
Abstract
The obesity pandemic has focused attention in recent years to the physiological and molecular mechanisms that regulate body weight and glucose metabolism. Key signaling pathways that regulate both parameters depend heavily on reversible phosphorylation of proteins on tyrosine residues, a process regulated by the opposing activities of tyrosine kinases and tyrosine phosphatases. Here we review the roles of protein tyrosine phosphatase epsilon (PTPe) in regulating the leptin and insulin signaling pathways and through them—body weight and glucose metabolism. Mice lacking PTPe are leptin-hypersensitive and are protected from weight gain that follows a high-fat diet. PTPe helps downregulate leptin receptor signaling in the hypothalamus by dephosphorylating JAK2 following activation of the leptin receptor, thus inhibiting the receptor post-activation. PTPe is induced to perform this function after undergoing leptin receptor-induced phosphorylation at its C-terminal Y695. Mice lacking PTPe are also insulin-hypersensitive, indicating that PTPe downregulates signaling by this receptor as well. Studies in muscle cells confirm that PTPe inhibits insulin receptor signaling, possibly by targeting the receptor itself. These studies identify PTPe as a physiological inhibitor of both signaling pathways and as a factor in supporting the resistance to leptin and insulin that is established in obesity and in type-II diabetes, respectively.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Protein Tyrosine Phosphatases
Reversible phosphorylation of proteins is one of the better-studied molecular mechanisms for regulation of protein structure and function in vivo. Although the majority of protein phosphorylation events occur in eukaryotic cells on serine or threonine residues, tyrosine phosphorylation plays a major and well-established role in regulating cellular processes [1, 2]. Tyrosine phosphorylation is a reversible process that is regulated by the generically opposing activities of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). The human genome contains 90 PTK genes and 107 genes that encode PTPs; of these, 85 and 81 genes, respectively, produce products that target protein substrates [3]. The numbers of PTKs and PTPs are then similar and are dwarfed by the much larger numbers of known and hypothesized tyrosine phosphorylation sites in cellular proteins. As a result, each PTK or PTP targets on average several substrates and fulfills multiple distinct physiological roles in different cell types.
The 38 members of the “classical” PTP family, which target exclusively phosphotyrosine residues in proteins, form the core of the PTP superfamily. Classical PTPs contain one or two PTP domains of approximately 240 amino acids each in their cytosolic regions and dephosphorylate their substrates by a two-step mechanism. In the first step, the PTP forms a covalent bond with the phosphate group of the substrate phosphotyrosine, displacing the phosphate group from this tyrosine in the process and releasing the dephosphorylated protein. The PTP-phosphate bond is then hydrolyzed, regenerating the active PTP and releasing the phosphate group [4]. Classical PTPs can be classified into two subgroups: the receptor-type PTPs (RPTPs), which number 21 genes, and the remaining 17 genes that encode non-RPTPs. Each subgroup can be subdivided further according to structural and sequence similarities. RPTPs contain an extracellular domain of varying length and structure, a membrane-spanning domain, and one or two cytosolic PTP domains. Although the structure of the extracellular domains of RPTPs resemble those of PTKs, few ligands of RPTPs are known and most are orphan receptors. Non-RPTPs are typically comprised of a single PTP domain that is flanked by other protein domains, which control the subcellular localization of the enzyme or regulate its activity [3, 5].
Recent studies have elucidated the molecular roles of specific PTPs in regulating discrete physiological processes such as regulation of body weight, glucose homeostasis, neural development, immune function, bone structure, and others. The reader is referred to several recent reviews that discuss the physiological roles of PTPs and their modes of regulation in detail [5–10]. Advances in understanding the molecular roles of discrete PTPs in physiology have led to attempts to inhibit PTPs for pharmaceutical use, either by design of specific inhibitors (e.g., [11, 12]) or by other means (e.g., [13]). Major challenges in the field include identifying PTPs that are suitable candidates for pharmaceutical intervention in a given context and designing inhibitors that can enter cells and function in vivo in a specific manner, without targeting related PTPs whose activities may be beneficial.
PTP Epsilon
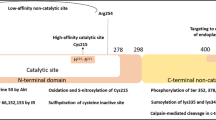
Protein tyrosine phosphatase epsilon (PTPe) exists as a small family of proteins, all produced from the single Ptpre gene (PTPRE in humans; Fig. 10.1a). PTPe was described originally as a receptor-type PTP (RPTPe, tm-PTPe, PTPepsilonM) that includes a short and heavily glycosylated extracellular domain and two cytosolic PTP domains of which only the first, membrane-proximal one, is active [14–16]. A second major form of PTPe was subsequently discovered. This cytosolic isoform of PTPe (cyt-PTPe) (=PTPepsilonC) contains a short hydrophilic sequence of 12 amino acids that replaces the membrane-spanning and extracellular domains of RPTPe [17, 18]. The two major PTPe proteins are products of distinct transcripts of the Ptpre gene; each transcript is expressed from a separate promoter and exhibits a unique pattern of expression among cells and tissues [17–19]. Both transcripts share most of their sequence and differ only at their 5′ end; as a result, their protein products RPTPe and cyt-PTPe differ only at their N termini (Fig. 10.1a, b). The Ptpre gene gives rise to two additional but less-abundant proteins, p67 and p65. p67 is transcribed from the RPTPe or cyt-PTPe mRNAs starting at an initiation codon that is located in the region common to both mRNAs. In contrast, p65 is produced by calpain-mediated post-translational processing of RPTPe, cyt-PTPe, or p67 proteins [20, 21] (Fig. 10.1a). The four PTPe proteins differ only at their amino termini (Fig. 10.1b), hence each has its own pattern of subcellular localization. RPTPe is exclusively membranal, cyt-PTPe is predominantly cytosolic, but is found to some extent associated with the membrane or within the cell nucleus, while p67 and p65 are exclusively cytosolic [18, 20–22].

