
Abstract
Insulin resistance is a major disorder that links obesity to type 2 diabetes mellitus (T2D). It involves defects in the insulin actions owing to a reduced ability of insulin to trigger key signaling pathways in major metabolic tissues. The pathogenesis of insulin resistance involves several inhibitory molecules that interfere with the tyrosine phosphorylation of the insulin receptor and its downstream effectors. Among those, growing interest has been developed toward the protein tyrosine phosphatases (PTPs), a large family of enzymes that can inactivate crucial signaling effectors in the insulin signaling cascade by dephosphorylating their tyrosine residues. Herein we briefly review the role of several PTPs that have been shown to be implicated in the regulation of insulin action, and then focus on the Src homology 2 (SH2) domain-containing SHP1 and SHP2 enzymes, since recent reports have indicated major roles for these PTPs in the control of insulin action and glucose metabolism. Finally, the therapeutic potential of targeting PTPs for combating insulin resistance and alleviating T2D will be discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Obesity, once considered an endemic medical condition of the rich or a featuring syndrome of several unfortunate genetic variants, has now become one of the greatest global pandemics of the 21st century. The prevalence of obesity has doubled between 1980 and 2008 with the WHO estimates of over 300 million clinically obese adults in 2004 [1], which poses a serious health threat with the associated non-communicable diseases, including atherosclerosis, metabolic syndrome, non-alcoholic fatty liver diseases (NAFLD), type 2 diabetes (T2D), and even some cancers. As a common link between obesity and type 2 diabetes [2–8], insulin resistance is believed to be a sine qua non in the pathogenesis of obesity-associated metabolic diseases [9].
The development of insulin resistance in obesity is due to various conditions, mostly initiated by adipose tissue dysfunction [10–13]. Elevated circulating levels of NEFA (non-esterified fatty acids) or free fatty acids (FFA) released in abnormally high quantity by the adipose tissue and decreased FFA clearance in obesity [14–16] has been shown to be one main cause of insulin resistance [17]. By attenuating insulin signaling, FFA impair insulin-mediated glucose uptake in muscles and fat [18–21], suppression of glucose production from the liver [13, 21–23], and pancreatic insulin secretion [24–26]. In addition, adipocyte-derived hormones, metabolites, and inflammatory cytokines released from the overly expanded adipose tissue also down-regulate insulin sensitivity in all major insulin-target cells [11, 13, 27]. Altogether with compensatory hyperinsulinemia and overactivation of nutrient sensing mechanisms such as the mTOR/S6K1 pathway [28], insulin resistance develops along with impaired insulin secretion, leading the way to T2D and other related metabolic disorders. In order to understand this syndrome, the insulin signaling pathways and their regulation have been investigated intensively to unravel this complex machinery. One crucial part of the regulation of insulin signaling pathways is the negative feedback mechanism provided by phosphatases, especially the protein tyrosine phosphatases (PTPs). With only a few PTPs being extensively investigated in the field of metabolism, the importance of many other PTPs remains in the shadow. In this review, we will briefly update the major insulin signaling pathways involved in metabolic regulation, and then discuss the role of PTPs in the modulation of insulin action and energy metabolism, with a particular focus on recent findings that have unraveled an unsuspected metabolic role for the SH2-domain-containing PTPs, namely SHP1 and SHP2.
2 Insulin action and signaling
2.1 Insulin’s metabolic actions
In clinical applications, the most important function of insulin is to maintain glucose homeostasis, as insulin is the most potent endogenous blood glucose-lowering endocrine factor. Regardless of the fasting, feeding, or intermediate states, blood glucose is controlled within a narrow window of 4 to 7 mM in normal individuals. This tight regulation of glucose results from a balance of intestinal absorption, hepatic storage and production, and uptake/metabolism in peripheral tissues, which is fine-tuned by insulin. Through stimulating the translocation of GLUT4 glucose transporters from intracellular locations to the cell surface, insulin induces glucose uptake in fat and skeletal muscle, the latter accounting for up to 75 % of whole-body glucose disposal in the post-absorptive state [29]. In many cell types, insulin promotes glucose storage by increasing glycogenesis and, in some organs such as the liver, by suppressing glucose output via inhibition of glycogenolysis and gluconeogenesis.
From an integrative physiological point of view, insulin exerts multiple functions in many tissues to promote anabolism and proper growth as well as differentiation [30–33]. Besides its anabolic actions in glucose metabolism, insulin also stimulates the uptake of fatty acids and amino acids. By increasing the expression and/or activity of enzymes responsible for glycogen, lipid, and protein synthesis and inhibiting the expression and/or activity of those that catalyze degradation in major tissues, insulin promotes the storage of nutrients and at the same time inhibits their release via its ability to suppress glycogenolysis, lipolysis, and proteolysis [34]. In terms of energy homeostasis, insulin regulates many proteins and enzymes involved in various metabolic pathways in part through inhibiting the release of glucagon [35], a catabolic hormone secreted by the pancreatic α cells. Insulin also directly acts in the CNS to regulate body weight, reproduction, peripheral glucose and lipid metabolism [36–41]. In cardiac muscle, insulin induces relaxation with a positive inotropic effect [42, 43]. Electrolyte homeostasis is also regulated by insulin, as it stimulates potassium uptake while inhibiting renal sodium excretion [44, 45]. In the vasculature, insulin acutely stimulates endothelial function, but when its levels are abnormally elevated, such as in insulin resistance states, it can also promote atherogenenesis through enhancement of vascular smooth muscle cell proliferation [46]. Insulin also plays an important role in aging by down-regulating autophagy [47]. Finally, insulin can regulate its own expression and secretion by an autocrine action in the pancreatic β-cells [48, 49] and by promoting GLP-1 secretion from the intestinal L-cells [50].
Given the vast functions of insulin in numerous sites of action, regulation of insulin signaling is hence a vital process that can be targeted by multiple pathways implicated in the development of metabolic disorders such as insulin resistance.
2.2 Insulin signaling
The insulin receptor (IR) is a member of a highly conserved subfamily of receptor tyrosine kinases (RTK), including the insulin-like growth factor (IGF)-I receptor and the insulin receptor-related receptor (IRR). Functional hybrids of IR, IGF-I receptor, and IRR also exist in many different cell types [51], and the activity of each receptor is dependent on the other partner within the hybrid [52]. In a stable tetrameric form with allosteric enzymatic functions, IR consists of an extracellular domain containing two α-subunits able to inhibit the intrinsic tyrosine kinase activity of the two transmembrane β-subunits with multiple tyrosine residues. Upon insulin binding to the α-subunits, the repression of the β-subunits is lifted and their kinase activity increases as they phosphorylate each other on specific tyrosine sites in an intramolecular reaction (transautophosphorylation) followed by a rapid conformational change [53, 54]. This is an important event, as it enables the insulin receptor to phosphorylate and activate docking effectors (IR substrates) for propagation of insulin signaling. Any reduction in IR tyrosine phosphorylation leads to decreased signal transduction downstream and results in down-regulation of its metabolic actions.
So far, at least nine immediate IR substrates have been identified. Four out of six identified members of the IRS protein family, IRS-1, -2, -3, and -4, have been confirmed to propagate IR signal transduction in known mechanisms. The other five IR substrates are Gab-1 (Grb2-associated-binding protein), p60Dok (60-kDa tyrosine phosphorylated protein), Cbl (Casitas B-lineage lymphoma), APS (adapter protein with a PH and SH2 domain), and isoforms of Shc (SH2-domain containing proteins) [34, 55–58].
A critical step in insulin signaling is the activation of the IRS proteins, especially IRS-1 and IRS-2 which are predominantly expressed in metabolic tissues. Although both isoforms are major signal transducers that are regulated in a similar fashion and involved in insulin-mediated functions, IRS-1 and IRS-2 exert specific functions in different tissues or cell types. For example, IRS-1 predominantly activates pathways leading to general cell growth and GLUT4 translocation in muscle cells whereas IRS-2 is more important in pancreatic β-cell survival/growth, neuronal cell growth, reproduction, food intake, and hepatic insulin action [59–63]. The role of IRS-2 in carbohydrate and lipid metabolism seems more crucial in comparison. IRS-2 knockout mice develop full-blown type 2 diabetes, while IRS-1 null mice are only mildly insulin-resistant with growth retardation, because its absence is partially compensated by IRS-2 [64–67]. Interestingly, the existence of a dynamic relay between IRS-1 and IRS-2 in hepatic insulin signaling has been proposed in which IRS-2 mainly functions during fasting and immediately after refeeding, while IRS-1 functions primarily after refeeding [68]. Not much is known of IRS-3 and IRS-4, but they have been shown to negatively regulate the IGF-1 signaling pathway by inhibiting the activation of IRS-1 and IRS-2, perhaps by competition [69]. The general structure of an IRS protein consists of a well conserved PH (pleckstrin homology) domain at the extreme N-terminus for membrane localization, a phosphotyrosine binding domain (PTB) for IR binding, the carboxy terminal rich in tyrosine phosphorylation motifs for activation, and special YMXM motifs for docking SH2 domain-containing downstream signaling molecules, such as PI3K and SHP2 (Fig. 1) [70].

Example scheme of IR-IRS-PI3K/SHP2 binding. Adapted from White [70]. The IRS-signalling system: a network of docking proteins that mediate insulin action. Mol Cell Biochem. 1998 May;182(1–2):3–11
Regulation of tyrosine phosphorylation, degradation, or binding of both IR and IRS proteins directly affects three main downstream insulin signaling pathways implicated in metabolic regulation, namely the phosphoinositol 3-kinase (PI3K) pathway, the mitogen activated protein kinase (MAPK) pathway, and the mammalian target of rapamycin (mTOR) pathway [34].
2.3 The major insulin signaling pathways
2.3.1 The PI3K pathway
As an important signaling molecule in many insulin-triggered metabolic processes, PI3K is activated via the catalytic p110 subunit after binding to IRS with its SH2-domain containing p85 regulatory subunit (Fig. 1) [71]. It then phosphorylates phosphotidylinositol-4,5-bisphosphate (PIP2) to produce phosphotidylinositol-3,4,5-triphosphate (PIP3), which binds to PH domains of various signaling molecules and alters their activity or localization [72]. In general, PIP3 activates three classes of signaling molecules, the AGC family of serine/threonine protein kinases, guanine nucleotide-exchange proteins of the Rho family of GTPase, and the Tec family of tyrosine kinases [34, 73–75]. The most well recognized serine/threonine kinase in the AGC family, Akt/PKB, interacts with PIP3 through its PH domain for better membrane localization in order to be catalytically activated upon threonine 308 (T308) phosphorylation by another family member, phosphoinositide-dependent kinase (PDK) 1 [76, 77]. For complete activation of Akt, serine 473 (S473) is phosphorylated by mTORC2, which can further facilitate T308 phosphorylation by PDK1 [78]. Active Akt can also positively reinforce IRS-1 signaling by direct phosphorylation on its PTB domain [79]. PDK-1 also activates the other AGC family members, atypical protein kinase C (aPKC) isoforms ζ and λ, leading to Akt-independent downstream signal transduction [80, 81]. Signaling by Akt and aPKC is essential for different biological responses, including insulin-stimulated translocation of GLUT4 and general protein synthesis [34, 70, 82]. PI3K-Akt signaling also mediates insulin-stimulated glycogenesis by phosphorylation-dependent deactivation of GSK3 (glycogen synthase kinase 3) [83] and relays IR signals to suppress gluconeogenesis by regulating gene expression of important enzymes via phosphorylation-dependent regulation of the Forkhead transcription factor (Fkhr/FoxO1) and PPARγ co-activator 1α (PGC-1α) [84–88].
