
Abstract
Acromegaly is a chronic, debilitating disease caused by chronic growth hormone (GH) hypersecretion which results in chronic medical comorbidities, poor quality of life and high mortality rates. Successful treatment can improve clinical signs and symptoms and normalize mortality rates. Over 95% of acromegaly is caused by a somatotroph adenoma of the pituitary, and the first-line treatment is generally transsphenoidal surgery, which can be curative in 50–60% of patients. Nonetheless, high rates of persistent acromegaly following surgery and the limited efficacy of radiation therapy necessitate chronic medical treatment for many patients. Somatostatin analogues have become the preferred first-line medical therapy for many practitioners, as they achieve better biochemical and direct tumor control than the dopamine agonists, and long-acting preparations make once monthly administration possible. Cabergoline, a dopamine agonist, offers a lower-cost option and may be effective in patients with a pituitary tumor that co-secretes GH and prolactin. Pegvisomant is a GH receptor antagonist that produces exceptional biochemical response rates but lacks any direct effects on the tumor, which may limit its effectiveness as life-long monotherapy. Combinations of these three drug classes have not been rigorously studied, and preliminary trials do not suggest improved clinical outcomes. While medical treatment options for acromegaly have significantly improved over the last 30 years, limitations remain, and a multi-specialty team approach is necessary for the effective long-term management of patients with acromegaly.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
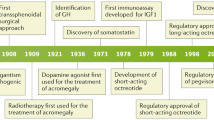
Acromegaly is a chronic, debilitating disease caused by chronic growth hormone (GH) hypersecretion. Over 95% of cases are caused by a pituitary somatotroph adenoma, with only rare cases of ectopic neoplasms producing GH or GH-releasing hormone (GHRH) [1, 2]. The onset of the somatic symptoms and physical features is insidious, often going unrecognized by patients and physicians until advanced stages of the disease. The condition is rare, with prevalence rates reported at 53 cases per million, and incidence of three to four new cases per million over 11 years [1, 3]. It has long been recognized that the amelioration of GH excess can reverse many of the untoward symptoms and associated co-morbid conditions, and diminish the risk of premature death. The mainstay of treatment has been surgical resection of the GH-secreting adenoma, which remains the only therapy that has a potential for cure. However, the high rate of surgical failure with persistent GH-hypersecretion frequently necessitates chronic medical treatment with pharmacologic agents that block the release of GH or its peripheral action at the GH-receptor. Recently, significant advancements in the understanding of central and peripheral neuroendocrine physiology and pharmacology have resulted in the development of new therapeutic agents; a growing body of published clinical trials have refined medical management and improved patient outcomes.
2 Comorbidity and mortality of acromegaly: rationale for treatment
Because of the gradual onset and non-specific nature of many of the clinical signs and symptoms of acromegaly, the delay to diagnosis averages 8–10 years from the onset of symptoms [3–5]. This delay in diagnosis is reflected in the size of the pituitary tumors that are a macroadenoma (>1 cm) at the time of diagnosis in at least 70% of patients [3, 6]. Macroadenomas often present with visual field loss because of optic chiasm compression, cranial-nerve palsies from cavernous sinus invasion, or non-specific headache syndromes. The somatotrophic effects of acromegaly are in part determined by the age of the patient. When the disease develops before the closure of epiphiseal growth plates, the result is gigantism, with both vertical and hypertrophic growth features. Acromegaly developing in adult patients often manifests as characteristic coarse facial features and enlargement of hands and feet from axial bone growth, soft tissue proliferation, and tissue edema. However, in older patients these physical traits can be more subtle or absent, making the condition more difficult to recognize in the presence of normal, age-related changes of facial features.
While recognition of the somatic changes of acromegaly is the most common trigger for appropriate diagnostic workup, the constellation of medical co-morbidities that develop with disease progression is equally characteristic. Hypertrophic arthropathy is common in the large joints of the knees and hips, as well as in the hands and feet, but can affect any joints in the skeleton. Severe osteoarthritis is common and often necessitates hip and knee surgery even after successful treatment [7]. Arthralgias are compounded by marked tissue edema in the hands and feet; a rapid post-operative reduction in hand arthralgias is often the first indication of biochemical remission. Soft tissue proliferation and third spacing of expanded plasma volume results in carpal tunnel syndrome [3]. Potential upper airway obstruction results from the typical hypertrophy of the tongue and soft palate predisposing these patients to obstructive sleep apnea (OSA), which occurs in as many as 90% of acromegalic patients who report snoring [8]. Interestingly, the same study that established these high rates of OSA also demonstrated that 33% of acromegalics with OSA suffered from central sleep apnea, rather than obstructive sleep apnea. Central sleep apnea was associated with higher GH and IGF-1 levels, suggesting that central hypothalamic pathways regulating involuntary respiration may be affected by GH hypersecretion [8]. Because OSA is an independent risk factor for hypertension, myocardial infarction, and stroke [9, 10], it has been suggested that underlying OSA may account for, or at least contribute to, the high cardiovascular morbidity and mortality seen in acromegaly [11].
However, many other common cardiovascular risk factors develop in acromegalic patients as a direct result of GH-hypersecretion. The counter-regulatory effect of GH leads to a spectrum of insulin resistance, impaired glucose tolerance, and type 2 diabetes mellitus. Hypertension is also common, and is poorly understood, though clearly multifactorial [3]. Basic science and clinical studies show conflicting results with variable degrees of contribution from insulin resistance, increased intravascular volume from the direct anti-natriuretic effects of GH and IGF-1 at renal tubules, and indirect GH effects that may be mediated through atrial natriuretic peptide, vascular endothelial growth factor, and increased sympathetic tone [3, 12, 13, 14–18]. Hypertension and insulin resistance likely contribute to increased cardiac morbidity, but may be outweighed by the direct GH effects on myocardium that lead to left ventricular diastolic dysfunction independent of the presence of hypertension and diabetes [3]. Diastolic dysfunction contributes to pulmonary hypertension and eventual right-sided heart failure, and in the advanced stages of disease a dilated cardiomyopathy can result. This constellation of GH related co-morbidities results in cardiovascular mortality in 60% of acromegalic patients, while pulmonary causes account for 25% and malignancy for 15% of overall mortality [19–26]. Patients who have cardiac disease at the time of diagnosis have a life expectancy of less than 15 years [26]. Hypertension, cardiac disease and high GH levels strongly predict negative mortality outcomes, with smaller contributions from diabetes and malignancy [23]. Bates et al. demonstrated that acromegalics with GH levels >5 μg/l had higher mortality rates than non-acromegalic subjects; these differences resolved if GH levels were maintained below 2 μg/l [27]. In a more recent analysis, morality curves did not normalize until GH levels were consistently <1 μg/l [28]. These data highlight the importance of aggressive, multi-modality treatment to achieve long-term remission or biochemical control to improve quality of life and reduce mortality in patients with persistent acromegaly.