The PTPe proteins. (a) Schematic outline of the four known forms of PTPe protein. The single Ptpre gene gives rise to two distinct mRNA species via separate promoters. The 5′ sequences of these mRNAs (white oval and rectangle) are unique, but downstream sequences are identical. Each mRNA produces a major protein—RPTPe or cyt-PTPe. Both mRNAs also produce p67 via initiation of translation at an ATG codon located in the sequences common to both mRNAs. Finally, all three PTPe proteins can be cleaved by calpain to generate p65. All PTPe proteins contain the same PTP domains (black rectangles); the N terminus of cyt-PTPe is unique to this form, (striped rectangle) Asterisk denote C-Terminal phosphorylation site. (b) N terminal sequences of all 4 forms of PTPe protein. Asterisks denote identical protein sequences
Physiological Roles of PTPe
In a manner similar to other PTPs that have several substrates, PTPe has been shown to regulate several divergent physiological systems; in some of these the role of PTPe is inhibitory, while in others it activates signaling events:
-
(a)
Breast cancer: Analysis of mammary tumors that were induced in mice due to expression of oncogenes in their mammary epithelium revealed that RPTPe is expressed specifically in tumors initiated by activated (V664E) Neu or activated (G12R) Ras, but not by c-Myc or several other onco-proteins [14]. Further studies indicated that RPTPe is not an oncogene in its own right [23], but that it supports the transformed phenotype induced by Neu in mammary epithelial tumor cells. RPTPe performs this role by linking Neu with its downstream effector Src; Neu phosphorylates RPTPe at its C-terminal Y695, which drives RPTPe to dephosphorylate and activate Src [24]. Accordingly, mammary tumor cells induced by Neu that lack RPTPe exhibit reduced Src activity and appear less transformed than similar cells that express RPTPe [25].
-
(b)
Peripheral nerve myelination: cyt-PTPe is expressed in Schwann cells, which carry out myelination of axons in the peripheral nervous system. The delayed-rectifier, voltage-gated potassium channels Kv2.1 and Kv1.2 are physiological substrates of cyt-PTPe in these cells [26]. Membrane depolarization triggers Kv2.1 and induces it to facilitate exit of potassium cations from the cell; this process is potentiated by phosphorylation of Kv2.1 at Y124 by Src or Fyn. Cyt-PTPe antagonizes the activity of Src or Fyn by dephosphorylating Y124 of Kv2.1, thus downregulating channel activity both in vitro and in vivo [27]. In agreement, mice that lack PTPe exhibit a severe delay in sciatic nerve myelination and Kv2.1 is hyper-phosphorylated in Schwann cells derived from these mice [26]. A similar mechanism may act in the central nervous system, since expression of an inactive “substrate trapping” mutant of RPTPe in transgenic mice delays optic nerve myelination [28].
-
(c)
Osteoclast activity: Female mice that lack PTPe exhibit mild osteopetrosis (gain of bone mass), which is due to reduced adhesion and activity of bone-resorbing osteoclasts. Osteoclasts express cyt-PTPe; lack of this PTP significantly disrupts the structure, cellular distribution, and stability of podosomes, the adhesion structures of these cells, leading to reduced bone-resorbing activity [29]. In agreement, mobilization of hematopoietic precursor cells from the bone marrow to the general circulation, in which osteoclasts participate, is defective in PTPe-deficient female mice [30]. At the molecular level, cyt-PTPe links signaling by integrins, the mechano-sensory receptors present on osteoclasts, with downstream activation of Src. Integrin activation induces phosphorylation of cyt-PTPe at its C-terminal Y638 (=Y695 in RPTPe), an event that is central in enabling cyt-PTPe to dephosphorylate and activate Src [31]. Accordingly, Src activity is reduced in osteoclasts that lack cyt-PTPe. As proper activity of Src is absolutely required for osteoclast-mediated bone resorption [32, 33], this strongly suggests that lack of cyt-PTPe disrupts osteoclast activity via reducing Src activity. Indeed, increasing Src activity in PTPe-deficient osteoclasts can correct the podosomal phenotype of PTPe-deficient osteoclasts [31].
-
(d)
Other roles: PTPe can downregulate mitogenic signaling induced by MAP kinase [34, 35] or by JAK-STAT signaling in M1 leukemia cells [36–38]. Macrophages from PTPe-deficient mice are impaired in their respiratory burst response and produce reduced amounts of cytokines in response to bacterial lipopolysaccharide [39], and PTPe negatively regulates proliferation of endothelial cells [40]. RPTPe also affects erythrocyte morphology and downregulates activity of Ca2+-activated potassium channels in these cells [41]. In what follows we describe a novel role for PTPe in regulating body mass and whole-body glucose homeostasis.