2.3.2 The MAPK pathway
The other branch of downstream signaling from IRS proteins is the MAPK pathway, leading mostly to the regulation of general gene expression. IRS-1 or Shc can directly interact with growth factor receptor-bound protein 2 (Grb2) to relocate SOS (Son-of-sevenless) to the cell membrane thus activating Ras, a small GTPase that initiates a cascade of serine kinase activation from Raf (proto-oncogene serine/threonine-protein kinase) to MEK (MAPK kinase), then to MAPK/ERK. Once MAPK/ERK is activated, it is translocated into the nucleus to modulate gene expression of various proteins via direct regulation of transcription factors (TF) [89, 90]. The activation of Ras also requires the stimulation of SHP2 (SH2 domain-containing protein tyrosine phosphatase 2), a known tyrosine phosphatase with multiple positive roles in conducting signal transduction in different pathways, including insulin signaling [34, 70, 91, 92].
2.3.3 The mTOR pathway
Major protein synthesis and degradation-repression actions of insulin, including its inhibition of autophagy [47, 93], are carried out by the activation of the mTOR pathway. The signaling molecule mTOR is a special member of the PI3K family with only serine/threonine kinase capabilities [94, 95]. The detailed activation and downstream signaling of the mTOR pathway is extensive and complex involving the formation, regulation, and downstream signaling of the two complexes, mTORC1 and mTORC2. Comprised of mTOR, GβL, PRAS40, deptor, and raptor, mTORC1 controls transcription, ribosome biogenesis, protein/lipid synthesis, nutrient transport, autophagy, and other processes related to cell growth, with the help of its downstream effectors including ribosomal protein S6 kinase (S6K) and eukaryotic initiation factor 4E binding protein (4E-BP). On the other hand, mTORC2, formed by mTOR, GβL, protor-1/2, mSIN1, deptor, and rictor, regulates cytoskeleton and cell survival via activation of Akt and PKCα [78, 95–98]. Both mTORC1 and mTORC2 are activated by insulin and other growth factors through the IRS-PI3K-Akt pathway, as mTOR itself is a direct target of Akt [99–102]. A key role of mTOR signaling in insulin action is to provide a negative feedback inhibition at the level of IRS-1. Chronic activation of the mTORC1/S6K1 signaling by nutrients or prolonged insulin treatment increases the inhibitory serine phosphorylation of IRS-1, leading to reduced IRS-1 function and impaired activation of the PI3K-Akt pathway [28, 103]. On the other hand, acute inhibition of mTORC1 by rapamycin restores insulin-stimulated activation of the PI3K-Akt pathway and sensitizes insulin-mediated glucose transport in muscle and adipose cells [104, 105].
2.4 Inhibition of insulin signaling and insulin resistance
Throughout evolution, almost all biological processes are efficiently maintained by both positive and negative feedback mechanisms, especially in the regulation of energy homeostasis, and this holds true for insulin-mediated metabolic processes. In normal physiological conditions in healthy individuals, the inhibitory regulations of insulin signaling is essential for protecting the system from over-stimulation and for fine-tuning the signal transduction to produce the appropriate physiological effects. In disease states such as obesity, however, this balance is disturbed as insulin resistance develops from the overactivation of the negative regulators by various internal and external stimuli, including nutrients and inflammatory cytokines such as FFA and TNFα (tumor necrosis factor-α), respectively [106, 107].
3 Phosphatases: Impact on insulin signaling
Contrary to the action of phosphorylases and kinases, phosphatases are enzymes that remove the phosphate entity from cellular substrates. Phosphatases dephosphorylate their substrates mostly via direct binding followed by hydrolyzing the target phosphate into a phosphate ion. Dephosphorylation by phosphatases is as important as phosphorylation, because it regulates normal cellular processes that impact all physiological systems. Phosphatases discovered to date are divided into different classes according to their substrate preferences. The major classes are histidine phosphatases, lipid phosphatases, serine/threonine phosphatases (STPs), protein tyrosine phosphatases (PTPs), and dual-specificity phosphatases that target both phospho-Tyr and phospho-Ser/Thr residues. In the insulin signaling pathways, phosphatases take the regulatory control in almost every step of the cascade.
3.1 Lipid phosphatases
Phospholipids and sphingolipids are signaling molecules whose activities are regulated by five groups of different lipid phosphatases. In the signal transduction from PI3K to Akt, the two best known negative regulators, lipid phosphatases PTEN (phosphatase and tensin homologue) and SHIP2 (SH2 domain-containing inositol-5-phosphatase 2) interfere by dephosphorylating PIP3 at the 3′ and 5′ positions, respectively [108–111]. PTEN has also been reported to directly dephosphorylate Akt [112]. In the mTOR pathway, Lipin 1, a phosphatase with the ability of a transcriptional coactivator, down-regulates mTORC1-stimulated expression of genes involved in de novo lipogenesis (SREBP-1, FAS, SCD), while promoting FA oxidation by up-regulating the expression of PGC-1α/PPARα target genes (ACO, PPARα, CPT-1, medium and very long chain ACD) [113, 114].
3.2 Serine/threonine phosphatases (STPs)
Several STPs have been shown to regulate insulin signaling pathways. PP1 is a positive mediator of insulin, as it inhibits glycogen phosphorylase activity and stimulates glycogen synthase activity as well as SREBP-1 mobilization [115, 116]. PP2A selectively impairs hepatic insulin glucoregulation through dephosphorylation-mediated activation of FoxO1 and deactivation of Akt, which can also occur through other STPs such as PHLPP1/2 and PP2B (calcineurin) [112, 117, 118]. Increased PHLPP levels are detected in obese subjects with insulin resistance [119]. Phosphorylation of Akt at T308 and S473 can be directly down-regulated by PP1, PP2A, and PHLPP [120–122].
3.3 Protein tyrosine phosphatases (PTPs)
In normal physiological conditions, the phosphorylation of tyrosyl residues on all signaling molecules is controlled with a balanced activity between the tyrosine kinases and phosphatases. Since the discovery of the zinc inhibition on a phospho-tyrosyl-protein phosphatase in 1981 [123], PTPs have been found to play an important role in this steady-state balance by catalyzing tyrosine dephosphorylation. PTPs can be divided into four groups based on their catalytic domains; class I, II, and III cysteine-based PTPs and class IV aspartate-based PTPs [124]. Characterized by a conserved active signature motif, the cysteine-based PTPs are subdivided into either classical phospho-tyrosine (pY)-specific PTPs or dual specificity PTPs; the classical PTPs are further classified as receptor-like PTPs (RPTP), non-RPTP, and cytosolic PTPs [125, 126]. Tyrosine phosphorylation is the key activation of IR and IRS proteins, but it is easily undone either directly or indirectly by PTPs, such as LAR, PTP1B, SHP1 and SHP2 [127–133]. The inhibitory regulation of insulin signaling by these PTPs will be discussed in details below.
4 Protein tyrosine phosphatases and insulin resistance
PTPs have been shown to play a role in the development of insulin resistance in classic metabolic tissues through inhibiting the activation of several molecules in the insulin signaling cascade. However, there is growing evidence that PTPs also modulate insulin sensitivity and metabolism through their actions in the central nervous system (CNS), the immune system, the gastrointestinal tract, particularly the intestinal microbiota [134–142]. These actions of several PTPs will be briefly reviewed in the following sections.
4.1 LAR and PTP1B: Historically featured PTPs
4.1.1 LAR and the family
Leukocyte common antigen related (LAR), encoded by PTPRF, is one of the first tyrosine phosphatases identified to directly associate with the insulin receptor to negatively regulate insulin signaling [127, 143]. Being the most abundant PTP in the liver and endothelial cells, LAR is also expressed in insulin target tissues, the skeletal muscles and adipose tissue, at low or moderate level, respectively [144]. The general reduction in LAR activity enhances insulin-stimulated insulin-receptor autophosphorylation and downstream substrate activation and signaling [133, 145, 146]. Although one murine model with complete LAR deficiency exhibits a paradoxical phenotype of heightened insulin sensitivities and secondary abnormalities resulting in resistance to insulin-stimulated glucose disposal and inhibition of hepatic glucose output [147], increased LAR in obesity has been observed, and its overexpression in muscle induces insulin resistance and IRS-2 deactivation [148–150]. In the CNS, LAR, together with its two other family members, PTPδ and PTPσ, have been recognized to be important synaptic adhesion molecules and responsible for basic neuronal functions [151], which can potentially affect the central control of energy homeostasis. LAR activity in the hypothalamus has been found to associate with central insulin resistance [152]. PTPσ is crucial for the growth and development of the hypothalamo-pituitary axis, a key regulatory center of glucose homeostasis [153–155].
4.1.2 PTP1B
Being the primary human PTP with hereditary variants associated with T2D and metabolic disorder [156], PTP1B, encoded by PTPN1 and expressed ubiquitously in almost all tissues [144], has been the most well studied PTP involved in the insulin signaling pathway. Increased activity and expression of PTP1B have been observed in skeletal muscle, liver and brain in obese and insulin-resistant animal models [157–159]. Systemic deficiency of PTP1B results in augmented insulin sensitivity along with a protection against diet-induced weight gain [160], as it is known to directly dephosphorylate IR and IRS-1 [129, 130]. In the liver, hepatocyte-specific PTP1B deficiency prevents diet-induced hepatic steatosis and insulin resistance [161, 162], and the inducible knockdown improves glucose and lipid homeostasis in adult mice [163]. In adipocytes, PTP1B modulates insulin-stimulated glucose uptake by scrambling insulin signaling and GLUT4 vesicle trafficking via Munc18c, an essential regulator of SNARE-mediated budding and fusion of GLUT4 vesicles [164]. PTP1B also negatively regulates adipocyte development and may mediate TNFα action to impair adipocyte differentiation in obesity [165]. However, mice with adipocyte-specific deletion of PTP1B exhibit mild glucose intolerance with larger adipocyte size and decreased leptin sensitivity [166]. In muscles, MCK-Cre driven depletion of PTP1B augments systemic insulin sensitivity and glucose homeostasis [167]. During obesity-associated chronic inflammation, PTP1B, with further induced expression [159, 168], either promotes or protects against insulin resistance through modulation of immune cell functions and cytokine production [169–171]. In the CNS, PTP1B activity is associated with hypothalamic insulin resistance and leptin signaling [152, 172–174], as neuronal deletion of PTP1B improves both leptin and insulin signaling as well as general energy homeostasis [175, 176]. Within the cells, PTP1B contributes to the development of insulin resistance by potentiating ER stress both peripherally and centrally [142, 177]. By controlling cellular filament organization via the regulation of keratin 8 solubility [178], PTP1B can also affect cellular trafficking signaled by insulin [140].
4.2 Other PTPs: Small but big
Although both LAR and PTP1B are well known regulators of insulin’s metabolic actions, insulin signaling is also regulated by many other PTPs, which are mostly known to be negative regulators in insulin target tissues (Table 1).