3 Limitations of surgical and radiation therapy
3.1 Surgery
Transsphenoidal surgery is the established first-line treatment for all pituitary adenomas, excluding prolactinomas. However, depending on the degree of suprasellar extension of these often massive GH-secreting macroadenomas, a temporal approach via craniotomy may be necessary to complete tumor debulking or resection. The long-term efficacy of surgery is determined by GH levels at diagnosis, tumor size, extrasellar invasion, and the experience of the surgeon [21, 29, 30]. For tumors that are less than 1 cm (less than 30% of patients), the surgical remission rates can be as high as 70–80% in the hands of an experienced surgeon, but success rates fall to less that 50% for a macroadenoma [21, 29]. For those with initial remission following surgery, long-term follow-up has consistently identified relapse rates ranging from 3–10% [31, 32], although Nomikos and colleagues recently published a series of 668 surgical cases and reported a relapse rate of only 0.4% at 10 years [30]. The result is that 40–60% of patients will experience persistent or recurrent acromegaly following surgery, necessitating adjuvant radiation or medical therapy [6, 21, 29, 30, 33].
3.2 Radiation therapy
Conventional, fractionated radiation therapy (RT) using approximately 40–54 Gy total dose has resulted in highly variable results, with no effect on IGF-1 levels in one study [34], to as many as two thirds of patients showing normalization of IGF-1 levels in other studies with longer term follow-up [35, 36]. Stereotactic radiosurgery (SRS), such as Gamma Knife® (Elekta Corporation, Stockholm, Sweden), has been reported to result in shorter response times than conventional RT [37], however, a recent SRS study using more rigorous definitions for biochemical remission showed only a 17% remission rate after a mean of 50 months of follow-up [38]. The delay in biochemical response with both RT and SRS necessitates at least short-term medical therapy to control GH-hypersecretion, and long-term management is necessary for the 50–80% of patients that do not respond to radiation treatment.
4 Medical therapies
Currently, there are three available medical options for the management of acromegaly. Somatostatin analogues and dopamine agonists are used to inhibit GH secretion by pituitary tumors, whereas the GH-receptor antagonist, pegvisomant, has no direct effect on the tumor, but blocks GH activity at peripheral GH-receptors. The goal of these current medical treatments is to control the chronic disease of acromegaly through the normalization of IGF-1 levels, thus mitigating symptoms, improving quality of life, and reducing long-term morbidity and mortality. None of the current medical treatment options are curative. Medical treatment can be used as primary therapy for patients for whom surgery is not an option, as short-term, pre-operative, or post-radiation adjuvant therapy. Medical therapy is most widely used as secondary treatment for persistent or recurrent acromegaly following unsuccessful surgery.
4.1 Somatostatin analogues
The somatostatin analogues are the most widely used medical therapy for acromegaly and include octreotide and lanreotide. Octreotide (Novartis, Cambridge, MA) was the first commercially available agent, and has potent affinity for the somatostatin receptor subtype 2 (sst2), and more modest affinity for receptor subtype 5 (sst5) [39]. Stimulation of sst2 has been shown to induce both cell cycle arrest and apoptosis [40, 41], whereas activation of sst5 appears to only induce cell cycle arrest [42, 43]. Somatostatin analogues may also affect tumor growth via antiangiogenic mechanisms, with direct inhibition of endothelial cell growth and possible indirect effects via inhibition of vascular endothelial growth factor (VEGF) [43, 44].
Octreotide is short acting, suppressing GH for about 5 hours, and is thus given as multiple daily subcutaneous (SC) injections (two to four injections per day), with rises in GH observed between injections [45]. A long-acting preparation, octreotide LAR is available; this formulation encapsulates octreotide in microspheres of a biodegradable polymer [46]. The LAR formulation produces peak levels of octreotide at seven to fourteen days after injection, and plateau levels are maintained for 20–30 days which allows for intramuscular (IM) injection of 10 to 30 mg every four weeks in most patients [46]; some patients with lower GH levels may be able to be treated with intervals longer then four weeks [47]. Lanreotide has a similar profile of somatostatin receptor affinity for subtypes 1–5 [48], and comes in an intermediate-acting preparation (Lanreotide SR©, Ipsen Limited, Slough, UK), which is also encapsulated in polymer microspheres that prolongs release over 10–14 days following IM injection [49]. Lanreotide SR is available only as a 30 mg dose, so dose adjustment is only achieved through modification of the dosage interval (7, 10, or 14 days) [5, 49]. A newer preparation, lanreotide Autogel (Lanreotide LAR©, Somatuline, Ipsen Limited, Slough, UK), is now available. After deep SC injection, the drug naturally congeals into a slow release aqueous gel that allows for once monthly administration of 60, 90, or 120 mg doses [39, 50]. The long-acting somatostatin analogues remain expensive, with the annual cost of octreotide LAR 20 mg in the USA around $18,000–25,000, and it requires a clinic visit for injection. Lanreotide Autogel is expected to be priced 10% higher than octreotide LAR to account for the logistical advantage of home injections.
4.1.1 Clinical studies
Somatostatin analogues are capable of reducing GH-levels and normalizing IGF-1 levels in at least 48 and 42% of acromegalic patients, respectively [5], and also reduce tumor size in a subset of patients [51–53]. This dual therapeutic action, along with the easy administration of the long-acting preparations has lead to the widespread use of these agents as adjuvant pre-operative therapy [54], secondary medical management for post-operative persistent or recurrent acromegaly [52], and as primary medical therapy in patients whom refuse surgery, or are otherwise poor candidates for surgery [39, 53].