Regulation of Body Weight by Protein Tyrosine Phosphatases
The obesity pandemic has focused attention on the physiological and molecular mechanisms that regulate body weight and on possible approaches to manipulate them for therapeutic gain. Leptin, a hormone that is produced in white adipose tissue, is a well-established regulator of energy balance and body weight [42]. Leptin affects body weight by activating its receptor in the hypothalamus; this leads to activation of the receptor-associated PTK JAK2, to subsequent phosphorylation of downstream effector molecules, and to activation of the STAT3, PI3-kinase, and extracellular signal-regulated kinase (ERK) signaling pathways (Fig. 10.2). Activation of the leptin receptor ultimately increases production of the anorexigenic (appetite-depressing) neuropeptide pro-opiomelanocortin (POMC) and inhibits production of the orexigenic (appetite-stimulating) neuropeptides agouti-related protein (AgRP) and neuropeptide Y. The combined effect of leptin is therefore to decrease appetite and stimulate energy expenditure, which is consistent with presence within the organism of sufficient energy deposits in the form of body fat [43–46]. Interestingly, obesity is often associated in humans with resistance to the effects of leptin that is caused by poor activation of hypothalamic leptin signaling by the hormone. As a result, obese individuals often display elevated concentrations of leptin in circulation and respond poorly in terms of weight loss to exogenously administered leptin.

Schematic outline of hypothalamic leptin receptor signaling. The hypothalamic leptin receptor is associated with the JAK2 PTK. Upon activation by leptin, the receptor activates JAK2, which autophosphorylates and trans-phosphorylates the receptor at tyrosines 985, 1077, and 1138. Phosphorylated tyrosines bind SHP2, STAT5, and STAT3, respectively, and activate the signaling pathways shown. Inhibition of leptin signaling is achieved by SOCS3, a target gene of STAT3, and by tyrosine phosphatases such as PTP1B, TC-PTP, and PTPe. PTPs are shown in boldface italic type
The central role of the JAK2 PTK in hypothalamic leptin signaling strongly suggests that tyrosine phosphorylation events are critical in regulation of body weight. In agreement, several PTPs have been shown to participate in regulation of leptin receptor signaling in this context (reviewed elsewhere within this volume). The non-receptor PTP PTP1B dephosphorylates and inactivates hypothalamic JAK2; accordingly, mice lacking PTP1B are hypersensitive towards the effects of leptin and exhibit reduced adiposity [47–49]. The closely related PTP TCPTP also inhibits leptin receptor signaling. Mice lacking TCPTP in neurons exhibit leptin hypersensitivity and are resistant to weight gain induced by high-fat food. In a manner distinct from PTP1B, TCPTP targets STAT3; accordingly, combined deletion of both PTPs induces stronger protection from diet-induced obesity than loss of either PTP alone [50]. The SH2 domain-containing PTP SHP2 affects leptin signaling in several ways. SHP2 promotes leptin signaling by binding the leptin receptor and activating downstream ERK signaling; however, it also inhibits JAK2 and STAT3 signaling, thereby downregulating receptor activity. The overall effect of SHP2 is to upregulate hypothalamic leptin receptor signaling, as shown by the increased adiposity, decreased leptin sensitivity, and decreased energy expenditure in mice that lack hypothalamic SHP2 [51, 52]. In agreement, expression of a dominant-active mutant of SHP2 in forebrain neurons of mice induced resistance to weight gain caused by high-fat food and improved whole-body glucose homeostasis [53]. Interestingly, this latter effect was detected in females, suggesting that SHP2 links estrogen and leptin signaling [53]. Inhibition of PTPs that downregulate leptin receptor signaling should improve the response of the receptor to its ligand and promote weight loss. However, such inhibition of PTPs should be specific and avoid inhibiting other PTPs, such as SHP2, that perform opposite roles in hypothalamic leptin receptor signaling [54] and whose inhibition would most likely increase body weight. In this context, we describe the roles of PTPe in regulation of hypothalamic leptin signaling.
Lack of PTPe Protects Mice from Weight Gain and Increases Basal Metabolic Rate
Mice that lack PTPe do not express any form of PTPe protein (PTPe-knockout [EKO] mice; [26]). EKO mice of both genders are born in Mendelian ratios and gain weight normally throughout development. However, when placed on a high-fat diet (HFD), female EKO mice gain less weight than their wild-type (WT) counterparts; after 11 weeks on fat-rich food, EKO female mice gained 55 % less weight than controls [55]. Significant protection from weight gain was observed also in mice that had been ovariectomized, a procedure that also results in massive weight gain in WT mice. Moreover, elderly female EKO mice (age >1 year) weighed significantly less than matched WT controls [55]. Reduced weight of EKO mice was associated with reduced mass of white adipose tissue deposits. Interestingly, protection from weight gain was detected in female EKO mice; little protection was observed in male EKO mice. Further studies by indirect calorimetry revealed that when fed regular lab chow (in which 18 % of calories are derived from fat), female EKO mice tended to produce more body heat, consume more oxygen, and produce more carbon dioxide, but this did not reach the level of statistical significance. In contrast, when mice were shifted to high-fat food (59 % of calories from fat), body heat production, oxygen consumption, and carbon dioxide production were significantly increased in EKO mice and were significantly higher than in control WT mice. In all of these studies EKO mice and matched controls consumed similar amounts of food and their physical activities were similar. It then appears that EKO female mice gain less weight when fed fat-rich food due to increased basal metabolic rate, which reduces the amount of excess food-derived energy stored as fat [55].