Aside from the direct negative roles in insulin signaling, these PTPs can also attenuate insulin sensitivity indirectly. Since the catalytic and adaptor functions of PTPα are required for IL-1β signaling, PTPα promotes the inflammation-associated deterioration of insulin sensitivity in obesity [196]. TC-PTP shares many similarities in activity and function with PTP1B, especially in the negative regulation of insulin signaling and glucose metabolism, which have been well reviewed [197]. Expressed in great abundance in the liver [144], TC-PTP, together with PTP1B, is known to modulate ER stress, which potentiates the development of obesity-induced insulin resistance [173]. In neuronal cells, similar to PTP1B, elevated expression of TC-PTP in obesity also contributes to central leptin resistance [198]. PTP-PEST binding to Shc down-regulates insulin-induced ERK activation in human cells [190].
Another class of PTP containing two tandem SH2 domains at the N-terminus and a catalytic phosphatase C-terminal domain is composed of only two family members, SHP1 and SHP2, which are discussed in details in the following section.
5 SHP1 and SHP2
Though sharing much similarity in structure and activity, SHP1 and SHP2 have been commonly believed to play opposing roles, with SHP1 being the general negative regulator and SHP2 the positive one, even in the modulation of insulin signaling pathways [131, 199–202]. However, more and more studies have challenged this simplistic view [203].
5.1 Structure, expression and activity
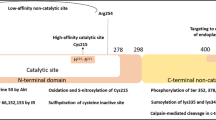
Also known as SH-PTP1, PTP1C, HCP and SHP, SHP1 is encoded by the PTPN6 gene. Alternative splicing results in two major SHP1 isoforms, the 68-kDa SHP1a (mouse) or SHP1-1 (human) form and the slightly longer form of SHP1b (mouse) or SHP1-2 (human) encompassing two extra amino acids, whose enzymatic activities are fairly comparable [204]. In human, an active 70-kd long form of SHP1, the SHP1L, also exists, although with limited substrate affinity and activity [202, 205]. SHP1 is abundantly expressed in hematopoietic cells, neuronal cells, astrocytes, oligodendrocytes, microglia, the epithelial cells of the prostate, and insulin-sensitive organs such as the liver and the skeletal muscle [131, 206–211], with the shorter form being predominant in hematopoietic cells and the longer form in cells of epithelial lineages [204]. SHP2, also known as SH-PTP2, PTP1D, PTP2C and SYP, is encoded by PTPN11 and only exists in one form [202]. Sharing 50 % similar sequences with SHP1 [202], SHP2 is however ubiquitously expressed [212].
Both SHP1 and SHP2 contain two SH2 domains at the N-terminus (N-SH2 and C-SH2) for substrate binding and the catalytic phosphatase domain at the C-terminus [213]. Inactive forms have a folded structure of the N-SH2 domain directly blocking the catalytic domain (Fig. 2a) [213, 214]. The phosphatase activity of both SHP1 and SHP2 is stimulated upon binding of pY-containing substrate to the SH2 domains (Fig. 2b), hence the substrate specificity determines the binding [213–215]. The active site of the catalytic domain contains important amino acid residues for substrate binding, stabilizing the negative charge of the phospho-tyrosine, and being the proton donor and acceptor in product release [216]. The catalytic pockets of SHP1 and SHP2 are only different in four residues, Arg352, Lys356, Arg358, and Asn359 in SHP1 versus Lys358, Arg362, Lys364, and Ser 365 in SHP2 [217]. The C-terminal end can also regulate PTP activity [202, 218, 219], as it can be phosphorylated on tyrosine residues, which are important for molecule recruitment that directly affects phosphatase activity upon various stimuli [202, 219, 220]. Both Src kinase and the insulin receptor have been shown to stimulate the phosphorylation of SHP1 and SHP2, which was the first observation linking these two PTPs with modulation of insulin signaling [203, 221, 222]. Both SHP1 and SHP2 can also be phosphorylated at certain serine residues in a PKCα-dependent manner [202], and their activity can be disrupted by NO-mediated S-nitrosylation at the active cysteine residue [223]. Interestingly, SHP1 can also exert autoregulation by binding to another SHP1 molecule to be inactivated (Fig. 2c) [224]. Most of the time, SHP1 and SHP2 are cytosolic PTPs, although they can signal from being bound to lipid rafts [202]. More interestingly, both PTPs also localize in the nucleus, indicating their potential role in the regulation of nuclear proteins and transcriptional regulation [225–229].

Protein structures and regulation of SHP1 and SHP2. a In the basal state, SHP1 and SHP2 are kept inactive, because the N-terminal SH2 domain (N-SH2) blocks the C-terminal catalytic domain by inserting into the catalytic cleft. b SHP1 and SHP2 are activated by binding of Tyr-phosphorylated substrates to the N- and C-terminal SH2-domains thereby releasing the internal inhibition. c The activity of SHP1 and SHP2 can also be modulated by forming homodimers via an intramolecular interaction between a N-terminal SH2 domain and a catalytic domain
5.2 Metabolic roles differentiate SHP1 and SHP2 functions
Although increased activity and expression of both SHP1 and SHP2 have been observed in major insulin target metabolic tissues in obese and insulin-resistant animal models [157, 158, 230], their substrate preference and specific pattern of expression and regulation enable them with distinct metabolic roles.
5.2.1 SHP1
The association of SHP1 with insulin signaling through the PI3K-Akt pathway empowers SHP1 as a regulator of insulin action. Under normal conditions, the p85 regulatory subunit of PI3K keeps the p110 catalytic subunit stabilized and inactivated. This inhibitory activity of p85 is relieved by binding of the N-terminal SH2 domain to Src family kinases, which is reversed by the association of SHP1 to maintain the inhibition of PI3K activity [231, 232]. Thus theoretically, in the absence of SHP1, insulin signaling activates the p110 subunit through the tyrosine phosphorylation of p85, allowing further propagation of signal transduction to activate Akt by phosphorylation. SHP1 also selectively binds and dephosphorylates PTEN, subsequently modulating signal transduction in the PI3K-Akt pathway by increasing PIP3 [233]. AT2 receptor-mediated SHP1 activation can lead to inhibition of IRS-2 activation upstream of the PI3K-Akt pathway [234]. However, the best evidence has come from the viable motheaten mice. These mice are markedly glucose tolerant and insulin sensitive as compared to their WT littermates. This has been found to be a result of enhanced IR signaling through IRS-PI3K-Akt in both liver and muscle [131]. Liver-targeted down-regulation of SHP1 expression and activity in normal mice by adenoviral expression of a catalytically inert mutant of SHP1 or by small hairpin RNA-mediated gene silencing reveals similar findings [131]. In addition, adenoviral introduction of DN-SHP1 containing a C453S mutation increases insulin-induced Akt phosphorylation and augments glucose uptake via GLUT4 up-regulation and glycogen synthesis in L6 myotubes and skeletal muscle tissue, confirming that SHP1 regulates insulin action in a cell-autonomous manner [235].
Interestingly, transcriptional activation of the SHP1 gene (Ptpn6) by overexpression of the homeodomain transcription factor Prep1 attenuates insulin signaling and glucose storage in hepatic cells, while Prep1-deficient mice show improved hepatic insulin action in association with reduced SHP1 expression [236], further supporting a key role for SHP1 in the control of liver glucose metabolism. As Prep1 expression is up-regulated in both diet-induced and genetically-driven obesity [236], this indirectly hints a possible increase in SHP1 expression in obesity. Accordingly, SHP1 gene and protein expression is found to be significantly up-regulated in metabolic tissues of mice with diet-induced obesity [230]. On the same diet regime, though hepatocyte-specific SHP1 knockout mice (Ptpn6 H-KO) develop obesity as their control WT littermates, they exhibit improved glucose tolerance and insulin sensitivity [230].
SHP1 also modulates insulin action and glucose homeostasis through its ability to inhibit insulin clearance by regulating the IR-associated complex of Cdk2/SHP1/β-catenin/CEACAM1 [237]. Being the primary organ of insulin clearance, liver is responsible for clearing 45–50 % of the insulin secreted from the pancreatic β-cells [238–240]. In the hepatocytes, like many ligand-receptor interactions, insulin-IR complexes are sequestered from the plasma membrane (PM) into the endosomes, where insulin-IR signal transduction takes place before insulin is dissociated from IR. When insulin signaling terminates upon insulin-IR dissociation, insulin is processed and IR is sorted then recycled back to the PM, or both are sent to lysosomes for degradation (Fig. 3) [241, 242].

Simplified scheme of IR-mediated insulin clearance in liver. Activation of IR by insulin binding induces insulin-IR-complex internalization through endocytosis in clathrin-coated vesicles. These complexes are passed from early to late endosomes, still retaining their signaling activity. During this process, insulin is dissociated from its receptor. Insulin degradation is initiated in the endosomes and finalized in the lysosomes, whereas IR can be either degraded after translocation to the lysosomes or recycled back to the plasma membrane
In general, IR-mediated insulin clearance has a positive role in subcellular insulin signaling and can be defective in abnormal conditions such as obesity, hepatic steatosis, and insulin resistance. Peripheral FFA infusion significantly alters hepatic insulin clearance rate, and portal FFA infusion reduces insulin clearance to an even greater extent [243]. Liver fat content also negatively regulates hepatic insulin clearance and action [244]. Prolonged insulin stimulation of IR and/or hyperinsulinemia also can augment lysosomal IR degradation, leading to IR down-regulation [245, 246]. IDE, the primary insulin degrading enzyme responsible for cellular insulin processing and degradation [247], is increased in insulin-resistant subjects [248]. The rate of IR-mediated insulin clearance is significantly reduced in obese patients, and can be a sign of insulin resistance [248]. In a mouse model of diet-induced obesity, insulin clearance rate is drastically deteriorated [249]. CEACAM1, a key regulator of insulin clearance and insulin action [249, 250], is a direct substrate of SHP1, which binds and dephosphorylates CEACAM1 on tyrosine residues located in the ITIM region of its long cytoplasmic domain [251]. In the viable motheaten mice and mice with a temporary SHP1 disruption in the liver, insulin clearance is significantly improved [131]. Ptpn6 H-KO mice with a hepatocyte-specific Shp1 deletion also display a more rapid insulin clearance rate [230].
Taken together, SHP1 negatively regulates glucose metabolism as an inhibitor of insulin clearance and signaling through the PI3K-Akt pathways in contribution to obesity-linked insulin resistance (Figs. 3 and 4 and Table 2).