4.1.2 Preoperative adjuvant therapy
Many studies have determined that GH levels at the time of diagnosis and tumor size are predictive of surgical outcomes, therefore, any therapy that decreases GH levels and tumor size before surgery would have a theoretical advantage [6, 21, 30, 33]. The largest clinical trial evaluating preoperative, adjuvant somatostatin analogue therapy was published by Lucas and colleagues who studied 104 newly diagnosed, previously untreated patients to evaluate the impact of preoperative lanreotide SR on tumor shrinkage, and short-term biochemical control [54]. Lanreotide SR 30 mg was administered IM every 10 days for 1 month (n = 84), 2 months (n = 13), or 3 months (n = 7) before transsphenoidal surgery. Reduction in tumor size of at least 20% on serial MRI studies was considered significant, and occurred in only 29% of patients. A total of 66% had some measurable tumor reduction, while only 18% experienced greater than 20% tumor growth during treatment. Rates of grade I and grade II extra-sellar extension increased by 5.2% during treatment; the rates of grade III and IV extension decreased by 2.1 and 3.2%, respectively. Of those patients with greater than 20% reduction in tumor size, 90% had a biochemical response to treatment. In contrast, among those with less than 20% shrinkage of the tumor, only 54.9% demonstrated biochemical response [54]. Biochemical response was the sole predictor of tumor shrinkage, with an odds ratio of 7.8 (95% confidence interval 1.6–37.1). Univariate analyses of surgical outcomes revealed that younger age, higher random GH and IGF-I levels at diagnosis, larger preoperative tumor volume, a tumor with more than one area of extrasellar extension, and the presence of cavernous sinus invasion were each predictive of persistent disease after surgery. However, the logistic regression model only found higher IGF-I levels at diagnosis and cavernous sinus invasion to be predictive of surgical failure [54]. Despite treatment, nine of 52 patients with no cavernous sinus involvement at baseline developed ‘doubtful’ or ‘certain’ cavernous sinus invasion at the end of treatment and none of the 29 patients with cavernous sinus invasion at baseline showed radiologic evidence for complete regression of tumor in the cavernous sinus [54]. Surgical remission rates were highly variable and were directly related to extrasellar extension: 55.8% remission for tumors limited to the sella, 32.6% remission for tumors with a single area of extrasellar extension, and 11.6% remission for tumors with more than one area of extrasellar extension (i.e. suprasellar and cavernous sinus involvement) [54]. The lack of a control group in this and many of the smaller trials makes the role of preoperative somatostatin analogue therapy difficult to determine. However, these respective remission rates suggest that the development of cavernous sinus involvement during treatment in 17% of patients with tumor previously limited to the sella would have a significant negative impact on surgical outcomes, and may outweigh the theoretical benefits of the tumor shrinkage observed in only 29% of the cohort [54]. Therefore, surgery should be performed as soon as possible, and the preoperative adjuvant use of somatostatin analogues can be considered for those patients in whom a delay in surgery is anticipated.
4.1.3 Secondary medical therapy
Newman et al. studied octreotide as secondary treatment in 81 patients with a history of persistent acromegaly at least 1 year after transsphenoidal surgery (n = 46), transsphenoidal surgery and radiation (n = 27), or radiation alone (n = 8) [52]. Patients were randomized to octreotide 100 or 250 μg administered by SC injection every 8 h for 6 months, after which time an extended open label treatment phase allowed for clinician directed dose titration to achieve biochemical control. The average daily dose of octreotide for the whole cohort at the end of the six-month, fixed-dose phase was 513 μg. Mean GH levels fell from 30.4 ± 10.1 to 7.1 ± 1.4 μg/l. After open label dose titration, the mean daily dose was 635 μg, and mean GH levels were further suppressed to 5.6 ± 1.1 μg/l by 3 months and remained suppressed throughout a mean follow-up of 34 months. The clinical and biochemical efficacy improved over the course of treatment, and at a mean follow-up of 3 years, 22% of patients had GH levels <2 μg/l on at least four visits, and IGF-1 levels declined to normal in 67% of patients [52].
More recently, a smaller study from Colao and colleagues evaluated octreotide LAR in 21 patients with persistent acromegaly after surgery [51]. All patients received octreotide LAR 20 mg IM every 28 days for three months, then dose titration occurred if GH levels were >5 μg/l; 15 patients were titrated to a dose of 30 mg every 28 days; at twelve months seven of these patients were further titrated to the maximum dose of 40 mg every 28 days. After 24 months, a normal IGF-1 level was achieved in 67% of subjects [51]. GH suppression was achieved in more patients than in the short-acting octreotide study with 15 of 21 patients (71%) achieving a GH nadir of 2.0 μg/l or less. Only nine patients were evaluated by MRI for tumor shrinkage; four patients (44%) had tumor shrinkage of >20% (range, 23–33%) [51].
A concern regarding the reported biochemical response rates in many of the somatostatin analogue trials has been the use of patient pre-selection in which GH-response to a single dose of octreotide is used to determine eligibility. In their meta-analysis of somatostatin analogues, Freda et al. evaluated GH and IGF-1 response rates among pre-selected and non-selected patients undergoing primary or secondary treatment for at least three months in 44 trials. In studies with octreotide LAR, efficacy for GH suppression was met by 58% of pre-selected patients, and in 54% of unselected patients (not statistically different), while IGF-1 normalization occurred in 68 and 63%, respectively (p = 0.04) [5]. The effects of patient pre-selection were the same in the trials with lanreotide SR, but this large meta-analysis found that the overall response rates were lower for these patients than in those receiving octreotide LAR regarding both GH suppression—50% for lanreotide SR, and 58% for octreotide LAR—and for IGF-1 normalization—56 vs 68%, respectively [5].