Loss of PTPe Induces Leptin Hypersensitivity in the Hypothalamus
Female mice lacking PTPe exhibit a strong trend for reduced concentrations of leptin in circulation [55]. When mice were subjected to a HFD, ovariectomy, or were analyzed at an advanced age—all paradigms in which EKO mice protected from weight gain—EKO mice exhibited significantly reduced levels of leptin in circulation. As was the case with weight gain protection, circulating levels of leptin were normal in male EKO mice; fat-rich food increased circulating levels of leptin significantly but similarly in both EKO and WT male mice. Reduced leptin in circulation could arise from a primary defect in adipose tissue that reduces leptin synthesis or secretion. Alternatively, the primary defect could reside in the hypothalamus: feedback from increased hypothalamic leptin sensitivity could reduce synthesis or secretion of leptin by adipose tissue in order to maintain normal overall hypothalamic leptin signaling. Strong data in favor of the latter explanation was provided when female EKO mice were shown to be leptin-hypersensitive when challenged with leptin in acute and in chronic leptin stimulation protocols. Acute injection of leptin to female EKO mice resulted in increased phosphorylation of hypothalamic STAT3, while chronic administration of leptin over several days resulted in a greater drop in food intake in these mice than in matched controls [55]. Coupled with high levels of RPTPe expression in the hypothalamus and very low PTPe expression in adipose tissue, these results indicate that lack of PTPe upregulates leptin receptor signaling in the hypothalamus. Indeed, further studies revealed co-localization of PTPe and leptin receptor mRNAs in neurons in the arcuate nucleus of the hypothalamus [55].
PTPe Dephosphorylates JAK2 and Downregulates Leptin Receptor Signaling
Molecular studies in heterologous systems indicated that RPTPe can reduce leptin-induced phosphorylation of JAK2 (at Y1007/1008) and STAT3 (at Y705). Moreover, RPTPe and the leptin receptor co-immunoprecipitate together with JAK2, and purified RPTPe can dephosphorylate purified JAK2. In all, the data indicate that RPTPe physically associates with the leptin receptor and JAK2, and that JAK2 is a substrate of RPTPe in the context of leptin receptor signaling [55]. It is then reasonable to conclude that lack of RPTPe prevents sufficient dephosphorylation of hypothalamic JAK2 following leptin stimulation, does not inhibit leptin signaling sufficiently, and results in leptin hypersensitivity.
Regulation of RPTPe by Leptin-Induced Phosphorylation
As indicated above, the abilities of both major forms of PTPe, RPTPe and cyt-PTPe, to carry out their physiological function can be affected by phosphorylation of their C-terminal tyrosine residue (Y695 in RPTPe = Y638 in cyt-PTPe, Fig. 10.1). Accordingly, phosphorylation of Y695 is critical for RPTPe to activate Src in Neu-induced mammary tumor cells [24], while similar phosphorylation of cyt-PTPe supports its activation of Src in osteoclasts. Phosphorylation of cyt-PTPe is also required for its EGF-induced association with tubulin, which inhibits the PTP [31, 56]. These findings suggest the existence of a signaling module, in which activation of a cellular receptor induces C-terminal phosphorylation of RPTPe or cyt-PTPe that induces the PTP to dephosphorylate a downstream molecule as part of its physiological function. Receptor-induced phosphorylation may be direct (most likely as is the case of Neu in mammary tumors, [24]) or indirect via a surrogate kinase (such as when activated integrins in osteoclasts partially activate Src to phosphorylate cyt-PTPe [31]). The molecular mechanism by which phosphorylation affects PTPe is unknown, but it most likely does not involve altering the kinetic parameters of its catalytic activity [24]. Rather, it might affect the ability of PTPe to interact with other molecules.
The above results suggested that C-terminal phosphorylation of RPTPe may play a role in regulating its activity in leptin signaling. In agreement with the above model, intra-peritoneal injection of leptin into female WT mice induced transient phosphorylation of RPTPe at Y695 (Fig. 10.3a) [55]. Feeding female EKO mice high-fat food induces phenotypes in these mice (higher basal metabolic rates, protection from diet-induced weight gain), suggesting that the importance of RPTPe function is increased when mice are challenged by fat-rich food. Leptin-induced phosphorylation of RPTPe was higher in mice fed fat-rich food than in mice fed regular lab chow (Fig. 10.3b), suggesting that RPTPe phosphorylation is important for its function. Further studies indicated that JAK2 can phosphorylate RPTPe at Y695 upon leptin stimulation in cells, and that purified JAK2 can phosphorylate purified RPTPe in vitro [55]. Importantly, the non-phosphorylatable mutant Y695F RPTPe is significantly impaired in its ability to dephosphorylate JAK2 following leptin stimulation, confirming the functional importance of this phosphorylation event [55]. In all, the data suggest a model (Fig. 10.4) by which RPTPe is associated with the leptin receptor and JAK2. Upon leptin binding to its receptor, JAK2 phosphorylates several downstream substrates, among them RPTPe at Y695. RPTPe then dephosphorylates and inactivates JAK2, thus participating in a negative-feedback regulatory loop that helps return leptin signaling to its pre-stimulation levels. PTPe auto-dephosphorylates at Y695, thus possibly limiting its own activity in this pathway. In this respect PTPe functions similarly to PTP1B and to the related TCPTP, which inhibit hypothalamic leptin receptor signaling; absence of any of these PTPs induces leptin hypersensitivity in mice [47–50]. Although both PTP1B and RPTPe target JAK2, only RPTPe appears to be regulated by C-terminal phosphorylation [48, 49, 55]. This indicates that PTP1B and RPTPe may inhibit leptin signaling in distinct physiological contexts. Phosphorylation-dependent inhibition of RPTPe also implies that RPTPe might play a role in shutting down the leptin receptor post-activation rather than in preventing its inappropriate activation beforehand. Finally, loss of RPTPe affects the body weight of females in particular. Estrogen signaling in the hypothalamus affects body weight, and it is possible that the physiological role of RPTPe is affected by both estrogen and leptin signaling events. Further studies are required to clarify this issue.