Known roles of cytosolic SHP1 in the regulation of insulin signaling and of IR-CEACAM1-mediated insulin clearance and glucose metabolism. SHP1 regulates insulin clearance and insulin signaling by targeting several Tyr-phosphorylated molecules in these pathways. It modulates CEACAM1 activity by dephosphorylating it on the plasma membrane thereby down-regulating insulin-IR-complex endocytosis, but also by dephosphorylating it after internalization, thereby preventing its lysosomal degradation and favouring its recycling to the cell surface. SHP1 negatively regulates insulin signaling at different steps. It dephosphorylates the IR both on the plasma membrane and after internalization into the endosomes. It also inhibits PI3K by dephosphorylating the p85 regulatory subunit
5.2.2 SHP2
SHP2 can be physically associated with the insulin receptor and IRS-1 [252, 253, 258, 259] and is believed to be a bona fide negative regulator of insulin signaling, not only by direct dephosphorylation but also by adapting other inhibitory signaling molecules such as JAK2 and PKC onto IRS [254, 260]. Interestingly, hepatic SHP2 protein expression changes with nutritional status, as it decreases during fasting and increases after feeding, especially after a high-fat diet [263]. Increased activity and expression of SHP2 have been observed in skeletal muscle and liver of obese and insulin-resistant animal models [157, 158], which suggests that this PTP may be implicated in the development of obesity-associated insulin resistance. However, SHP2 was initially thought to positively regulate insulin signaling. Transgenic mice expressing a dominant-negative mutant lacking the PTP catalytic domain (ΔPTP) and inhibiting the insulin-induced association of endogenous SHP2 with IRS-1 show impaired insulin-stimulated glucose uptake and general glucose intolerance, as the activation of the IRS-1/PI3K/Akt pathway is attenuated [255]. This led to the conclusion that the inhibition of endogenous SHP2 function may diminish insulin sensitivity. However further loss-of-function studies, conducted by the generation of tissue-specific SHP2 KO animals further confirmed the negative function of SHP2 in the control of insulin action and glucose metabolism. Indeed, liver-specific SHP2 KO mice (LSHKO) exhibit improved insulin sensitivity and increased glucose tolerance compared with control mice on standard diet. Acute SHP2 deletion by tail-vein injection of adenovirus carrying Ad5-Cre yield comparable results, with improved hepatic insulin signaling via the PI3K/Akt pathway [262]. In diet-induced obesity, LSHKO show enhanced suppression of hepatic glucose production by insulin and general protection against the development of obesity-linked insulin resistance. Unlike mice with hepatic SHP1 deletion (Ptpn6 H-KO), LSHKO exhibit less weight gain, increased energy expenditure, and less hepatic steatosis [263]. A potential positive role for the PTP in insulin signaling is, however, suggested by another study which shows that SHP2-deficient mice are insulin-resistant and exhibit impaired muscle glucose uptake, which is associated with defective activation of atypical PKCz/l and AMPK [261]. However, these mice also experience severe cardiomyopathy, reflected by multiple cardiovascular disturbances, which may lead to secondary changes in skeletal muscle insulin signaling and attenuated glucose metabolism. In adipose tissue, one study of adipose-specific SHP2 deletion by adiponectin-Cre reports no alteration in systemic glucose homeostasis [264], while another study confirms the important role of SHP2 in the promotion of adipogenesis, as mice with adipocyte-specific deletion of SHP2 suffer from premature death, lack of white fat, low blood pressure, compensatory erythrocytosis, and hepatic steatosis [265]. Most recently, a role of SHP2 in the regulation of lipid synthesis has been found, as ubiquitination-mediated degradation of FAS can be facilitated by SHP2, the key adapter PTP for the binding of p38-activated COP1 to FAS in the FAS-SHP2-COP1 complex [266].
6 Role of PTPs in transcription regulation
In many cases, signaling pathways lead to a regulation of transcriptional processes by phosphorylation and dephosphorylation of transcription factors. Although many transcription factors have been shown to be regulated by serine and threonine phosphorylation, tyrosine phosphorylation has not been implicated in the control of many transcriptional processes with one exception. The signal transducer and activator of transcription (STAT) proteins are the prime example for a family of transcription factors whose activity is regulated by tyrosine-phosphorylation [267, 268]. In general, tyrosine-phosphorylation of STATs, which is triggered by cytokines and growth factors via their respective receptors and associated Janus kinase (JAK) proteins, results in dimerization of STAT proteins, translocation to the nucleus and ultimately in activation of transcription. This activation of the JAK-STAT pathway is counteracted by several protein tyrosine phosphatases including SHP1, SHP2, PTP1B and TC-PTP. Although this regulation of the JAK-STAT pathway by PTPs has been extensively studied in hematopoetic and cancer cells and well characterized in the leptin signaling pathway of the CNS [197, 269, 270], very little is known about PTP-mediated control of tyrosine-phosphorylation of STATs in peripheral insulin target tissues. Tyrosine-phosphorylation of STAT3 (Y705) is increased in livers of high fat-fed mice heterozygous for TC-PTP-deletion (Ptpn2+/−) and IL-6 treated primary hepatocytes from these mice or in primary hepatocytes where TC-PTP has been knocked-down by shRNA, implying the attenuation of STAT3 by TC-PTP [193]. Tyrosine-phosphorylation of STAT3 (Y705) is also elevated in livers from mice with a liver-specific SHP2 knock-out (Ptpn11−/−) fed a high-fat diet, suggesting redundancy in the dephosphorylation of STAT3 in hepatocytes [263].
Independent from STAT3 association, several other transcriptional regulators have been identified as substrates of SHP1 and SHP2. Parafibromin/Cdc73, a core subunit of the nuclear RNA polymerase II-associated factor (PAF) complex, is dephosphorylated by SHP2 and thereby converts into an inducer of Wnt target genes by stably interacting with β-catenin [271]. β-catenin itself is a target for SHP1 in epithelial cells, whose transcriptional activity is negatively regulated by SHP1-mediated dephosphorylation [272, 273]. SHP1 has also been implicated in the regulation of transcriptional processes in insulin target tissues. Down-regulation of SHP1 in muscle cells increases GLUT4 gene expression, but the exact molecular details of this augmentation still have to be elucidated [235]. In a recent study, we found that SHP1 inhibits PPARγ-dependent transcription and thereby regulates expression of genes involved in lipid metabolism (unpublished data). Indirectly, SHP2 has been proposed to regulate adipogenesis in white adipose tissue via a SHP2-TAO2-p38-p300-PPARγ pathway, which can contribute to defective adipogenesis in obesity [265].
7 PTPs as therapeutic targets for insulin resistance
Some PTP inhibitors may be considered for combating insulin resistance and other disturbances of the metabolic syndrome. Cryptotanshinone, a common CVD drug, has been identified to inhibit SHP2 with 1.76 fold more selectivity than for SHP1 in direct binding to their catalytic pocket [217]. Cembrane diterpenoids from the soft coral Sarcophyton trocheliophorum Marenzeller are a new class of PTP1B inhibitors [274]. Ursolic acid and its derivatives, competitive inhibitors of PTP1B and SHP2, can improve insulin-stimulated glucose uptake by L6 myotubes in vitro [275]. PTP1B, SHP1 and SHP2, but not TC-PTP, can be inhibited by S-nitrosylation through NO, which leads to enhancement of insulin responsiveness with increased insulin signaling [276]. The structurally modified phospho-tyrosine mimetic, phosphonodifluoromethyl phenylalanine, a new PTP-MEG2 inhibitor possessing highly efficacious cellular activity to augment insulin signaling and regulation of glucose homeostasis in diet-induced obese mice has been characterized [277].
Whereas it may be worthwhile to develop novel PTP inhibitors for alleviating insulin resistance and T2D, another potential strategy is to identify selective substrates and partners of individual PTP and explore their potential for representing more selective therapeutic targets. Although the function of PTPs in glucose and lipid metabolism has been well established, very few substrates of these PTPs have been identified in insulin target tissues. Given that the nuclear localization of several PTPs has been known for 10 or more years, we are especially missing nuclear targets that are regulated by tyrosine phosphorylation. Three of the PTPs described in this review can shuttle between the cytoplasm and the nucleus. TC-PTP exists as two splice variants, a nuclear 45kD isoform and an endoplasmic reticulum-targeted 48 kD isoform [191, 278]. SHP1 and SHP2 localize to the nucleus in non-hematopoetic cells [228, 272, 279, 280]. We need mass spectrometric approaches to detect new substrates of these PTPs in insulin target tissues. One approach could be to identify the phospho-tyrosine proteome in insulin target cells or tissues with deletions of PTPs to enrich specific targets. Another way would be to identify interaction partners of PTPs in insulin target cells using substrate-trapping mutants of these PTPs in an affinity-purification mass spectrometric approach. A combination of these approaches should give us a comprehensive collection of potential PTP substrates and partners both in the cytoplasm and nucleus. First attempts have been made to detect novel proteins involved in the insulin signaling pathway, but none of them has specifically targeted PTPs [281, 282]. PhosphoSitePlus gives us a collection of many phospho-tyrosine sites in proteins including transcription factors with functions in glucose and lipid metabolism that have been identified by mass spectrometry from cells treated with general PTP inhibitors [283]. Most of these sites have no assigned function, but it could be used as a starting point to elucidate the molecular mechanisms and physiological roles of these tyrosine phosphorylation events in insulin target cells and tissues.
8 Conclusion
We have reviewed how PTPs can inactivate crucial signaling effectors in the insulin signaling cascade by dephosphorylating their tyrosine residues, and their potential implication in the development of insulin resistance and T2D. We have reviewed recent evidence implicating both SHP1 and SHP2 as new potential therapeutic targets. Clearly, we need to further advance our understanding of the mechanisms by which these PTPs regulate insulin signaling and glucose metabolism, and this will be markedly enhanced by the identification of selective partners of these enzymes in key insulin target metabolic tissues.
