JP (1986) Formation of methane and carbon-dioxide from dimethylselenide in anoxic sediments and by a methanogenic bacterium. Appl Environ Microbiol 52:1031–1036
Oremland RS, Hollibaugh JT, Maest AS, Presser TS, Miller LG, Culbertson CW (1989) Selenate reduction to elemental selenium by anaerobic-bacteria in sediments and culture—biogeochemical significance of a novel, sulfate-independent respiration. Appl Environ Microbiol 55:2333–2343
Oremland RS, Steinberg NA, Maest AS, Miller LG, Hollibaugh JT (1990) Measurement of in situ rates of selenate removal by dissimilatory bacterial reduction in sediments. Environ Sci Technol 24:1157–1164
Oremland RS, Blum JS, Culbertson CW, Visscher PT, Miller LG, Dowdle P, Strohmaier FE (1994) Isolation, growth, and metabolism of an obligately anaerobic, selenate-respiring bacterium, strain Ses-3. Appl Environ Microbiol 60:3011–3019
Oremland RS, Herbel MJ, Blum JS, Langley S, Beveridge TJ, Ajayan PM, Sutto T, Ellis AV, Curran S (2004) Structural and spectral features of selenium nanospheres produced by se-respiring bacteria. Appl Environ Microbiol 70:52–60
Papelis C, Brown GE, Parks GA, Leckie JO (1995) X-ray-absorption spectroscopic studies of cadmium and selenite adsorption on aluminum-oxides. Langmuir 11:2041–2048
Parida KM, Gorai B, Das NN, Rao SB (1997) Studies on ferric oxide hydroxides 3. Adsorption of selenite (SeO3 2−) on different forms of iron oxyhydroxides. J Colloid Interf Sci 185:355–362
Peak D (2006) Adsorption mechanisms of selenium oxyanions at the aluminum oxide/water interface. J Colloid Interf Sci 303:337–345
Peak D, Sparks DL (2002) Mechanisms of selenate adsorption on iron oxides and hydroxides. Environ Sci Technol 36:1460–1466
Peak D, Saha UK, Huang PM (2006) Selenite adsorption mechanisms on pure and coated montmorillonite: an EXAFS and XANES spectroscopic study. Soil Sci Soc Am J 70:192–203
Pearce CI, Coker VS, Charnock JM, Pattrick RAD, Mosselmans JFW, Law N, Beveridge TJ, Lloyd JR (2008) Microbial manufacture of chalcogenide-based nanoparticles via the reduction of selenite using Veillonella atypica: an in situ EXAFS study. Nanotechnology 19:13
Pilon-Smits EAH, de Souza MP, Hong G, Amini A, Bravo RC, Payabyab ST, Terry N (1999) Selenium volatilization and accumulation by twenty aquatic plant species. J Environ Qual 28:1011–1018
Pointeau I, Hainos D, Coreau N, Reiller P (2006) Effect of organics on selenite uptake by cementitious materials. Waste Manage 26:733–740
Popov AI (1976) Correlation between molecular-structure and properties of amorphous selenium. J Phys C Solid State Phys 9:L675–L677
Presser TS (1994) The Kesterson effect. Environ Manage 18:437–454
Presser TS, Ohlendorf HM (1987) Biogeochemical cycling of selenium in the San Joaquin valley, California, USA. Environ Manage 11:805–821
Presser TS, Piper DZ (1998) Mass balance approach to selenium cycling through the San Joaquin valley: from source to river to bay. In: Frankenberger WT, Engberg RA (eds) Environmental chemistry of selenium, 1st edn. CRC Press, New York, pp 153–182
Presser TS, Swain WC (1990) Geochemical evidence for Se mobilization by the weathering of pyritic shale, San Joaquin valley, California, USA. Appl Geochem 5:703–717
Ranjard L, Prigent-Combaret C, Nazaret S, Cournoyer B (2002) Methylation of inorganic and organic selenium by the bacterial thiopurine methyltransferase. J Bacteriol 184:3146–3149
Rayman MP (2000) The importance of selenium to human health. Lancet 356:233–241
Rayman MP (2004) The use of high-selenium yeast to raise selenium status: how does it measure up? Br J Nutr 92:557–573
Reeder RJ, Lamble GM, Lee JF, Staudt WJ (1994) Mechanism of SeO4 2− substitution in calcite—an XAFS study. Geochim Cosmochim Acta 58:5639–5646