Lanreotide Autogel (also referred to as lanreotide LAR or Somatuline) has been shown to have near identical efficacy to lanreotide SR in a few studies, including a four-year follow-up study reported by Gutt et al. [47, 55, 56]. Lanreotide Autogel has also been compared with octreotide LAR in two small cross-over studies, with no clear evidence of superiority for either drug [57, 58]. Lanreotide SR and Autogel preparations may result in higher rates of diarrhea, abdominal pain, and flatulence than octreotide LAR [59], while rates of local injection-site reactions may be lower for lanreotide Autogel [58].
4.1.4 Primary medical therapy
Surgical resection is the only therapy that offers a chance for cure, so primary medical therapy should be reserved for those patients who have contraindications to surgery, or for those patients with macroadenomas exhibiting extrasellar extension to regions that make complete surgical resection unfeasible. The use of short-acting octreotide as primary treatment for 26 newly diagnosed acromegaly patients was studied in parallel with the secondary treatment of 81 patients in the three phase trial by Newman and colleagues described above [52]. Patients were randomized to 100 or 250 μg octreotide SC every eight hours for 6 months, then dose titration was used to attempt GH levels <2 μg/l; the mean dose of octreotide after titration was 777 μg/day in the primary treatment group vs 635 μg/day in the secondary treatment group. There was a higher percentage of responders in the primary treatment group (82 vs 67%), and mean GH levels in responders were the same in both groups at 3 years (3.7 ± 0.9 vs 3.8 ± 1.2 μg/l, respectively) [52], suggesting that somatostatin analogues offer similar biochemical outcomes in both primary and secondary treatment.
Because primary medical therapy is generally reserved for patients who are poor surgical candidates, avoiding the need for surgery for symptoms of local tumor encroachment on critical structures (i.e. optic chiasm, cranial nerves in the cavernous sinus), becomes paramount. A recent critical review of the literature evaluating tumor response during primary medical therapy found that among 14 studies (n = 424 patients), 36.6% of patients experienced significant shrinkage (>20%); within this group the mean reduction in tumor size was 45% [60]. The wide range of tumor response rates reported results from a lack of uniformity in definitions of clinically significant shrinkage (range 10–45%), a lack of uniform measurement (one dimensional, versus three dimensional volume averaging), and the lack of controls in these studies [60]. This review found no difference between short-acting and long-release preparations of somatostatin analogues regarding tumor response rates (34.0 vs 37.8%) or mean tumor volume shrinkage (49.5 vs 50.0%, respectively) [60]. The short-term (1 month for most patients), preoperative lanreotide study by Lucas et al, described above, reported that the GH-response was the sole predictor of tumor shrinkage [54]; however, all four studies that have evaluated this relationship in long-term primary treatment have shown no correlation between the GH-response and tumor shrinkage [47, 51, 53, 61]. In contrast, these studies and others have consistently shown that macroadenomas have a higher tumor response rate and greater reduction in size than microadenomas [47, 51, 53, 61]. Nonetheless, there are reports of radiologic disappearance of microadenomas after 6–16 months of long-acting somatostatin analog treatment [51, 62].
While much attention and debate has been paid to the degree and significance of tumor shrinkage with somatostatin analogues, the primary therapeutic goals of primary and secondary medical treatment should be control of GH-hypersecretion and inhibition of continued tumor growth in order to prevent the need for further surgical intervention. Tumor shrinkage is not a primary goal of treatment, and it has been shown that even for highly responsive tumors in which biochemical control and shrinkage are achieved, discontinuation of medical therapy results in the rapid return to pretreatment GH-hypersecretion and tumor growth [52, 62]. This is dramatically illustrated in the case report from Livadas et al, which shows complete radiologic disappearance of a 17 × 19 × 14 mm pituitary adenoma with suprasellar extension after 16 months of lanreotide SR treatment. There was no evidence of tumor on MRI scans at 24, 40, 51, and 62 weeks (at which point lanreotide was discontinued). Within 6 months of stopping lanreotide SR treatment, the GH and IGF-1 levels were 50 and 80% of pretreatment levels and the adenoma had grown to 4 × 5 × 4.5 mm. These data raise questions about the clinical implications of tumor shrinkage in the primary and secondary treatment of acromegaly. While it is perhaps psychologically reassuring to patients and physicians when it occurs, tumor shrinkage does not consistently predict biochemical response, nor does it affect long-term prognosis or the need for life-long suppressive treatment. Because biochemical control and the avoidance of surgery are the primary goals of medical treatment, the prevention of tumor growth is more clinically relevant than tumor shrinkage. Long-term somatostatin analogue trials have consistently shown that tumor growth is very rare during treatment; Bevans et al. reviewed 34 trials (n = 921 patients), which studied tumor shrinkage as a primary outcome, and among biochemical responders only one patient experienced tumor growth (tumor growth appeared to be the result of cystic expansion) [39]. Additionally, among all 921 patients (including both biochemical responders and non-responders), there were only 20 (3%) who had tumor growth, with tumor control or shrinkage in 97% of patients, suggesting that direct tumor effects may occur in the majority of patients receiving a somatostatin analogue, regardless of biochemical efficacy [39].
Given their potential for long-term biochemical suppression and tumor control, somatostatin analogue treatment plays an important role in the secondary management of persistent acromegaly, and in the primary management of patients without optic nerve compression or opthalmoplegia at presentation who are poor candidates for surgery [2, 39, 52, 60]. The disadvantages of somatostatin analogues include the high cost, a persistent 20–40% non-response rate, and side effects including nausea, fatigue, orthostatic hypotension and cholelithiasis [2]. In addition, these agents also inhibit the secretion of insulin, glucagon, and several gastrointestinal hormones [43]. While there is a lack of head-to-head trials of sufficient size to determine if distinct clinical advantages exist for one somatostatin analogue over another, the above trials and the meta-analysis from Freda et al. suggest that response rates may be higher with octreotide LAR than with short-acting octreotide and lanreotide SR [5, 51, 52]. To date there are no data to suggest significant differences in clinical outcomes for lanreotide Autogel and octreotide LAR [57, 58]. In considering therapeutic outcomes for the somatostatin analogues in general, reported rates of IGF-1 normalization may be exaggerated because of the widespread enrollment of pre-selected patients in clinical trials [5, 51, 52].