Abbreviations
- aPKC:
-
Atypical protein kinase C
- ABC:
-
ATP binding cassette transporter
- ACD:
-
Acyl CoA dehydrogenases
- ACO:
-
Acyl CoA oxidase
- AGC:
-
Protein kinase A G, and C
- Akt:
-
Protein kinase B
- AP:
-
Adaptor protein
- aPKC:
-
Atypical protein kinase C
- AT2:
-
Angiotensin II subtype 2 receptor
- Cbl:
-
Casitas B-lineage lymphoma
- Cdk:
-
Cyclin-dependent kinase
- CEA:
-
Carcinoembryonic antigen
- CEACAM:
-
CEA-related adhesion molecules
- CNS:
-
Central nervous system
- COP1:
-
Caspase recruitment domain-containing protein 16
- CPT:
-
Carnitine palmitoyl transferase
- CVD:
-
Cardiovascular diseases
- ER:
-
Endoplasmic reticulum
- FA:
-
Fatty acids
- FAS:
-
Fatty acid synthase
- FFA/NEFA:
-
Free fatty acids/non-esterified FA
- Fkhr/FoxO1:
-
Forkhead transcription factor
- G6Pase:
-
Glucose-6-phosphatase
- Gab:
-
Grb2-associated-binding protein
- GLP:
-
Glucagon-like peptide
- GLUT:
-
Glucose transporter
- Grb2:
-
Growth factor receptor-bound protein 2
- GSK3:
-
Glycogen synthase kinase 3
- IGF:
-
Insulin growth factor
- IL:
-
Interleukin
- IR:
-
Insulin receptor
- IRR:
-
Insulin receptor-related receptor
- IRS:
-
Insulin-receptor substrate
- JAK:
-
Janus kinase
- KO:
-
Knockout
- LAR:
-
Leukocyte common antigen-related (phosphatase)
- LMW-PTP:
-
Low molecular weight PTP
- MAPK/ERK:
-
Mitogen activated protein kinase/Extracellular signal-regulated kinase
- MCK:
-
Muscle creatine kinase
- MEK:
-
MAPK kinase
- mRNA:
-
Messenger ribonucleic acid
- mTORC:
-
Mammalian TOR complex
- NAFLD:
-
Non-alcoholic fatty liver disease
- NEFA:
-
Non-esterified fatty acids
- NO:
-
Nitric oxide
- p60dok :
-
60-kDa tyrosine phosphorylated protein
- PAF:
-
RNA polymerase II associated factor
- PDK:
-
PDH kinase or Phosphoinositide-dependent kinase
- PGC-1:
-
PPARγ co-activator 1
- PH:
-
Pleckstrin homology
- PHLPP:
-
PH-domain leucine-rich repeat protein phosphatase
- PI3K:
-
Phosphatidylinositol 3-kinase
- PIP2:
-
Phosphotidylinositol-4,5-bisphosphate
- PIP3:
-
Phosphotidylinositol-3,4,5-triphosphate
- PKB:
-
Protein kinase B (also known as Akt)
- PM:
-
Plasmic membrane
- PP:
-
Protein phosphatase
- PPAR:
-
Peroxisome proliferator-activated receptor
- Prep:
-
PBX-regulating protein
- PTB:
-
Phospho-tyrosine binding domain
- PTEN:
-
Phosphatase with sequence homology to protein-tyrosine phosphatases and the cytoskeleton protein tensin
- PTP:
-
Protein tyrosine phosphatase
- Raf:
-
Proto-oncogene serine/threonine-protein kinase
- Ras:
-
G-protein and/or a family of related proteins discovered from rat sarcoma
- RPTP:
-
Receptor-like PTP
- RTK:
-
Receptor tyrosine kinases
- S6K1:
-
S6 kinase 1
- SCD:
-
Stearoyl-CoA desaturase
- SH:
-
Src homology
- Shc:
-
SH2-domain containing proteins
- SHP:
-
2-SH2-domain containing protein tyrosine phosphatase
- SNARE:
-
Soluble NSF attachment protein receptor
- SOS:
-
Son-of-sevenless exchange protein
- SREBP:
-
Sterol response element binding protein
- STAT:
-
Signal transducer and activator of transcription
- STP:
-
Serine/threonine phosphatase
- T2D:
-
Type 2 diabetes
- TAO:
-
Thousand and one amino acid kinase
- TC-PTP:
-
T-cell PTP
- TF:
-
Transcription factors
- TNF:
-
Tumor necrosis factor
- TOR:
-
Target of rapamycin
- WHO:
-
World Health Organization
- Wnt:
-
Wingless and int-1
References
Waxman A. WHO global strategy on diet, physical activity and health. Food Nutr Bull. 2004;25(3):292–302.
O’Sullivan JB, Mahan CM. Blood sugar levels, glycosuria, and body weight related to development of diabetes mellitus. The Oxford epidemiologic study 17 years later. JAMA. 1965;194(6):587–92.
Wilson PW, McGee DL, Kannel WB. Obesity, very low density lipoproteins, and glucose intolerance over fourteen years: the Framingham study. Am J Epidemiol. 1981;114(5):697–704.
Weyer C et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. 2001;86(5):1930–5.
McKenney RL, Short DK. Tipping the balance: the pathophysiology of obesity and type 2 diabetes mellitus. Surg Clin N Am. 2011;91(6):1139–48. vii.
McCarthy MI. Genomics, type 2 diabetes, and obesity. N Engl J Med. 2010;363(24):2339–50.
Smyth S, Heron A. Diabetes and obesity: the twin epidemics. Nat Med. 2006;12(1):75–80.
DeFronzo RA, Ferrannini E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14(3):173–94.
Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–71.
Goossens GH. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiol Behav. 2008;94(2):206–18.
Qatanani M, Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev. 2007;21(12):1443–55.
Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007;87(2):507–20.
Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest. 2000;106(4):473–81.
Opie LH, Walfish PG. Plasma free fatty acid concentrations in obesity. N Engl J Med. 1963;268:757–60.
Bjorntorp P, Bergman H, Varnauskas E. Plasma free fatty acid turnover rate in obesity. Acta Med Scand. 1969;185(4):351–6.
Karpe F, Dickmann JR, Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes. 2011;60(10):2441–9.
Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes. 2011;18(2):139–43.
Randle PJ et al. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1(7285):785–9.
Pan DA et al. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes. 1997;46(6):983–8.
Roden M et al. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1996;97(12):2859–65.
Boden G et al. FFA cause hepatic insulin resistance by inhibiting insulin suppression of glycogenolysis. Am J Physiol Endocrinol Metab. 2002;283(1):E12–9.
Lam TK et al. Mechanisms of the free fatty acid-induced increase in hepatic glucose production. Am J Physiol Endocrinol Metab. 2003;284(5):E863–73.
Wu N et al. Taurine prevents free fatty acid-induced hepatic insulin resistance in association with inhibiting JNK1 activation and improving insulin signaling in vivo. Diabetes Res Clin Pract. 2010;90(3):288–96.
Morita S, et al. Effect of exposure to non-esterified fatty acid on progressive deterioration of insulin secretion in patients with Type 2 diabetes: a long-term follow-up study. Diabet Med. 2012;29(8):980–5.
Koyama K et al. Tissue triglycerides, insulin resistance, and insulin production: implications for hyperinsulinemia of obesity. Am J Physiol. 1997;273(4 Pt 1):E708–13.
Oprescu AI et al. Free fatty acid-induced reduction in glucose-stimulated insulin secretion: evidence for a role of oxidative stress in vitro and in vivo. Diabetes. 2007;56(12):2927–37.
Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793–801.
Tremblay F et al. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci U S A. 2007;104(35):14056–61.
Klip A, Paquet MR. Glucose transport and glucose transporters in muscle and their metabolic regulation. Diabetes Care. 1990;13(3):228–43.
Heuson JC, Coune A, Heimann R. Cell proliferation induced by insulin in organ culture of rat mammary carcinoma. Exp Cell Res. 1967;45(2):351–60.
Wang W et al. Mediator MED23 links insulin signaling to the adipogenesis transcription cascade. Dev Cell. 2009;16(5):764–71.
Pause A et al. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature. 1994;371(6500):762–7.
Fulzele K et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. 2010;142(2):309–19.
Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806.
Buchanan KD, Mawhinney WA. Insulin control of glucagon release from insulin-deficient rat islets. Diabetes. 1973;22(11):801–3.
Koch L et al. Central insulin action regulates peripheral glucose and fat metabolism in mice. J Clin Invest. 2008;118(6):2132–47.
Konner AC et al. Role for insulin signaling in catecholaminergic neurons in control of energy homeostasis. Cell Metab. 2011;13(6):720–8.
Bruning JC et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289(5487):2122–5.
Fisher SJ et al. Insulin signaling in the central nervous system is critical for the normal sympathoadrenal response to hypoglycemia. Diabetes. 2005;54(5):1447–51.
Hill JW et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010;11(4):286–97.
Konner AC et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5(6):438–49.
Lee JC, Downing SE. Effects of insulin on cardiac muscle contraction and responsiveness to norepinephrine. Am J Physiol. 1976;230(5):1360–5.
Maier S et al. Stimulation of L-type Ca2+ current in human atrial myocytes by insulin. Cardiovasc Res. 1999;44(2):390–7.
Gupta AK, Clark RV, Kirchner KA. Effects of insulin on renal sodium excretion. Hypertension. 1992;19(1 Suppl):I78–82.
Alvestrand A et al. Insulin-mediated potassium uptake is normal in uremic and healthy subjects. Am J Physiol. 1984;246(2 Pt 1):E174–80.
Breen DM, Giacca A. Effects of insulin on the vasculature. Curr Vasc Pharmacol. 2011;9(3):321–32.
Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–95.
Muller D et al. Identification of insulin signaling elements in human beta-cells: autocrine regulation of insulin gene expression. Diabetes. 2006;55(10):2835–42.
Kulkarni RN et al. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96(3):329–39.
Lim GE et al. Insulin regulates glucagon-like peptide-1 secretion from the enteroendocrine L cell. Endocrinology. 2009;150(2):580–91.
Kovacina KS, Roth RA. Characterization of the endogenous insulin receptor-related receptor in neuroblastomas. J Biol Chem. 1995;270(4):1881–7.
Butler AA, LeRoith D. Minireview: tissue-specific versus generalized gene targeting of the igf1 and igf1r genes and their roles in insulin-like growth factor physiology. Endocrinology. 2001;142(5):1685–8.
Patti ME, Kahn CR. The insulin receptor–a critical link in glucose homeostasis and insulin action. J Basic Clin Physiol Pharmacol. 1998;9(2–4):89–109.
De Meyts P, Whittaker J. Structural biology of insulin and IGF1 receptors: implications for drug design. Nat Rev Drug Discov. 2002;1(10):769–83.
Lizcano JM, Alessi DR. The insulin signalling pathway. Curr Biol. 2002;12(7):R236–8.
Zhang-Sun G et al. A 60-kilodalton protein in rat hepatoma cells overexpressing insulin receptor was tyrosine phosphorylated and associated with Syp, phophatidylinositol 3-kinase, and Grb2 in an insulin-dependent manner. Endocrinology. 1996;137(7):2649–58.
Moodie SA, Alleman-Sposeto J, Gustafson TA. Identification of the APS protein as a novel insulin receptor substrate. J Biol Chem. 1999;274(16):11186–93.
Pronk GJ et al. Insulin-induced phosphorylation of the 46- and 52-kDa Shc proteins. J Biol Chem. 1993;268(8):5748–53.
Huang C et al. Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in l6 myotubes. J Biol Chem. 2005;280(19):19426–35.
Previs SF et al. Contrasting effects of IRS-1 versus IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo. J Biol Chem. 2000;275(50):38990–4.
Kido Y et al. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest. 2000;105(2):199–205.
Rhodes CJ, White MF. Molecular insights into insulin action and secretion. Eur J Clin Invest. 2002;32 Suppl 3:3–13.
Sadagurski M et al. Insulin receptor substrate 2 plays diverse cell-specific roles in the regulation of glucose transport. J Biol Chem. 2005;280(15):14536–44.
Withers DJ et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391(6670):900–4.
Yamauchi T et al. Insulin signalling and insulin actions in the muscles and livers of insulin-resistant, insulin receptor substrate 1-deficient mice. Mol Cell Biol. 1996;16(6):3074–84.
Tamemoto H et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature. 1994;372(6502):182–6.
Araki E et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372(6502):186–90.
Kubota N et al. Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metab. 2008;8(1):49–64.
Tsuruzoe K et al. Insulin receptor substrate 3 (IRS-3) and IRS-4 impair IRS-1- and IRS-2-mediated signaling. Mol Cell Biol. 2001;21(1):26–38.
White MF. The IRS-signalling system: a network of docking proteins that mediate insulin action. Mol Cell Biochem. 1998;182(1–2):3–11.
Backer JM et al. Insulin stimulation of phosphatidylinositol 3-kinase activity maps to insulin receptor regions required for endogenous substrate phosphorylation. J Biol Chem. 1992;267(2):1367–74.
Lietzke SE et al. Structural basis of 3-phosphoinositide recognition by pleckstrin homology domains. Mol Cell. 2000;6(2):385–94.
Peterson RT, Schreiber SL. Kinase phosphorylation: keeping it all in the family. Curr Biol. 1999;9(14):R521–4.