Activation of the leptin receptor induces phosphorylation of hypothalamic RPTPe at Y695. (a) Four-month-old female WT mice were injected intraperitoneally with leptin (5 μg/g body weight). Hypothalami were isolated and analyzed with a pY695-PTPe-specific antibody. (b) Bar diagram showing increased phosphorylation of RPTPe at Y695 at t = 60 min following leptin injection in mice fed chow diet (CD) or fatty food (HFD). N = 7–10 mice per bar; Asterisks, p ≤ 0.0065 by Student’s t-test. Reprinted from Cell Metabolism, 13(5), Rousso-Noori et al., Protein tyrosine phosphatase epsilon affects body weight by downregulating leptin signaling in a phosphorylation-dependent manner, pp. 562–572, Copyright 2011, with permission from Elsevier

Inhibition of hypothalamic leptin receptor signaling by RPTPe. In this schematic model, prior to initiation of leptin signaling the leptin receptor, JAK2 and RPTPe form part of a molecular complex (a). Following binding of leptin to its receptor, JAK2 undergoes autophosphorylation (b) and trans-phosphorylates RPTPe at Y695 (c). RPTPe then dephosphorylates JAK2 and inactivates it (d), after which RPTPe auto-dephosphorylates (e) and returns the system to its original state (a). Model scheme adapted from Cell Metabolism, 13(5), Rousso-Noori et al., Protein tyrosine phosphatase epsilon affects body weight by downregulating leptin signaling in a phosphorylation-dependent manner, pp. 562–572, Copyright 2011, with permission from Elsevier
PTPe as an Inhibitor of Insulin Receptor Signaling
Some of the physiological functions of the receptors for insulin and for leptin are closely related. Obesity and diabetes are both part of the metabolic syndrome and, if left to follow its clinical course, obesity is often followed by disruptions to glucose homeostasis and diabetes. Moreover, both insulin and leptin play similar roles in the hypothalamus with respect to regulation of body weight [57]. Leptin is believed to function predominantly in regulating body weight of females, while insulin is more dominant in this respect in males [58, 59]. Several studies have suggested that PTPe inhibits insulin receptor signaling. A screen of Baby Hamster Kidney (BHK) cells that overexpressed the insulin receptor identified RPTPe as a phosphatase that can dephosphorylate the receptor and downregulate its activity [60]. In this system the non-receptor form of PTPe, cyt-PTPe, did not affect insulin receptor signaling, although subsequent studies in other systems indicate that cyt-PTPe can also target the receptor (see below). Later studies in primary hepatocytes confirmed that RPTPe targets the insulin receptor and can inhibit downstream signaling events, such as activation of protein kinase B (PKB), ERK, and glycogen synthase kinase 3 (GSK3), as well as insulin-induced glycogen synthesis and insulin-induced suppression of phosphoenolpyruvate carboxykinase expression (PEPCK) [61]. Adenoviral-induced expression of RPTPe in mice also reduced insulin-induced inhibition of PEPCK expression in the liver [61].
More recent studies have shown that PTPe, in particular cyt-PTPe, can inhibit insulin signaling in skeletal muscle cells. The L6 rat skeletal muscle cell line expresses predominantly cyt-PTPe. Expression of GFP-tagged cyt-PTPe in L6 cells revealed that after stimulating the cells with insulin, cyt-PTPe co-localizes with the internalized receptor [62]. Over-expression of cyt-PTPe in these cells reduced phosphorylation of the insulin receptor at tyrosines 972 and 1162/1163; cyt-PTPe also inhibited phosphorylation of IRS1, PKB, and GSK and reduced insulin-induced glucose uptake into the cells. As expected, downregulation of cyt-PTPe expression by RNAi induced the opposite results [62]. Increased phosphorylation of the insulin receptor and IRS1 were also observed when primary skeletal muscle cells from EKO mice were stimulated with insulin [62]. In all, it appears that both RPTPe and cyt-PTPe can inhibit insulin receptor phosphorylation and downregulate downstream signaling events in cultured cells.
These conclusions were borne out in vivo when whole-body glucose homeostasis and insulin signaling were evaluated in EKO mice. Fasting glucose and fasting insulin levels were normal in both male and female EKO mice that had been fed regular chow. However, male EKO mice that had been fed a HFD exhibited reductions of 21 % and 45 % in their circulating levels of glucose and insulin, respectively, after an overnight fast [55]. Improved performance in regulating blood glucose levels in the presence of reduced circulating insulin indicates that EKO mice are insulin-hypersensitive. In agreement, male EKO mice showed better control of their blood glucose levels than WT controls when injected with a bolus of glucose; similar results were obtained in both lean and obese mice [55]. Interestingly, male EKO mice presented a stronger phenotype in this respect than female EKO mice. Injection of insulin to mice fed regular lab chow induced increased hyperphosphorylation of PKB in muscles and in livers of EKO mice compared to control mice, providing additional evidence in favor of PTPe inhibiting insulin receptor signaling in vivo [55]. In all, the results presented here indicate that both major forms of PTPe, RPTPe and cyt-PTPe, inhibit insulin receptor signaling. Although the precise mechanism by which PTPe exerts this effect is not proven, the fact that phosphorylation of the insulin receptor itself is affected by changes in PTPe expression (e.g., [62]) strongly suggests that the receptor is a substrate of PTPe.