4.2 Dopamine agonists
Somatotroph tumors are derived from plurihormonal acidophil cells which also give rise to prolactin-secreting cells. About one third of GH-secreting tumors co-secrete prolactin, although, generally at much lower concentrations than in a true prolactinoma [63]. This shared cell lineage and the frequency of hormonal co-secretion spurred clinical trials of dopamine agonists for the management of acromegaly in the mid 1970’s and they continue to provide a therapeutic option for secondary treatment. Currently, bromocriptine and cabergoline are the only two dopamine agonists widely available for the management of acromegaly. Bromocriptine is an oral ergot derivative that has been used for over thirty years in the management of Parkinson’s disease and prolactinomas. The short half-life of the drug usually requires three to four daily doses to achieve therapeutic efficacy. Produced only in 2.5 mg tablets or 5 mg capsules, bromocriptine regimens can be laborious for patients, as effective daily doses in acromegaly trials are as high as 20–60 mg [64]. In contrast, cabergoline is a newer oral ergot derivative with a very long half-life, allowing for a starting dose of weekly or biweekly administration of the 0.5 mg tablet, and dose titration can be achieved with increasing dose frequency—with maximum doses of 0.5–1 mg daily used in acromegaly trials [65]. Trials of IM injections with a long-acting bromocriptine LAR preparation and the apomorphine derivatives such as quinagolide have been evaluated in clinical trials, but poor response rates have discouraged their widespread use [66, 67]. The clear advantage of the dopamine agonist is cost: in the USA the annual cost of cabergoline twice weekly dosing with 0.5 mg is about $2,000, while bromocriptine 20 mg daily costs about $3,500.
4.2.1 Clinical trials
Dopamine agonists, particularly bromocriptine, have been in clinical use for the medical management of persistent acromegaly for more than 30 years. However, the utility of bromocriptine has been limited by disappointing rates of biochemical response, and unacceptable side effects including nausea, vomiting, diarrhea, fatigue, and orthostatic hypotension, which may occur with the high doses (40–60 mg daily) required to achieve GH suppression, [64].
4.2.2 Secondary treatment
Bromocriptine became the first viable medical therapy for acromegaly following the landmark studies of Liuzzi et al. [68] and Thorner et al. [69], both of which confirmed GH suppressive effects of the drug. However, subsequent studies reported relatively low rates of GH response with only 20–40% of patients achieving GH levels <5 μg/l, and only 10–20% achieving normalization of IGF-1 levels, despite subjective clinical improvement in 50% of patients [65, 70, 71].
These therapeutic limitations were highlighted in a 1981 study evaluating the short-term efficacy of bromocriptine in a double-blinded, placebo controlled, cross-over study of eighteen patients with acromegaly. The study failed to show a significant difference in the GH response to the oral glucose tolerance test (OGTT) following three months of either placebo or bromocriptine, and patient-reported reductions in clinical symptoms were nearly identical for placebo and bromocriptine [72]. A follow-up study showed that one of the flaws of this negative study may have been a delay between the last dose of bromocriptine and the OGTT, demonstrating OGTT GH suppression at 1 h after the last dose, but not at 10 h [73]. This finding emphasized the transient effectiveness of bromocriptine with the high likelihood of suboptimal therapeutic action even between doses of the standard four times daily regimen. Furthermore, a subsequent report on the long-term outcomes of 235 patients treated with bromocriptine suggested that among responders, successful GH suppression is often short lived [74]. These shortcomings of bromocriptine led to the investigation of longer-acting dopamine agonists as they became available in the 1990’s. Secondary treatment with cabergoline 1.0–1.75 mg/week was evaluated in a multi-center, open-label trial involving 64 patients, including a subgroup of sixteen patients with tumors that co-secreted GH and prolactin (PRL) as identified by immunohistochemistry on surgical pathology specimens and elevated serum PRL levels [65]. With a starting dose of cabergoline 0.5 mg twice weekly, the dose was titrated to 0.5 mg every other day (1.75 mg/week; n = 28) in an effort to suppress IGF-1 to less than 300 μg/l; further dose titration to 0.5 mg daily in seven non-responders and 0.5 mg twice daily in one patient did not show any effect. Treatment duration ranged from 3 to 40 months. Overall, 39% of patients had a reduction of IGF-1 to less than 300 μg/l and 46% achieved GH levels <2 μg/l [65]. This large study identified subsets of patients with significantly higher response rates: among patients with initial IGF-1 levels < 750 μg/l (n = 40), 53% achieved normalization of IGF-1 and 60% achieved GH levels <2 μg/l; within the subset of GH/PRL tumors (n = 16), 80% achieved both normalization of IGF-1 and GH <2 μg/l. Serial MRI scans in 12 patients showed ‘distinct’ tumor shrinkage in five patients (none achieved a 50% reduction), some shrinkage in four others, and no change in the remaining three [65]. Cabergoline was well tolerated with mild nausea and orthostatic hypotension noted in a few patients at the start of treatment; only two patients (both receiving 1.75 mg/week) discontinued treatment due to nausea.
While the long-acting somatostatin analogues have significantly improved clinical outcomes and captured much of the market for medical treatment of persistent or inoperable acromegaly, the high cost and the potential side effects may deter their use in some patients. Cabergoline offers an alternative with substantial cost savings, a simple oral regimen, and minimal side effects. Relatively high rates of therapeutic response can be expected in those patients who have IGF-1 levels of <750 μg/l, and/or have tumors that co-secrete GH and PRL. The use of cabergoline as primary therapy for acromegaly has not been studied as rigorously as somatostatin analogues; the small studies suggest less dramatic effects on tumor shrinkage. A potential concern with long-term cabergoline therapy has recently arisen with the recognition of cardiac valvulopathy occurring in Parkinson’s patients treated with much higher doses (3.8 mg daily) of cabergoline [75]. To date, there has been no indication that the doses used in the management of pituitary tumors cause cardiac valve changes, but some practitioners have adopted routine echocardiogram evaluation for patients on chronic treatment.