Reeder RJ, Schoonen MAA, Lanzirotti A (2006) Metal speciation and its role in bioaccessibility and bioavailability. In: Rosso JJ (ed) The emergent field of medical mineralogy and geochemistry, 1st edn. Mineralogical Society of America and the Geochemical Society, USA, pp 59–113
Reilly C (1997) Selenium in food and health. Blackie Academic & Professional, London
Riveros G, Lincot D, Guillemoles JF, Henriquez R, Schrebler R, Cordova R, Gomez H (2003) Redox and solution chemistry of the SeSO3 2−Zn-EDTA(2-) system and electrodeposition behavior of ZnSe from alkaline solutions. J Electroanal Chem 558:9–17
Robbins CW, Carter DL (1970) Selenium concentrations in phosphorus fertilizer materials and associated uptake by plants. Soil Sci Soc Am Proc 34:506
Roman-Ross G, Cuello GJ, Turrillas X, Fernandez-Martinez A, Charlet L (2006) Arsenite sorption and co-precipitation with calcite. Chem Geol 233:328–336
Rosenfeld I, Beath OA (1964) Selenium: geobotany, biochemistry toxicity and nutrition. Academic Press, New York
Ruby MV, Davis A, Schoof R, Eberle S, Sellstone CM (1996) Estimation of lead and arsenic bioavailability using a physiologically based extraction test. Environ Sci Technol 30:422–430
Ruby MV, Schoof R, Brattin W, Goldade M, Post G, Harnois M, Mosby DE, Casteel SW, Berti W, Carpenter M, Edwards D, Cragin D, Chappell W (1999) Advances in evaluating the oral bioavailability of inorganics in soil for use in human health risk assessment. Environ Sci Technol 33:3697–3705
Taylor SR, McLennan SM (1985) The continental crusts: its composition and evolution. Blackwell, Oxford
Saeki K, Matsumoto S, Tatsukawa R (1995) Selenite adsorption by manganese oxides. Soil Sci 160:265–272
Sarathchandra SU, Watkinson JH (1981) Oxidation of elemental selenium to selenite by Bacillus megaterium. Science 211:600–601
Sarin L, Yan A, Sanchez V, Kane A, Hurt RH (2007) Carbon nanoparticles as a vehicle for cell delivery of nano selenium. Proceedings of the American Chemical Society 233rd Meeting, Symposium “Carbon Nanoparticles and Nanotubes”, Chicago
Sarret G, Avoscan L, Carriere M, Collins R, Geoffroy N, Carrot F, Coves J, Gouget B (2005) Chemical forms of selenium in the metal-resistant bacterium Ralstonia metallidurans CH34 exposed to selenite and selenate. Appl Environ Microbiol 71:2331–2337
Scheidegger AM, Grolimund D, Cui D, Devoy J, Spahiu K, Wersin P, Bonhoure I, Janousch M (2003) Reduction of selenite on iron surfaces: a micro-spectroscopic study. J Phys Iv 104:417–420
Scheinost AC, Charlet L (2008) Selenite reduction by mackinawite, magnetite and siderite: XAS characterization of nanosized redox products. Environ Sci Technol 42:1984–1989
Scheinost AC, Kirsch R, Banerjee D, Fernandez-Martinez A, Zaenker H, Funke H, Charlet L (2008). X-ray absorption spectroscopy investigation of selenite reduction by Fe(II)-bearing minerals. J Contam Hydrol 102:228–245
Schlekat CE, Dowdle PR, Lee BG, Luoma SN, Oremland RS (2000) Bioavailability of particle-associated Se to the bivalve Potamocorbula amurensis. Environ Sci Technol 34:4504–4510
Schrauzer GN (2000) Selenomethionine: a review of its nutritional significance, metabolism and toxicity. J Nutr 130:1653–1656
Schulthess CP, Hu ZQ (2001) Impact of chloride anions on proton and selenium adsorption by an aluminum oxide. Soil Sci Soc Am J 65:710–718
Schwarz K, Foltz CM (1957) Selenium as an integral part of factor 3 against dietary necrotic liver degeneration. Soc 79:3292–3293
Seby F, Gautier MP, Lespes G, Astruc M (1997) Selenium speciation in soils after alkaline extraction. Sci Total Environ 207:81–90
Seby F, Potin-Gautier M, Giffaut E, Borge G, Donard OFX (2001) A critical review of thermodynamic data for selenium species at 25°C. Chem Geol 171:173–194
Seiler RL (1998) Prediction of lands susceptible to irrigation-induced selenium contamination of water. CRC Press, New York
Selim HM, Sparks D (2001) Heavy metals release in soils. CRC Press, Boca Raton