Mackay DJ, Hall A. Rho GTPases. J Biol Chem. 1998;273(33):20685–8.
Ziegler SF et al. Molecular cloning and characterization of a novel receptor protein tyrosine kinase from human placenta. Oncogene. 1993;8(3):663–70.
Alessi DR et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7(4):261–9.
Stephens L et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279(5351):710–4.
Sarbassov DD et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–101.
Paz K et al. Phosphorylation of insulin receptor substrate-1 (IRS-1) by protein kinase B positively regulates IRS-1 function. J Biol Chem. 1999;274(40):28816–22.
Standaert ML et al. Protein kinase C-zeta as a downstream effector of phosphatidylinositol 3-kinase during insulin stimulation in rat adipocytes. Potential role in glucose transport. J Biol Chem. 1997;272(48):30075–82.
Chou MM et al. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr Biol. 1998;8(19):1069–77.
Scott PH et al. Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1998;95(13):7772–7.
Cross DA et al. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785–9.
Li X et al. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447(7147):1012–6.
Nakae J, Park BC, Accili D. Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem. 1999;274(23):15982–5.
Cook SA et al. Transcriptional effects of chronic Akt activation in the heart. J Biol Chem. 2002;277(25):22528–33.
Yoon JC et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413(6852):131–8.
Puigserver P et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423(6939):550–5.
Boulton TG et al. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65(4):663–75.
Lazar DF et al. Mitogen-activated protein kinase kinase inhibition does not block the stimulation of glucose utilization by insulin. J Biol Chem. 1995;270(35):20801–7.
Noguchi T et al. Role of SH-PTP2, a protein-tyrosine phosphatase with Src homology 2 domains, in insulin-stimulated Ras activation. Mol Cell Biol. 1994;14(10):6674–82.
Sasaoka T et al. Evidence for a functional role of Shc proteins in mitogenic signaling induced by insulin, insulin-like growth factor-1, and epidermal growth factor. J Biol Chem. 1994;269(18):13689–94.
Moscat J, Diaz-Meco MT. Feedback on fat: p62-mTORC1-autophagy connections. Cell. 2011;147(4):724–7.
Raught B, Gingras AC, Sonenberg N. The target of rapamycin (TOR) proteins. Proc Natl Acad Sci U S A. 2001;98(13):7037–44.
Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–84.
Peterson TR et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873–86.
Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136(4):731–45.
Alessi DR, Pearce LR, Garcia-Martinez JM. New insights into mTOR signaling: mTORC2 and beyond. Sci Signal. 2009;2(67):pe27.
Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009;19(22):R1046–52.
Avruch J et al. Amino acid regulation of TOR complex 1. Am J Physiol Endocrinol Metab. 2009;296(4):E592–602.
Manning BD, Cantley LC. Rheb fills a GAP between TSC and TOR. Trends Biochem Sci. 2003;28(11):573–6.
Nave BT et al. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344(Pt 2):427–31.
Tzatsos A, Kandror KV. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol. 2006;26(1):63–76.
Tremblay F, Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem. 2001;276(41):38052–60.
Tremblay F et al. Activation of the mammalian target of rapamycin pathway acutely inhibits insulin signaling to Akt and glucose transport in 3T3-L1 and human adipocytes. Endocrinology. 2005;146(3):1328–37.
Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106(2):171–6.
Hotamisligil GS. The role of TNFalpha and TNF receptors in obesity and insulin resistance. J Intern Med. 1999;245(6):621–5.
Ogg S, Ruvkun G. The C. elegans PTEN homolog, DAF-18, acts in the insulin receptor-like metabolic signaling pathway. Mol Cell. 1998;2(6):887–93.
Wada T et al. Role of the Src homology 2 (SH2) domain and C-terminus tyrosine phosphorylation sites of SH2-containing inositol phosphatase (SHIP) in the regulation of insulin-induced mitogenesis. Endocrinology. 1999;140(10):4585–94.
Lazar DF, Saltiel AR. Lipid phosphatases as drug discovery targets for type 2 diabetes. Nat Rev Drug Discov. 2006;5(4):333–42.
Sigal YJ, McDermott MI, Morris AJ. Integral membrane lipid phosphatases/phosphotransferases: common structure and diverse functions. Biochem J. 2005;387(Pt 2):281–93.
Mistafa O et al. Purinergic receptor-mediated rapid depletion of nuclear phosphorylated Akt depends on pleckstrin homology domain leucine-rich repeat phosphatase, calcineurin, protein phosphatase 2A, and PTEN phosphatases. J Biol Chem. 2010;285(36):27900–10.
Finck BN et al. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006;4(3):199–210.
Peterson TR et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146(3):408–20.
Armstrong CG, Doherty MJ, Cohen PT. Identification of the separate domains in the hepatic glycogen-targeting subunit of protein phosphatase 1 that interact with phosphorylase a, glycogen and protein phosphatase 1. Biochem J. 1998;336(Pt 3):699–704.
Wong RH, Sul HS. Insulin signaling in fatty acid and fat synthesis: a transcriptional perspective. Curr Opin Pharmacol. 2010;10(6):684–91.
Galbo T et al. Free fatty acid-induced PP2A hyperactivity selectively impairs hepatic insulin action on glucose metabolism. PLoS One. 2011;6(11):e27424.
Yan L, et al. The B55α-containing PP2A holoenzyme dephosphorylates FOXO1 in islet β-cells under oxidative stress. Biochem J. 2012;444(2):239–47.
Andreozzi F et al. Increased levels of the Akt-specific phosphatase PH domain leucine-rich repeat protein phosphatase (PHLPP)-1 in obese participants are associated with insulin resistance. Diabetologia. 2011;54(7):1879–87.
Xiao L et al. Protein phosphatase-1 regulates Akt1 signal transduction pathway to control gene expression, cell survival and differentiation. Cell Death Differ. 2010;17(9):1448–62.
Andjelkovic M et al. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc Natl Acad Sci U S A. 1996;93(12):5699–704.
Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18(1):13–24.
Brautigan DL, Bornstein P, Gallis B. Phosphotyrosyl-protein phosphatase. Specific inhibition by Zn. J Biol Chem. 1981;256(13):6519–22.
Schaapveld R, Wieringa B, Hendriks W. Receptor-like protein tyrosine phosphatases: alike and yet so different. Mol Biol Rep. 1997;24(4):247–62.
Alonso A et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117(6):699–711.
Koren S, Fantus IG. Inhibition of the protein tyrosine phosphatase PTP1B: potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract Res Clin Endocrinol Metab. 2007;21(4):621–40.
Hashimoto N et al. Insulin receptor protein-tyrosine phosphatases. Leukocyte common antigen-related phosphatase rapidly deactivates the insulin receptor kinase by preferential dephosphorylation of the receptor regulatory domain. J Biol Chem. 1992;267(20):13811–4.
Frangioni JV et al. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell. 1992;68(3):545–60.
Egawa K et al. Protein-tyrosine phosphatase-1B negatively regulates insulin signaling in l6 myocytes and Fao hepatoma cells. J Biol Chem. 2001;276(13):10207–11.
Goldstein BJ et al. Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein-tyrosine phosphatase 1B. Possible facilitation by the formation of a ternary complex with the Grb2 adaptor protein. J Biol Chem. 2000;275(6):4283–9.
Dubois MJ et al. The SHP-1 protein tyrosine phosphatase negatively modulates glucose homeostasis. Nat Med. 2006;12(5):549–56.
Kuhne MR et al. Dephosphorylation of insulin receptor substrate 1 by the tyrosine phosphatase PTP2C. J Biol Chem. 1994;269(22):15833–7.
Kulas DT et al. Insulin receptor signaling is augmented by antisense inhibition of the protein tyrosine phosphatase LAR. J Biol Chem. 1995;270(6):2435–8.
Coleman DL. Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia. 1978;14(3):141–8.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–7.
Plum L, Belgardt BF, Bruning JC. Central insulin action in energy and glucose homeostasis. J Clin Invest. 2006;116(7):1761–6.
Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489(7415):242–9.
Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900–17.
Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8(2):92–103.
Patel N, Huang C, Klip A. Cellular location of insulin-triggered signals and implications for glucose uptake. Pflugers Arch. 2006;451(4):499–510.
Filippi BM et al. Insulin activates Erk1/2 signaling in the dorsal vagal complex to inhibit glucose production. Cell Metab. 2012;16(4):500–10.
Ozcan L et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9(1):35–51.
Ahmad F, Goldstein BJ. Functional association between the insulin receptor and the transmembrane protein-tyrosine phosphatase LAR in intact cells. J Biol Chem. 1997;272(1):448–57.
Norris K et al. Expression of protein-tyrosine phosphatases in the major insulin target tissues. FEBS Lett. 1997;415(3):243–8.
Zhang WR et al. Modulation of insulin signal transduction by eutopic overexpression of the receptor-type protein-tyrosine phosphatase LAR. Mol Endocrinol. 1996;10(5):575–84.
Li PM, Zhang WR, Goldstein BJ. Suppression of insulin receptor activation by overexpression of the protein-tyrosine phosphatase LAR in hepatoma cells. Cell Signal. 1996;8(7):467–73.
Ren JM et al. Transgenic mice deficient in the LAR protein-tyrosine phosphatase exhibit profound defects in glucose homeostasis. Diabetes. 1998;47(3):493–7.
Zabolotny JM et al. Overexpression of the LAR (leukocyte antigen-related) protein-tyrosine phosphatase in muscle causes insulin resistance. Proc Natl Acad Sci U S A. 2001;98(9):5187–92.
Ahmad F, Considine RV, Goldstein BJ. Increased abundance of the receptor-type protein-tyrosine phosphatase LAR accounts for the elevated insulin receptor dephosphorylating activity in adipose tissue of obese human subjects. J Clin Invest. 1995;95(6):2806–12.
Ahmad F, Goldstein BJ. Increased abundance of specific skeletal muscle protein-tyrosine phosphatases in a genetic model of insulin-resistant obesity and diabetes mellitus. Metabolism. 1995;44(9):1175–84.
Um JW, Ko J. LAR-RPTPs: synaptic adhesion molecules that shape synapse development. Trends Cell Biol. 2013;23(10):465–75.
Garcia-San Frutos M et al. Involvement of protein tyrosine phosphatases and inflammation in hypothalamic insulin resistance associated with ageing: effect of caloric restriction. Mech Ageing Dev. 2012;133(7):489–97.
Elchebly M et al. Neuroendocrine dysplasia in mice lacking protein tyrosine phosphatase sigma. Nat Genet. 1999;21(3):330–3.
Wallace MJ et al. Neuronal defects and posterior pituitary hypoplasia in mice lacking the receptor tyrosine phosphatase PTPsigma. Nat Genet. 1999;21(3):334–8.
Lam CK, Chari M, Lam TK. CNS regulation of glucose homeostasis. Physiology (Bethesda). 2009;24:159–70.
Hendriks WJ, Pulido R. Protein tyrosine phosphatase variants in human hereditary disorders and disease susceptibilities. Biochim Biophys Acta. 2013;1832(10):1673–96.
Tagami S et al. Troglitazone ameliorates abnormal activity of protein tyrosine phosphatase in adipose tissues of Otsuka Long-Evans Tokushima Fatty rats. Tohoku J Exp Med. 2002;197(3):169–81.