4.3 GH Receptor antagonist
Treatment failures with dopamine agonists and the somatostatin analogues, and the high morbidity and mortality associated with uncontrolled acromegaly, has left a demand for novel medical therapies. This led to the development of pegvisomant (Somavert®, Pfizer, New York, NY), the first in its class of GH receptor antagonists. Pegvisomant is a modified GH molecule that functions as a GH receptor antagonist, with avid binding to cell surface GH receptors, thus blocking the binding of native GH and inhibiting GH receptor functional dimerization [76–78]. This GH receptor blockade inhibits hepatic production of downstream mediators of GH’s somatotrophic effects, including IGF-1, IGF-BP3 and acid-labile subunit (ALS) [76, 77]. Because there is no direct effect on the tumor or on GH secretion, GH levels increase with pegvisomant treatment; serum GH concentrations return to pretreatment baseline following cessation of the drug [79]. Pegvisomant is usually administered as a daily SC injection of 10, 20, or 30 mg. Peak plasma concentrations occur 48–72 h after initial injection and the average half-life is about 6 days. Clearance of pegvisomant decreases with escalating doses, resulting in a disproportionate rise in serum levels with higher doses: after 12 weeks of therapy with daily doses of 10, 15, and 20 mg, mean serum levels were 6,600 ± 1,330, 16,000 ± 2,200, and 27,000 ± 3,100 ng/ml, respectively [78]. The annual wholesale cost of pegvisomant 20 mg/day is approximately $65,000 and $97,000 for 30 mg/day.
4.3.1 Clinical trials
Trainer et al. reported the initial multi-center, placebo controlled clinical trial evaluating the safety and efficacy of pegvisomant in patients with acromegaly. The study involved 93 patients who had undergone transsphenoidal surgery, 57 of whom had also received conventional radiation, 6 patients who had only received radiation, 9 patients who had only been treated with other drug therapies and 4 who had not received any treatment [78]. Patients were randomized to receive daily SC injections of 10, 15, or 20 mg of pegvisomant, or placebo for twelve weeks. Normalization of IGF-1 occurred in 10% in the placebo group and in 54, 81, and 89% of patients, receiving 10, 15, or 20 mg of pegvisomant, respectively (p < 0.001 for each dose vs placebo) [78]. Three patients discontinued the study within the first week for issues not related to pegvisomant; only one patient left the study because of an adverse event, which involved an isolated transaminitis with a serum alanine aminotransferase concentration of 904 U/l (normal, 0–47) and a serum aspartate aminotransferase concentration of 389 U/l (normal, 0–37) after eight weeks of treatment. Transaminase levels returned to normal within 4 weeks of discontinuation, and recurred with drug rechallenge with pegvisomant 10 mg daily [78].
These rates of biochemical response proved to be an underestimation of the long-term efficacy, as demonstrated in a subsequent multi-center trial that reported the results in 152 patients receiving pegvisomant for an average of 465 days [79]. Normalization of IGF-1 occurred in 87 of 90 (97%) patients treated for 12 months or more. To document the consistent long-term efficacy, a subgroup of 38 patients who were treated for at least 83 weeks achieved a serum IGF-1 level within the age-adjusted normal range in 572 of 624 (91.7%) of study visits. Similar to the results of Trainer et al., this study demonstrated a progressive dose- and time-related increase in GH levels during pegvisomant treatment. Baseline GH levels differed significantly among the three cohorts which were defined by duration of treatment: pretreatment mean GH levels were 10.9 μg/l (±1.5), 13.2 μg/l (±2.1), and 19.2 μg/l (±4.3) in the 6, 12, and 18 month cohorts. Pegvisomant treatment increased mean GH levels by 12 to 14 μg/l in all three cohorts to 23.1 μg/l (±3.1), 25.6 μg/l (±4.3), and 33.8 μg/l (±8.6), respectively (p < 0.05 for within-cohort, baseline vs. final comparisons) [79]. In a subgroup of patients who were withdrawn from pegvisomant and not placed on alternative medical therapy for at least one month (n = 45), mean GH concentrations were 8.0 μg/l (±2.5) at baseline, 15.2 μg/l (±2.4) at the last visit before withdrawal, and reduced to 8.3 μg/l (±2.7) within 30 days of withdrawal (p = 0.67 vs. baseline) [79]. Metabolic profiles during the study suggested that these high levels of circulating GH were metabolically inactive during pegvisomant treatment with a decline in baseline fasting insulin levels from 23.3 to 12.4 mU/l after 18 months of pegvisomant (p = 0.03); fasting glucose levels declined from 98.4 to 90.4 mg/dl during pegvisomant therapy [79]. All measures of clinical signs and symptoms, including heat intolerance, headache, ring size, and blood pressure showed statistically significant improvement over 12 months of treatment [79].
This long-term study also addressed the concern that these elevated GH levels may act as a growth factor for the pituitary adenoma, thus predisposing patients to further need for surgery. Paired sets of the baseline and post-treatment MRI scans were available for 131 patients: in 78 patients previously treated with RT the mean tumor volume decreased by 0.126 cm3 (±0.071) over 12 months (p = 0.21), while in 53 patients who had not received RT the mean tumor volume increased by 0.103 cm3 (±0.093) over 10 months (p = 0.95) [79].
Overall, pegvisomant was well tolerated, with infections being the most frequent adverse event, reported in 33% of patients, most of which were non-serious, upper-respiratory-tract infections (seven cases of pneumonia), a gluteal abscess and a case of urosepsis [79]. In the 12-week trial, upper respiratory infections were also the most common reported side effect, but did not differ significantly from placebo [78]. Two patients in the long-term trial had increased concentrations of ALT and AST of more than tenfold of the upper limit of normal within 12 weeks of beginning pegvisomant therapy and as a consequence were withdrawn from the study.