Shongsheng J, Jingru G, Shan J, Chunsheng L, Anzhi C, Ming H, Shaoyong W, Shilin L (1997) Determination of the half-life of 79Se with the accelerator mass spectrometry technique. Nucl Instrum Methods B 123:405–409
Shrift A (1964) Selenium cycle in nature. Nature 201:1304–1305
Sieber F (2003) Selenium in oxidation state zero is a potent and selective anti-leukemia/lymphoma agent. Exp Hematol 31:119–120
Sieber F, Gunther WHH, Daziano JP, Krieg-Kowald M, Bousbaa J, RJ Bula (2007) Patent: method of making and the use of cytotoxic agents containing elemental selenium. QB, LLP, USA
Sirichakwal PP, Puwastien P, Polngam J, Kongkachuichai R (2005) Selenium content of Thai foods. J Food Compost Anal 18:47–59
Sneddon IR, Garelick H, Valsami-Jones E (2005) An investigation into arsenic(V) removal from aqueous solutions by hydroxylapatite and bone-char. Mineral Mag 69:769–780
Sors TG, Ellis DR, Salt DE (2005) Selenium uptake, translocation, assimilation and metabolic fate in plants. Photosynth Res 86:373–389
Spallholz JE (1994) On the nature of selenium toxicity and carcinostatic activity. Free Radic Biol Med 17:45–64
Sposito G (2004) The surface chemistry of natural particles. Oxford University Press, New York
Steinberg NA, Oremland RS (1990) Dissimilatory selenate reduction potentials in a diversity of sediment types. Appl Environ Microbiol 56:3550–3557
Stolz JF, Oremland RS (1999) Bacterial respiration of arsenic and selenium. FEMS Microbiol Rev 23:615–627
Stolz JE, Basu P, Santini JM, Oremland RS (2006) Arsenic and selenium in microbial metabolism. Annu Rev Microbiol 60:107–130
Su CM, Suarez DL (2000) Selenate and selenite sorption on iron oxides: an infrared and electrophoretic study. Soil Sci Soc Am J 64:101–111
Sundberg-Jones SE, Hassan SM (2007) Macrophyte sorption and bioconcentration of elements in a pilot constructed wetland for flue gas desulfurization wastewater treatment. Water Air Soil Pollut 183:187–200
Sutton R, Sposito G (2005) Molecular structure in soil humic substances: the new view. Environ Sci Technol 39:9009–9015
Tam SC, Chow A, Hadley D (1995) Effects of organic-component on the immobilization of selenium on iron oxyhydroxide. Sci Total Environ 164:1–7
Tamari Y, Ogawa H, Fukumoto Y, Tsuji H, Kusaka Y (1990) Selenium content and its oxidation-state in igneous rocks, rock-forming minerals, and a reservoir sediment. Bull Chem Soc Jpn 63:2631–2638
Tastet L, Schaumloffel D, Lobinski R (2008) ICP-MS-assisted proteomics approach to the identification of selenium-containing proteins in selenium-rich yeast. J Anal At Spectrom 23:309–317
Terry N, Zayed AM, de Souza MP, Tarun AS (2000) Selenium in higher plants. Annu Rev Plant Physiol Plant Mol Biol 51:401–432
Tessier E, Amouroux D, Abril G, Lemaire E, Donard OFX (2002) Formation and volatilisation of alkyl-iodides and -selenides in macrotidal estuaries. Biogeochemistry 59:183–206
Thayer JS (2002) Biological methylation of less-studied elements. Appl Organomet Chem 16:677–691
Virtamo J, Valkeila E, Alfthan G, Punsar S, Huttunen JK, Karvonen MJ (1985) Serum selenium and the risk of coronary heart-disease and stroke. Am J Epidemiol 122:276–282
Virupaksha TK, Shrift A (1965) Biochemical differences between selenium accumulator and non-accumulator Astragalus species. Biochimica Biophysica Acta 107:69–80
Wang MC, Chen HM (2003) Forms and distribution of selenium at different depths and among particle size fractions of three Taiwan soils. Chemosphere 52:585–593
Wang ZJ, Gao YX (2001) Biogeochemical cycling of selenium in Chinese environments. Appl Geochem 16:1345–1351
Wang ZJ, Xu Y, Peng A (1996) Influences of fulvic acid on bioavailability and toxicity. Biol Trace Elem Res 55:147–162
Watts CA, Ridley H, Condie KL, Leaver JT, Richardson DJ, Butler CS (2003) Selenate reduction by Enterobacter cloacae SLD1a-1 is catalysed by a molybdenum-dependent membrane-bound enzyme that is distinct from the membrane-bound nitrate reductase. FEMS Microbiol Lett 228:273–279