Gender Effects
An intriguing aspect of the effects of PTPe on leptin and on insulin signaling is their gender specificity. Loss of PTPe affects hypothalamic leptin signaling and protects from weight gain induced by a variety of physiological stimuli primarily in females, while it is mainly male EKO mice that enjoy improved glucose homeostasis. The basis for these effects is not clear at present. It has been suggested that, while both leptin and insulin signal in the hypothalamus to affect body weight, leptin is dominant in this respect in females while insulin is dominant in males [59]. This may occur due to gender-specific effects on signaling processes (e.g., by sex hormones) or to gender-specific variations in co-expression of PTPe and receptors for leptin or insulin in subsets of neurons. These interpretations are consistent with the fact that loss of hypothalamic PTPe increases leptin sensitivity and results in a female-specific body weight phenotype. The fact that a similar phenotype is not observed in EKO male mice may indicate that insulin receptor signaling is not affected in hypothalamic neurons. Similarly, the finding that EKO male mice display more significant improvement in glucose homeostasis than EKO females suggests that insulin receptor signaling in the periphery is affected by loss of PTPe predominantly in males. Further studies are required to challenge these interpretations and to uncover their molecular bases.
Future Directions
Studies outlined in this chapter indicate that PTPe plays a role in regulating signaling by the leptin and insulin receptors, thus affecting body weight and glucose homeostasis. While the basic role of PTPe in these signaling pathways is clear, further work is required to understand how PTPe functions in each tissue and cell type, and how these distinct effects combine in a whole organism to generate the overall picture observed in whole-body EKO mice described here.
From a broader perspective studies of PTPe and of other PTPs demonstrate that PTPs in general play pivotal roles in regulating leptin and insulin signaling and affect body weight and whole-body glucose homeostasis. The increasing spread of the metabolic syndrome pandemic makes it extremely tempting to design specific inhibitors of select PTPs to help regain control of leptin and insulin signaling in vivo for therapeutic gain. Demonstrated successes in designing specific inhibitors of PTKs for treatment of diseases in the past decade make this approach even more appealing. For this to occur, more complete basic understanding of the roles of various PTPs in regulating the relevant physiological pathways in vivo needs to be obtained. The difficulties encountered in designing PTP inhibitors that are specific and effective in vivo are, unfortunately, not trivial. However, studies such as those summarized here help lay the scientific foundation for this, by identifying specific PTPs as well as target cells and tissues where targeting should occur for maximal gain.
Abbreviations
- AgRP:
-
Agouti-related protein
- BHK:
-
Baby hamster kidney
- CD:
-
Chow diet
- Cyt-PTPe:
-
Cytosolic isoform of PTPe
- EKO:
-
PTPe-knockout
- ERK:
-
Extracellular signal-regulated kinase
- GSK:
-
Glycogen synthase kinase
- HFD:
-
High-fat diet
- PEPCK:
-
Phosphoenolpyruvate carboxykinase
- PKB:
-
Protein kinase B
- POMC:
-
Pro-opiomelanocortin
- PTK:
-
Protein tyrosine kinase
- PTP:
-
Protein tyrosine phosphatase
- PTPe:
-
Protein tyrosine phosphatase epsilon
- RPTPe:
-
Receptor-type isoform of PTPe
- RPTPs:
-
Receptor-type PTPs
- WT:
-
Wild-type
References
Hunter T (2000) Signaling—2000 and beyond. Cell 100(1):113–127
Lemmon MA, Schlessinger J (2010) Cell signaling by receptor tyrosine kinases. Cell 141(7):1117–1134
Alonso A, Sasin J, Bottini N, Friedberg I, Osterman A, Godzik A et al (2004) Protein tyrosine phosphatases in the human genome. Cell 117(6):699–711
Barford D, Das AK, Egloff MP (1998) The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu Rev Biophys Biomol Struct 27:133–164
Tonks NK (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol 7(11):833–846
Navis AC, van den Eijnden M, Schepens JT, Hooft van Huijsduijnen R, Wesseling P, Hendriks WJ (2010) Protein tyrosine phosphatases in glioma biology. Acta Neuropathol 119(2): 157–175
Vang T, Miletic AV, Arimura Y, Tautz L, Rickert RC, Mustelin T (2008) Protein tyrosine phosphatases in autoimmunity. Annu Rev Immunol 26:29–55
Hendriks WJ, Elson A, Harroch S, Stoker AW (2008) Protein tyrosine phosphatases: functional inferences from mouse models and human diseases. FEBS J 275(5):816–830
Pulido R, Hooft van Huijsduijnen R (2008) Protein tyrosine phosphatases: dual-specificity phosphatases in health and disease. FEBS J 275(5):848–866
den Hertog J, Ostman A, Bohmer FD (2008) Protein tyrosine phosphatases: regulatory mechanisms. FEBS J 275(5):831–847