These two pegvisomant trials represent some of the largest prospective clinical trials in the medical management of acromegaly; the 12-week placebo-controlled study offers the largest database of placebo treatment outcomes. The 10% normalization of IGF-1 in the placebo group likely represents delayed treatment effect of radiation treatment, which had previously been administered to 63 of the 93 patients and highlights the difficulty of determining drug efficacy in secondary treatment trials [79]. With very high rates (89–97%) of IGF-1 normalization and the excellent metabolic and clinical responses to long-term therapy, pegvisomant has been established as the most effective drug for biochemical control of acromegaly, but it lacks a direct tumor effect to control long-term tumor growth. Some concerns persist that the rise in GH which occurs with pegvisomant therapy may have some deleterious effects on tumor growth [80], however, the rapidly growing body of clinical experience is reassuring; Schreiber et al. has recently published the clinical outcomes for 229 patients treated for a mean of 51.8 weeks, with tumor growth verified in only 3.1% of patients [81]. Safety concerns aside, the cost of pegvisomant remains the primary barrier to its widespread use, which was the same issue with bromocriptine 25 years ago [70]. Currently, pegvisomant offers a highly effective, last-line medical treatment for patients who have failed lower-cost options.
4.3.2 Combination therapies
Each of the three drug classes currently available for the medical management of acromegaly offers unique mechanisms of action and therapeutic profiles; a single ideal agent has yet to be identified and treatment failures are a problem. As a result, the use of combination therapies may offer hope for therapeutic success when monotherapy has failed. Economic issues aside, the combination of a somatostatin analogue, offering the potential for potent direct tumor effects, and pegvisomant, with its effective biochemical control has obvious theoretical benefits. Feenstra et al. completed a 42-week, dose-finding study administering once weekly pegvisomant in combination with octreotide LAR 30 mg or lanreotide Autogel 120 mg monthly injections [80]. At 18 weeks, 21 of 26 patients (81%) had normal IGF-1 levels and at 42 weeks 18 of 19 patients (95%) had a normal IGF-1. While none of the 19 patients had tumor growth, only 3 of the 19 (16%) experienced tumor shrinkage [80]. Furthermore, the combination therapy seemed to exacerbate pegvisomant-related liver enzyme abnormalities, with 38% of patients developing mild elevations of transaminase levels. Similar results were recently reported by Neggers et al. who studied 32 patients treated with combination therapy for a mean of 138 weeks [82]. This study reported mild liver enzyme abnormalities in 11 of 32 patients (34%), but these changes were found to be transient, more often occurring early in treatment, and did not necessitate discontinuation or dose reduction of the pegvisomant [82]. The authors also report that the risk for liver enzyme abnormalities was much higher in diabetic patients [OR 5.1 (95% CI 1.02–25.54); p < 0.05) [82]. In both studies, the effort was made to reduce cost with once weekly pegvisomant, but the average dose of weekly pegvisomant required to normalize IGF-1 levels was 60 mg in both trials, and when combined with the cost of the somatostatin analogues, annual projected cost did not differ from daily pegvisomant monotherapy [80]. A lower-cost combination is being studied in a large, multi-center pharmaceutical sponsored trial (Novartis, Basel, Switzerland) which is enrolling patients in Germany to evaluate the addition of cabergoline to octreotide LAR for patients with partial response to previous monotherapy (http://clinicaltrials.gov).
5 Conclusions
Over the last 30 years significant advancements have been made in the medical treatment of acromegaly, with recent studies suggesting that pegvisomant is effective in achieving biochemical control in up to 97% of patients [79] while long-acting somatostatin analogues have been shown to prevent tumor growth in up to 97% of patients [39]. Unfortunately, the cost of pegvisomant remains prohibitive and a preliminary study of combination therapy does not suggest additive or synergistic therapeutic effects, instead resulting in a much lower rate of tumor shrinkage than with a somatostatin analogue alone [80]. Meanwhile, the lower-cost treatment strategies with the somatostatin analogues and dopamine agonists result in 30 to 80% non-response rates. For these reasons, a multi-disciplinary treatment strategy involving experienced neurosurgeons, radiation oncologists, and endocrinologists is necessary to determine the most efficacious and cost-effective approach for each patient with acromegaly.
6 Future directions and unanswered questions
Novartis (Basel, Switzerland) has developed a new somatostatin receptor agonist, SOM230, which has been shown to bind all of the somatostatin receptors except sst4, and binds sst5 with an affinity 40 times greater than does octreotide [83, 84]. In a proof-of-concept, single dose pilot study, 12 acromegalic patients received a single SC injection of 100 μg octreotide, 100 μg SOM230, or 250 μg SOM230 in a randomized, double-blinded, crossover fashion with a minimum six day washout between treatments [83]. A comparable suppression of GH levels by octreotide 100 μg and SOM230 250 μg was observed in eight patients (−65 ± 7 vs −72 ± 7%, respectively). Three patients were octreotide resistant, achieving only a 17% reduction in GH; whereas, 250 μg SOM230 was significantly more efficacious with a mean decrease of 70% in GH (p < 0.01) [83]. One patient showed better GH-lowering effect with octreotide than with SOM230 [83]. Subsequent in vitro analysis of surgical specimens from responders vs. non-responders demonstrated that sst5 rather than sst2 was the predominant somatostatin receptor in tumors that were more sensitive to SOM230, whereas in the tumors with octreotide sensitivity, sst2 was the predominant tumor receptor. Octreotide has a 2.5-fold higher affinity for the sst2 receptor than SOM230, which explains why GH response was similar for the octreotide 100 μg and the SOM230 250 μg doses in sst2 predominant tumors [83]. The results of phase 2 studies are not yet available, but this preliminary study suggests that SOM230 may offer a therapeutic benefit in a select minority who would need to be identified via drug challenge or analysis of surgical specimens to define predominant sst subtypes.
A potentially more significant breakthrough in the understanding and approach to medical treatment of pituitary tumors is reflected in the development of chimeric compounds that are capable of activating both dopamine and somatostatin receptors. The recent discovery that cell membrane G-protein-coupled receptors are capable of dimerization has created a shift in thinking regarding the degree of molecular cross-talk that occurs between cell surface receptors [85–87]. The implication is that a chimeric molecule that retains structural components of both dopamine and somatostatin may be capable of simultaneous, dual receptor activation, potentially inducing a unique, synergistic cellular and therapeutic response [86, 87]. As such, the sst5 receptor exhibits heterodimerization with the D2 dopamine receptor (D2R), and the sst5/D2R heterodimer displays enhanced signaling and unique pharmacologic properties [87, 88]. Clinical studies are now under way with a couple of these compounds; these chimeric compounds have yet to demonstrate that they can outperform combined therapies of existing somatostatin analogues and dopamine agonists, which as noted above, have been shown to perform less well than monotherapies [80, 87]. Nonetheless, as we better understand the complex in vivo interactions of the cellular and orphan nuclear receptors that regulate cell maturation and programmed cell death, we come closer to the next paradigm shift in the management of acromegaly: a medical cure.