Gonzalez-Rodriguez A et al. Essential role of protein tyrosine phosphatase 1B in obesity-induced inflammation and peripheral insulin resistance during aging. Aging Cell. 2012;11(2):284–96.
Zabolotny JM et al. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem. 2008;283(21):14230–41.
Elchebly M et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283(5407):1544–8.
Delibegovic M et al. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes. 2009;58(3):590–9.
Agouni A, et al. Liver-specific deletion of protein tyrosine phosphatase (PTP) 1B improves obesity- and pharmacologically induced endoplasmic reticulum stress. Biochem J. 2011;438(2):369–78.
Owen C et al. Inducible liver-specific knockdown of protein tyrosine phosphatase 1B improves glucose and lipid homeostasis in adult mice. Diabetologia. 2013;56(10):2286–96.
Bakke J et al. Regulation of the SNARE-interacting protein Munc18c tyrosine phosphorylation in adipocytes by protein-tyrosine phosphatase 1B. Cell Commun Signal. 2013;11(1):57.
Song DD et al. Protein tyrosine phosphatase 1B inhibits adipocyte differentiation and mediates TNFalpha action in obesity. Biochim Biophys Acta. 2013;1831(8):1368–76.
Owen C et al. Adipocyte-specific protein tyrosine phosphatase 1B deletion increases lipogenesis, adipocyte cell size and is a minor regulator of glucose homeostasis. PLoS One. 2012;7(2):e32700.
Delibegovic M et al. Improved glucose homeostasis in mice with muscle-specific deletion of protein-tyrosine phosphatase 1B. Mol Cell Biol. 2007;27(21):7727–34.
Picardi PK et al. Modulation of hypothalamic PTP1B in the TNF-alpha-induced insulin and leptin resistance. FEBS Lett. 2010;584(14):3179–84.
Zhang J et al. Protein tyrosine phosphatase 1B deficiency ameliorates murine experimental colitis via the expansion of myeloid-derived suppressor cells. PLoS One. 2013;8(8):e70828.
Nasimian A et al. Protein tyrosine phosphatase 1B (PTP1B) modulates palmitate-induced cytokine production in macrophage cells. Inflamm Res. 2013;62(2):239–46.
Xu H et al. Phosphatase PTP1B negatively regulates MyD88- and TRIF-dependent proinflammatory cytokine and type I interferon production in TLR-triggered macrophages. Mol Immunol. 2008;45(13):3545–52.
Pandey NR et al. The LIM domain only 4 protein is a metabolic responsive inhibitor of protein tyrosine phosphatase 1B that controls hypothalamic leptin signaling. J Neurosci. 2013;33(31):12647–55.
Bettaieb A et al. Differential regulation of endoplasmic reticulum stress by protein tyrosine phosphatase 1B and T cell protein tyrosine phosphatase. J Biol Chem. 2011;286(11):9225–35.
White CL et al. HF diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and -independent mechanisms. Am J Physiol Endocrinol Metab. 2009;296(2):E291–9.
Bence KK et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12(8):917–24.
Banno R et al. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J Clin Invest. 2010;120(3):720–34.
Gu F et al. Protein-tyrosine phosphatase 1B potentiates IRE1 signaling during endoplasmic reticulum stress. J Biol Chem. 2004;279(48):49689–93.
Snider NT, Park H, Omary MB. A conserved rod domain phosphotyrosine that is targeted by the phosphatase PTP1B promotes keratin 8 insolubility and filament organization. J Biol Chem. 2013;288(43):31329–37.
Chiarugi P et al. LMW-PTP is a negative regulator of insulin-mediated mitotic and metabolic signalling. Biochem Biophys Res Commun. 1997;238(2):676–82.
Pandey SK et al. Reduction of low molecular weight protein-tyrosine phosphatase expression improves hyperglycemia and insulin sensitivity in obese mice. J Biol Chem. 2007;282(19):14291–9.
Cho CY et al. Identification of the tyrosine phosphatase PTP-MEG2 as an antagonist of hepatic insulin signaling. Cell Metab. 2006;3(5):367–78.
Moller NP et al. Selective down-regulation of the insulin receptor signal by protein-tyrosine phosphatases alpha and epsilon. J Biol Chem. 1995;270(39):23126–31.
Lammers R, Moller NP, Ullrich A. The transmembrane protein tyrosine phosphatase alpha dephosphorylates the insulin receptor in intact cells. FEBS Lett. 1997;404(1):37–40.
Cong LN et al. Overexpression of protein tyrosine phosphatase-alpha (PTP-alpha) but not PTP-kappa inhibits translocation of GLUT4 in rat adipose cells. Biochem Biophys Res Commun. 1999;255(2):200–7.
Kapp K et al. The protein tyrosine phosphatase alpha modifies insulin secretion in INS-1E cells. Biochem Biophys Res Commun. 2003;311(2):361–4.
Arnott CH et al. Use of an antisense strategy to dissect the signaling role of protein-tyrosine phosphatase alpha. J Biol Chem. 1999;274(37):26105–12.
Le HT, Ponniah S, Pallen CJ. Insulin signaling and glucose homeostasis in mice lacking protein tyrosine phosphatase alpha. Biochem Biophys Res Commun. 2004;314(2):321–9.
Andersen JN et al. Comparative study of protein tyrosine phosphatase-epsilon isoforms: membrane localization confers specificity in cellular signalling. Biochem J. 2001;354(Pt 3):581–90.
Aga-Mizrachi S et al. Cytosolic protein tyrosine phosphatase-epsilon is a negative regulator of insulin signaling in skeletal muscle. Endocrinology. 2008;149(2):605–14.
Faisal A et al. Serine/threonine phosphorylation of ShcA. Regulation of protein-tyrosine phosphatase-pest binding and involvement in insulin signaling. J Biol Chem. 2002;277(33):30144–52.
Galic S et al. Regulation of insulin receptor signaling by the protein tyrosine phosphatase TCPTP. Mol Cell Biol. 2003;23(6):2096–108.
Xu J et al. Effects of small interference RNA against PTP1B and TCPTP on insulin signaling pathway in mouse liver: evidence for non-synergetic cooperation. Cell Biol Int. 2007;31(1):88–91.
Fukushima A et al. T-cell protein tyrosine phosphatase attenuates STAT3 and insulin signaling in the liver to regulate gluconeogenesis. Diabetes. 2010;59(8):1906–14.
Loh K et al. T cell protein tyrosine phosphatase (TCPTP) deficiency in muscle does not alter insulin signalling and glucose homeostasis in mice. Diabetologia. 2012;55(2):468–78.
Zee T et al. T-cell protein tyrosine phosphatase regulates bone resorption and whole-body insulin sensitivity through its expression in osteoblasts. Mol Cell Biol. 2012;32(6):1080–8.
Rajshankar D et al. Role of PTPalpha in the destruction of periodontal connective tissues. PLoS One. 2013;8(8):e70659.
Tiganis T. PTP1B and TCPTP–nonredundant phosphatases in insulin signaling and glucose homeostasis. FEBS J. 2013;280(2):445–58.
Loh K et al. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab. 2011;14(5):684–99.
Tsui HW et al. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet. 1993;4(2):124–9.
Shultz LD et al. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell. 1993;73(7):1445–54.
Qu CK. Role of the SHP-2 tyrosine phosphatase in cytokine-induced signaling and cellular response. Biochim Biophys Acta. 2002;1592(3):297–301.
Poole AW, Jones ML. A SHPing tale: perspectives on the regulation of SHP-1 and SHP-2 tyrosine phosphatases by the C-terminal tail. Cell Signal. 2005;17(11):1323–32.
Chong ZZ, Maiese K. The Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2: diversified control of cell growth, inflammation, and injury. Histol Histopathol. 2007;22(11):1251–67.
Banville D, Stocco R, Shen SH. Human protein tyrosine phosphatase 1C (PTPN6) gene structure: alternate promoter usage and exon skipping generate multiple transcripts. Genomics. 1995;27(1):165–73.
Jin YJ, Yu CL, Burakoff SJ. Human 70-kDa SHP-1L differs from 68-kDa SHP-1 in its C-terminal structure and catalytic activity. J Biol Chem. 1999;274(40):28301–7.
Yi TL, Cleveland JL, Ihle JN. Protein tyrosine phosphatase containing SH2 domains: characterization, preferential expression in hematopoietic cells, and localization to human chromosome 12p12-p13. Mol Cell Biol. 1992;12(2):836–46.
Horvat A et al. A novel role for protein tyrosine phosphatase shp1 in controlling glial activation in the normal and injured nervous system. J Neurosci. 2001;21(3):865–74.
Lurie DI et al. Tyrosine phosphatase SHP-1 immunoreactivity increases in a subset of astrocytes following deafferentation of the chicken auditory brainstem. J Comp Neurol. 2000;421(2):199–214.
Massa PT et al. Expression and function of the protein tyrosine phosphatase SHP-1 in oligodendrocytes. Glia. 2000;29(4):376–85.
Kim HY et al. Raft-mediated Src homology 2 domain-containing proteintyrosine phosphatase 2 (SHP-2) regulation in microglia. J Biol Chem. 2006;281(17):11872–8.
Valencia AM et al. Identification of a protein-tyrosine phosphatase (SHP1) different from that associated with acid phosphatase in rat prostate. FEBS Lett. 1997;406(1–2):42–8.
Vogel W et al. Activation of a phosphotyrosine phosphatase by tyrosine phosphorylation. Science. 1993;259(5101):1611–4.
Yang J et al. Crystal structure of human protein-tyrosine phosphatase SHP-1. J Biol Chem. 2003;278(8):6516–20.
Plutzky J, Neel BG, Rosenberg RD. Isolation of a src homology 2-containing tyrosine phosphatase. Proc Natl Acad Sci U S A. 1992;89(3):1123–7.
Ren L et al. Substrate specificity of protein tyrosine phosphatases 1B, RPTPalpha, SHP-1, and SHP-2. Biochemistry. 2011;50(12):2339–56.
Yang J et al. Crystal structure of the catalytic domain of protein-tyrosine phosphatase SHP-1. J Biol Chem. 1998;273(43):28199–207.
Liu W, et al. Identification of cryptotanshinone as an inhibitor of Oncogenic protein tyrosine phosphatase SHP2 (PTPN11). J Med Chem. 2013;56(18):7212–21.
Pei D et al. Intramolecular regulation of protein tyrosine phosphatase SH-PTP1: a new function for Src homology 2 domains. Biochemistry. 1994;33(51):15483–93.
Zhao Z et al. Purification and characterization of a protein tyrosine phosphatase containing SH2 domains. J Biol Chem. 1993;268(4):2816–20.
Bennett AM et al. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc Natl Acad Sci U S A. 1994;91(15):7335–9.
Frank C et al. Effective dephosphorylation of Src substrates by SHP-1. J Biol Chem. 2004;279(12):11375–83.
Uchida T et al. Insulin stimulates the phosphorylation of Tyr538 and the catalytic activity of PTP1C, a protein tyrosine phosphatase with Src homology-2 domains. J Biol Chem. 1994;269(16):12220–8.
Hsu MF, Meng TC. Enhancement of insulin responsiveness by nitric oxide-mediated inactivation of protein-tyrosine phosphatases. J Biol Chem. 2009;285(11):7919–28.