Barr AJ (2010) Protein tyrosine phosphatases as drug targets: strategies and challenges of inhibitor development. Future Med Chem 2(10):1563–1576
Scott LM, Lawrence HR, Sebti SM, Lawrence NJ, Wu J (2010) Targeting protein tyrosine phosphatases for anticancer drug discovery. Curr Pharm Des 16(16):1843–1862
Haque A, Andersen JN, Salmeen A, Barford D, Tonks NK (2011) Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell 147(1): 185–198
Elson A, Leder P (1995) Protein-tyrosine phosphatase epsilon. An isoform specifically expressed in mouse mammary tumors initiated by v-Ha-ras OR neu. J Biol Chem 270(44):26116–26122
Krueger NX, Streuli M, Saito H (1990) Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO J 9:3241–3252
Lim KL, Lai DS, Kalousek MB, Wang Y, Pallen CJ (1997) Kinetic analysis of two closely related receptor-like protein-tyrosine-phosphatases, PTP alpha and PTP epsilon. Eur J Biochem 245(3):693–700
Nakamura K, Mizuno Y, Kikuchi K (1996) Molecular cloning of a novel cytoplasmic protein tyrosine phosphatase PTP epsilon. Biochem Biophys Res Commun 218:726–732
Elson A, Leder P (1995) Identification of a cytoplasmic, phorbol ester-inducible isoform of protein tyrosine phosphatase epsilon. Proc Natl Acad Sci U S A 92(26):12235–12239
Tanuma N, Nakamura K, Kikuchi K (1999) Distinct promoters control transmembrane and cytosolic protein tyrosine phosphatase epsilon expression during macrophage differentiation. Eur J Biochem 259:46–54
Gil-Henn H, Volohonsky G, Elson A (2001) Regulation of protein-tyrosine phosphatases alpha and epsilon by calpain-mediated proteolytic cleavage. J Biol Chem 276(34):31772–31779
Gil-Henn H, Volohonsky G, Toledano-Katchalski H, Gandre S, Elson A (2000) Generation of novel cytoplasmic forms of protein tyrosine phosphatase epsilon by proteolytic processing and translational control. Oncogene 19(38):4375–4384
Kraut J, Volohonsky G, Toledano-Katchalski H, Elson A (2002) Nuclear localization of non-receptor protein tyrosine phosphatase epsilon is regulated by its unique N-terminal domain. Exp Cell Res 281(2):182–189
Elson A (1999) Protein tyrosine phosphatase epsilon increases the risk of mammary hyperplasia and mammary tumors in transgenic mice. Oncogene 18(52):7535–7542
Berman-Golan D, Elson A (2007) Neu-mediated phosphorylation of protein tyrosine phosphatase epsilon is critical for activation of Src in mammary tumor cells. Oncogene 26(49): 7028–7037
Gil-Henn H, Elson A (2003) Tyrosine phosphatase-epsilon activates Src and supports the transformed phenotype of Neu-induced mammary tumor cells. J Biol Chem 278(18): 15579–15586
Peretz A, Gil-Henn H, Sobko A, Shinder V, Attali B, Elson A (2000) Hypomyelination and increased activity of voltage-gated K(+) channels in mice lacking protein tyrosine phosphatase epsilon. EMBO J 19:4036–4045
Tiran Z, Peretz A, Attali B, Elson A (2003) Phosphorylation-dependent regulation of Kv2.1 Channel activity at tyrosine 124 by Src and by protein-tyrosine phosphatase epsilon. J Biol Chem 278(19):17509–17514
Muja N, Lovas G, Romm E, Machleder D, Ranjan M, Gallo V et al (2004) Expression of a catalytically inactive transmembrane protein tyrosine phosphatase epsilon (tm-PTP epsilon) delays optic nerve myelination. Glia 48(4):278–297
Chiusaroli R, Knobler H, Luxenburg C, Sanjay A, Granot-Attas S, Tiran Z et al (2004) Tyrosine phosphatase epsilon is a positive regulator of osteoclast function in vitro and in vivo. Mol Biol Cell 15(1):234–244
Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y et al (2006) Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med 12(6):657–664
Granot-Attas S, Luxenburg C, Finkelshtein E, Elson A (2009) PTP epsilon regulates integrin-mediated podosome stability in osteoclasts by activating Src. Mol Biol Cell 20(20):4324–4334
Boyce BF, Yoneda T, Lowe C, Soriano P, Mundy GR (1992) Requirement of pp 60c-src expression for osteoclasts to form ruffled borders and resorb bone in mice. J Clin Invest 90(4):1622–1627
Destaing O, Sanjay A, Itzstein C, Horne WC, Toomre D, De Camilli P et al (2008) The tyrosine kinase activity of c-Src regulates actin dynamics and organization of podosomes in osteoclasts. Mol Biol Cell 19(1):394–404
Toledano-Katchalski H, Kraut J, Sines T, Granot-Attas S, Shohat G, Gil-Henn H et al (2003) Protein tyrosine phosphatase epsilon inhibits signaling by mitogen-activated protein kinases. Mol Cancer Res 1(7):541–550
Wabakken T, Hauge H, Finne EF, Wiedlocha A, Aasheim H (2002) Expression of human protein tyrosine phosphatase epsilon in leucocytes: a potential ERK pathway-regulating phosphatase. Scand J Immunol 56(2):195–203
Tanuma N, Nakamura K, Shima H, Kikuchi K (2000) Protein-tyrosine phosphatase PTPepsilon C inhibits Jak-STAT signaling and differentiation induced by interleukin-6 and leukemia inhibitory factor in M1 leukemia cells. J Biol Chem 275:28216–28221