Wen HJ, Carignan J (2007) Reviews on atmospheric selenium: emissions, speciation and fate. Atmos Environ 41:7151–7165
Wessjohann LA, Schneider A, Abbas M, Brandt W (2007) Selenium in chemistry and biochemistry in comparison to sulfur. Biol Chem 388:997–1006
White AF, Benson SM, Yee AW, Wollenberg HA, Flexser S (1991) Groundwater contamination at the Kesterson reservoir, California 2. Geochemical parameters influencing selenium mobility. Water Resour Res 27:1085–1098
White PJ, Bowen HC, Parmaguru P, Fritz M, Spracklen WP, Spiby RE, Meacham MC, Mead A, Harriman M, Trueman LJ, Smith BM, Thomas B, Broadley MR (2004) Interactions between selenium and sulphur nutrition in Arabidopsis thaliana. J Exp Bot 55:1927–1937
Wijnja H, Schulthess CP (2000a) Interaction of carbonate and organic anions with sulfate and selenate adsorption on an aluminum oxide. Soil Sci Soc Am J 64:898–908
Wijnja H, Schulthess CP (2000b) Vibrational spectroscopy study of selenate and sulfate adsorption mechanisms on Fe and Al (hydr)oxide surfaces. J Colloid Interf Sci 229:286–297
Yamada H, Kase Y, Usuki M, Kajiyama S, Yonebayashi K (1999) Selective determination and formation of elemental selenium in soils. Soil Sci Plant Nutr 45:403–408
Yan R, Gauthier D, Flamant G, Peraudeau G (2001) Fate of selenium in coal combustion: volatilization and speciation in the flue gas. Environ Sci Technol 35:1406–1410
Yannopoulos SN, Andrikopoulos KS (2004) Raman scattering study on structural and dynamical features of noncrystalline selenium. J Chem Phys 121:4747–4758
Yu TR (1997) Chemistry of variable charge soils. Oxford University Press, New York
Yudovich YE, Ketris MP (2006) Selenium in coal: a review. Int J Coal Geol 67:112–126
Zawislanski PT, Benson SM, Terberg R, Borglin SE (2003) Selenium speciation, solubility, and mobility in land-disposed dredged sediments. Environ Sci Technol 37:2415–2420
Zehr JP, Oremland RS (1987) Reduction of selenate to selenide by sulfate-respiring bacteria-experiments with cell-suspensions and estuarine sediments. Appl Environ Microbiol 53:1365–1369
Zhang YQ, Frankenberger WT (1999) Effects of soil moisture, depth, and organic amendments on selenium volatilization. J Environ Qual 28:1321–1326
Zhang YQ, Moore JN (1996) Selenium fractionation and speciation in a wetland system. Environ Sci Technol 30:2613–2619
Zhang PC, Sparks DL (1990) Kinetics of selenate and selenite adsorption desorption at the goethite water interface. Environ Sci Technol 24:1848–1856
Zhang JS, Gao XY, Zhang LD, Bao YP (2001) Biological effects of a nano red elemental selenium. Biofactors 15:27–38
Zhang YQ, Zahir ZA, Frankenberger WT (2003) Factors affecting reduction of selenate to elemental selenium in agricultural drainage water by Enterobacter taylorae. J Agric Food Chem 51:7073–7078
Zhang YQ, Zahir ZA, Frankenberger WT (2004) Fate of colloidal-particulate elemental selenium in aquatic systems. J Environ Qual 33:559–564
Zhang ES, Wang HL, Yan XX, Zhang LD (2005) Comparison of short-term toxicity between Nano-Se and selenite in mice. Life Sci 76:1099–1109
Zieve R, Peterson PJ (1981) Factors influencing the volatilization of selenium from soil. Sci Total Environ 19:277–284
Zuyi T, Taiwei C, Jinzhou D, XiongXin D, Yingjie G (2000) Effect of fulvic acids on sorption of U(VI), Zn, Yb, I and Se(IV) onto oxides of aluminum, iron and silicon. Appl Geochem 15:133–139
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fernández-Martínez, A., Charlet, L. Selenium environmental cycling and bioavailability: a structural chemist point of view. Rev Environ Sci Biotechnol 8, 81–110 (2009). https://doi.org/10.1007/s11157-009-9145-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11157-009-9145-3