Ozawa T et al. Negative autoregulation of Src homology region 2-domain-containing phosphatase-1 in rat basophilic leukemia-2H3 cells. Int Immunol. 2007;19(9):1049–61.
Tenev T et al. Perinuclear localization of the protein-tyrosine phosphatase SHP-1 and inhibition of epidermal growth factor-stimulated STAT1/3 activation in A431 cells. Eur J Cell Biol. 2000;79(4):261–71.
Ram PA, Waxman DJ. Interaction of growth hormone-activated STATs with SH2-containing phosphotyrosine phosphatase SHP-1 and nuclear JAK2 tyrosine kinase. J Biol Chem. 1997;272(28):17694–702.
Yang W, Tabrizi M, Yi T. A bipartite NLS at the SHP-1 C-terminus mediates cytokine-induced SHP-1 nuclear localization in cell growth control. Blood Cells Mol Dis. 2002;28(1):63–74.
Craggs G, Kellie S. A functional nuclear localization sequence in the C-terminal domain of SHP-1. J Biol Chem. 2001;276(26):23719–25.
Watanabe N, et al. Heterogeneous nuclear ribonucleoprotein Q (hnRNP Q) is a novel substrate of SH2 domain-containing phosphatase-2 (SHP2). J Biochem. 2013;154(5):475–80.
Xu E, et al. Hepatocyte-specific Ptpn6 deletion protects from obesity-linked hepatic insulin resistance. Diabetes. 2012;61(8):1949–58.
Yu Z et al. SHP-1 associates with both platelet-derived growth factor receptor and the p85 subunit of phosphatidylinositol 3-kinase. J Biol Chem. 1998;273(6):3687–94.
Cuevas BD et al. Tyrosine phosphorylation of p85 relieves its inhibitory activity on phosphatidylinositol 3-kinase. J Biol Chem. 2001;276(29):27455–61.
Lu Y et al. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J Biol Chem. 2003;278(41):40057–66.
Cui TX et al. Angiotensin II subtype 2 receptor activation inhibits insulin-induced phosphoinositide 3-kinase and Akt and induces apoptosis in PC12W cells. Mol Endocrinol. 2002;16(9):2113–23.
Bergeron S et al. Inhibition of the protein tyrosine phosphatase SHP-1 increases glucose uptake in skeletal muscle cells by augmenting insulin receptor signaling and GLUT4 expression. Endocrinology. 2011;152(12):4581–8.
Oriente F et al. Prep1 controls insulin glucoregulatory function in liver by transcriptional targeting of SHP1 tyrosine phosphatase. Diabetes. 2011;60(1):138–47.
Fiset A et al. Compartmentalized CDK2 is connected with SHP-1 and beta-catenin and regulates insulin internalization. Cell Signal. 2011;23(5):911–9.
Kahn CR et al. Quantitative aspects of the insulin-receptor interaction in liver plasma membranes. J Biol Chem. 1974;249(7):2249–57.
Jaspan JB et al. Hepatic metabolism of glucagon in the dog: contribution of the liver to overall metabolic disposal of glucagon. Am J Physiol. 1981;240(3):E233–44.
Sato H et al. Receptor-recycling model of clearance and distribution of insulin in the perfused mouse liver. Diabetologia. 1991;34(9):613–21.
Burgess JW et al. Pharmacological doses of insulin equalize insulin receptor phosphotyrosine content but not tyrosine kinase activity in plasmalemmal and endosomal membranes. Biochem Cell Biol. 1992;70(10–11):1151–8.
Bergeron JJ et al. Uptake of insulin and other ligands into receptor-rich endocytic components of target cells: the endosomal apparatus. Annu Rev Physiol. 1985;47:383–403.
Yoshii H et al. Effects of portal free fatty acid elevation on insulin clearance and hepatic glucose flux. Am J Physiol Endocrinol Metab. 2006;290(6):E1089–97.
Kotronen A et al. Effect of liver fat on insulin clearance. Am J Physiol Endocrinol Metab. 2007;293(6):E1709–15.
Doherty 2nd JJ et al. Selective degradation of insulin within rat liver endosomes. J Cell Biol. 1990;110(1):35–42.
Backer JM, Kahn CR, White MF. The dissociation and degradation of internalized insulin occur in the endosomes of rat hepatoma cells. J Biol Chem. 1990;265(25):14828–35.
Di Guglielmo GM et al. Insulin receptor internalization and signalling. Mol Cell Biochem. 1998;182(1–2):59–63.
Valera Mora ME et al. Insulin clearance in obesity. J Am Coll Nutr. 2003;22(6):487–93.
Xu E et al. Targeted disruption of carcinoembryonic antigen-related cell adhesion molecule 1 promotes diet-induced hepatic steatosis and insulin resistance. Endocrinology. 2009;150(8):3503–12.
Najjar SM. Regulation of insulin action by CEACAM1. Trends Endocrinol Metab. 2002;13(6):240–5.
Huber M, Izzi L, Grondin P, Houde C, Kunath T, Veillette A, et al. The carboxyl-terminal region of biliary glycoprotein controls its tyrosine phosphorylation and association with protein-tyrosine phosphatases SHP-1 and SHP-2 in epithelial cells. J Biol Chem. 1999;274(1):335–44.
Rocchi S et al. Interaction of SH2-containing protein tyrosine phosphatase 2 with the insulin receptor and the insulin-like growth factor-I receptor: studies of the domains involved using the yeast two-hybrid system. Endocrinology. 1996;137(11):4944–52.
Walchli S et al. Identification of tyrosine phosphatases that dephosphorylate the insulin receptor. A brute force approach based on “substrate-trapping” mutants. J Biol Chem. 2000;275(13):9792–6.
Maegawa H et al. SHPTP2 serves adapter protein linking between Janus kinase 2 and insulin receptor substrates. Biochem Biophys Res Commun. 1996;228(1):122–7.
Arrandale JM et al. Insulin signaling in mice expressing reduced levels of Syp. J Biol Chem. 1996;271(35):21353–8.
Maegawa H et al. Expression of a dominant negative SHP-2 in transgenic mice induces insulin resistance. J Biol Chem. 1999;274(42):30236–43.
Fukunaga K et al. Requirement for protein-tyrosine phosphatase SHP-2 in insulin-induced activation of c-Jun NH(2)-terminal kinase. J Biol Chem. 2000;275(7):5208–13.
Tanaka S et al. Biological effects of human insulin receptor substrate-1 overexpression in hepatocytes. Hepatology. 1997;26(3):598–604.
Hayashi K et al. Insulin receptor substrate-1/SHP-2 interaction, a phenotype-dependent switching machinery of insulin-like growth factor-I signaling in vascular smooth muscle cells. J Biol Chem. 2004;279(39):40807–18.
Mussig K et al. Shp2 is required for protein kinase C-dependent phosphorylation of serine 307 in insulin receptor substrate-1. J Biol Chem. 2005;280(38):32693–9.
Princen F et al. Deletion of Shp2 tyrosine phosphatase in muscle leads to dilated cardiomyopathy, insulin resistance, and premature death. Mol Cell Biol. 2009;29(2):378–88.
Matsuo K et al. Altered glucose homeostasis in mice with liver-specific deletion of Src homology phosphatase 2. J Biol Chem. 2010;285(51):39750–8.
Nagata N et al. Hepatic Src homology phosphatase 2 regulates energy balance in mice. Endocrinology. 2012;153(7):3158–69.
Bettaieb A et al. Adipose-specific deletion of Src homology phosphatase 2 does not significantly alter systemic glucose homeostasis. Metabolism. 2011;60(8):1193–201.
He Z et al. Nonreceptor tyrosine phosphatase Shp2 promotes adipogenesis through inhibition of p38 MAP kinase. Proc Natl Acad Sci U S A. 2013;110(1):E79–88.
Yu J et al. Modulation of fatty acid synthase degradation by concerted action of p38 MAP kinase, E3 ligase COP1, and SH2-tyrosine phosphatase Shp2. J Biol Chem. 2013;288(6):3823–30.
Levy DE, Darnell Jr JE. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3(9):651–62.
Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006;6(8):602–12.
Xu D, Qu CK. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci. 2008;13:4925–32.
Tsou RC, Bence KK. Central regulation of metabolism by protein tyrosine phosphatases. Front Neurosci. 2012;6:192.
Takahashi A et al. SHP2 tyrosine phosphatase converts parafibromin/Cdc73 from a tumor suppressor to an oncogenic driver. Mol Cell. 2011;43(1):45–56.
Duchesne C et al. Negative regulation of beta-catenin signaling by tyrosine phosphatase SHP-1 in intestinal epithelial cells. J Biol Chem. 2003;278(16):14274–83.
Simoneau M et al. SHP-1 inhibits beta-catenin function by inducing its degradation and interfering with its association with TATA-binding protein. Cell Signal. 2011;23(1):269–79.
Liang LF et al. Cembrane diterpenoids from the soft coral Sarcophyton trocheliophorum Marenzeller as a new class of PTP1B inhibitors. Bioorg Med Chem. 2013;21(17):5076–80.
Zhang W et al. Ursolic acid and its derivative inhibit protein tyrosine phosphatase 1B, enhancing insulin receptor phosphorylation and stimulating glucose uptake. Biochim Biophys Acta. 2006;1760(10):1505–12.
Hsu MF, Meng TC. Enhancement of insulin responsiveness by nitric oxide-mediated inactivation of protein-tyrosine phosphatases. J Biol Chem. 2010;285(11):7919–28.
Zhang S et al. A highly selective and potent PTP-MEG2 inhibitor with therapeutic potential for type 2 diabetes. J Am Chem Soc. 2012;134(43):18116–24.
Lorenzen JA, Dadabay CY, Fischer EH. COOH-terminal sequence motifs target the T cell protein tyrosine phosphatase to the ER and nucleus. J Cell Biol. 1995;131(3):631–43.
Chughtai N et al. Prolactin induces SHP-2 association with Stat5, nuclear translocation, and binding to the beta-casein gene promoter in mammary cells. J Biol Chem. 2002;277(34):31107–14.
Wu TR et al. SHP-2 is a dual-specificity phosphatase involved in Stat1 dephosphorylation at both tyrosine and serine residues in nuclei. J Biol Chem. 2002;277(49):47572–80.
Wang Y et al. Proteomic analysis reveals novel molecules involved in insulin signaling pathway. J Proteome Res. 2006;5(4):846–55.
Kruger M et al. Dissection of the insulin signaling pathway via quantitative phosphoproteomics. Proc Natl Acad Sci U S A. 2008;105(7):2451–6.
Hornbeck PV et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40(Database issue):D261–70.
Acknowledgments
This review was written based on a literature search and some studies performed by the authors and that were supported by a grant from the Canadian Institutes of Health Research.
Conflict of interest
Authors declare no conflict of interest or financial relationship with the organization that sponsored some of the research described in this review article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xu, E., Schwab, M. & Marette, A. Role of protein tyrosine phosphatases in the modulation of insulin signaling and their implication in the pathogenesis of obesity-linked insulin resistance. Rev Endocr Metab Disord 15, 79–97 (2014). https://doi.org/10.1007/s11154-013-9282-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-013-9282-4