Tanuma N, Shima H, Nakamura K, Kikuchi K (2001) Protein tyrosine phosphatase epsilonC selectively inhibits interleukin-6- and interleukin- 10-induced JAK-STAT signaling. Blood 98:3030–3034
Tanuma N, Shima H, Shimada S, Kikuchi K (2003) Reduced tumorigenicity of murine leukemia cells expressing protein-tyrosine phosphatase, PTPepsilon C. Oncogene 22(12): 1758–1762
Sully V, Pownall S, Vincan E, Bassal S, Borowski AH, Hart PH et al (2001) Functional abnormalities in protein tyrosine phosphatase epsilon-deficient macrophages. Biochem Biophys Res Commun 286:184–188
Thompson LJ, Jiang J, Madamanchi N, Runge MS, Patterson C (2001) PTP-epsilon, a tyrosine phosphatase expressed in endothelium, negatively regulates endothelial cell proliferation. Am J Physiol Heart Circ Physiol 281:H396–H403
De Franceschi L, Biondani A, Carta F, Turrini F, Laudanna C, Deana R et al (2008) PTPepsilon has a critical role in signaling transduction pathways and phosphoprotein network topology in red cells. Proteomics 8(22):4695–4708
Spiegelman BM, Flier JS (2001) Obesity and the regulation of energy balance. Cell 104(4):531–543
Wauman J, Tavernier J (2011) Leptin receptor signaling: pathways to leptin resistance. Front Biosci 17:2771–2793
Myers MG, Cowley MA, Munzberg H (2008) Mechanisms of leptin action and leptin resistance. Annu Rev Physiol 70:537–556
Myers MG Jr, Munzberg H, Leinninger GM, Leshan RL (2009) The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metab 9(2):117–123
Morris DL, Rui L (2009) Recent advances in understanding leptin signaling and leptin resistance. Am J Physiol Endocrinol Metab 297(6):E1247–E1259
Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG et al (2006) Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med 12(8):917–924
Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ et al (2002) Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell 2(4):497–503
Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y et al (2002) PTP1B regulates leptin signal transduction in vivo. Dev Cell 2(4):489–495
Loh K, Fukushima A, Zhang X, Galic S, Briggs D, Enriori PJ et al (2011) Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab 14(5):684–699
Krajewska M, Banares S, Zhang EE, Huang X, Scadeng M, Jhala US et al (2008) Development of diabesity in mice with neuronal deletion of Shp2 tyrosine phosphatase. Am J Pathol 172(5):1312–1324
Zhang EE, Chapeau E, Hagihara K, Feng GS (2004) Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc Natl Acad Sci U S A 101(45):16064–16069
He Z, Zhang SS, Meng Q, Li S, Zhu HH, Raquil MA et al (2012) Shp2 controls female body weight and energy balance by integrating leptin and estrogen signals. Mol Cell Biol 32(10):1867–1878
Banno R, Zimmer D, De Jonghe BC, Atienza M, Rak K, Yang W et al (2010) PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J Clin Invest 120(3):720–734
Rousso-Noori L, Knobler H, Levy-Apter E, Kuperman Y, Neufeld-Cohen A, Keshet Y et al (2011) Protein tyrosine phosphatase epsilon affects body weight by downregulating leptin signaling in a phosphorylation-dependent manner. Cell Metab 13(5):562–572
Sines T, Granot-Attas S, Weisman-Welcher S, Elson A (2007) Association of tyrosine phosphatase epsilon with microtubules inhibits phosphatase activity and is regulated by the epidermal growth factor receptor. Mol Cell Biol 27(20):7102–7112
Belgardt BF, Bruning JC (2010) CNS leptin and insulin action in the control of energy homeostasis. Ann N Y Acad Sci 1212:97–113
Clegg DJ, Riedy CA, Smith KA, Benoit SC, Woods SC (2003) Differential sensitivity to central leptin and insulin in male and female rats. Diabetes 52(3):682–687
Shi H, Seeley RJ, Clegg DJ (2009) Sexual differences in the control of energy homeostasis. Front Neuroendocrinol 30(3):396–404
Andersen JN, Elson A, Lammers R, Romer J, Clausen JT, Moller KB et al (2001) Comparative study of protein tyrosine phosphatase-epsilon isoforms: membrane localization confers specificity in cellular signalling. Biochem J 354(Pt 3):581–590
Nakagawa Y, Aoki N, Aoyama K, Shimizu H, Shimano H, Yamada N et al (2005) Receptor-type protein tyrosine phosphatase epsilon (PTPepsilonM) is a negative regulator of insulin signaling in primary hepatocytes and liver. Zoolog Sci 22(2):169–175
Aga-Mizrachi S, Brutman-Barazani T, Jacob AI, Bak A, Elson A, Sampson SR (2008) Cytosolic protein tyrosine phosphatase-epsilon is a negative regulator of insulin signaling in skeletal muscle. Endocrinology 149(2):605–614
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Elson, A. (2013). Protein Tyrosine Phosphatase Epsilon as a Regulator of Body Weight and Glucose Metabolism. In: Bence, K. (eds) Protein Tyrosine Phosphatase Control of Metabolism. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-7855-3_10
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7855-3_10
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-7854-6
Online ISBN: 978-1-4614-7855-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)