References
Alexander L, Appleton D, Hall R, Ross W, Wilkinson R. Epidemiology of acromegaly in the Newcastle region. Clin Endocrinol 1980;12(1):71–9.
Katznelson L. Drug Insight: primary medical therapy of acromegaly. Nat Clin Pract Endocrinol Metab 2006;2:109–17.
Colao A, Ferone D, Marzullo P, Lombardi G. Systemic Complications of Acromegaly: Epidemiology, Pathogenesis, and Management. Endocr Rev 2004;25(1):102–52.
Melmed S. Acromegaly. N Engl J Med 1990;322(14):941–1012.
Freda PU, Katznelson L, van der Lely AJ, Reyes CM, Zhao S, Rabinowitz D. Long-acting somatostatin analog therapy of acromegaly: a meta-analysis. J Clin Endocrinol Metab 2005;90(8):4465–73.
Sheaves R, Jenkins P, Blackburn P, Huneidi AH, Afshar F, Medbak S, et al. Outcome of transsphenoidal surgery for acromegaly using strict criteria for surgical cure. Clin Endocrinol 1996;45(4):407–13.
Dons RF, Rosselet P, Pastakia B, Doppman J, Gorden P. Arthropathy in acromegalic patients before and after treatment: a long-term follow-up study. Clin Endocrinol 1988;28(5):515–24.
Grunstein R, Ho K, Sullivan C. Sleep apnea in acromegaly. Ann Int Med 1991;115(7):527–32.
Hoffstein V, Chan CK, Slutsky AS. Sleep apnea and systemic hypertension: A causal association review. Am J Med 1991;91(2):190–6.
Palomäki H, Partinen M, Erkinjuntti T, Kaste M. Snoring, sleep apnea syndrome, and stroke. Neurology 1992;42 7 Suppl 6:75–81.
Melmed S, Ho K, Klibanski A, Reichlin S, Thorner M. Clinical review 75: Recent advances in pathogenesis, diagnosis, and management of acromegaly. J Clin Endocrinol Metab 1995;80(12):3395–402.
Soszynski P, Slowinska-Srzednicka J, Zgliczynski S. Plasma concentrations of atrial natriuretic hormone in acromegaly: relationship to hypertension. Acta Endocrinol (Copenh) 1991;125(3):268–72.
Van Loon GR. Abnormal plasma catecholamine responses in acromegalics. J Clin Endocrinol Metab 1979;48(5):784–9.
Feld S, Hirschberg R. Growth hormone, the insulin-like growth factor system, and the kidney. Endocr Rev 1996;17(5):423–80.
Moller J. Effects of growth hormone on fluid homeostasis. Clinical and experimental aspects. Growth Horm IGF Res 2003;13(2–3):55–74.
Bondanelli M, Ambrosio MR, Franceschetti P, Margutti A, Trasforini G, degli Uberti EC. Diurnal Rhythm of Plasma Catecholamines in Acromegaly. J Clin Endocrinol Metab 1999;84(7):2458–67.
Hansen TK, Moller J, Thomsen K, Frandsen E, Dall R, Jorgensen JO, et al. Effects of growth hormone on renal tubular handling of sodium in healthy humans. Am J Physiol Endocrinol Metab 2001;281(6):E1326–332.
Bondanelli M, Ambrosio MR, degli Uberti EC. Pathogenesis and prevalence of hypertension in acromegaly. Pituitary 2001;4(4):239–49.
Bates AS, Hoff WV, Jones JM, Clayton RN. An audit of outcome of treatment in acromegaly. QJM 1993;86(5):293–9.
Etxabe J, Gaztambide S, Latorre P, Vazquez JA. Acromegaly: an epidemiological study. 1993;16(3):181–7.
Swearingen B, Barker FG II, Katznelson L, Biller BMK, Grinspoon S, Klibanski A, et al. Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegaly. J Clin Endocrinol Metab 1998;83(10):3419–26.
Orme SM, McNally RJQ, Cartwright RA, Belchetz PE. Mortality and cancer incidence in acromegaly: a retrospective cohort study. J Clin Endocrinol Metab 1998;83(8):2730–4.
Melmed S. Clinical perspective: acromegaly and cancer: not a problem. J Clin Endocrinol Metab 2001;86(7):2929–34.
Colao A, Marzullo P, Di Somma C, Lombardi G. Growth hormone and the heart. Clin Endocrinol 2001;54(2):137–54.
Lopez-Velasco R, Escobar-Morreale HF, Vega B, Villa E, Sancho JM, Moya-Mur JL, et al. Cardiac involvement in acromegaly: specific myocardiopathy or consequence of systemic hypertension. J Clin Endocrinol Metab 1997;82(4):1047–53.
Abosch A, Tyrrell JB, Lamborn KR, Hannegan LT, Applebury CB, Wilson CB. Transsphenoidal microsurgery for growth hormone-secreting pituitary adenomas: initial outcome and long-term results. J Clin Endocrinol Metab 1998;83(10):3411–8.
Bates A. Does treatment of acromegaly affect life expectancy. Metabolism 1995;44(11):1–5.
Holdaway IM, Rajasoorya RC, Gamble GD. Factors influencing mortality in acromegaly. J Clin Endocrinol Metab 2004;89(2):667–74.
Barker FG II, Klibanski A, Swearingen B. Transsphenoidal surgery for pituitary tumors in the United States, 1996–2000: mortality, morbidity, and the effects of hospital and surgeon volume. J Clin Endocrinol Metab 2003;88(10):4709–19.
Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical ‘cure’. Eur J Endocrinol 2005;152(3):379–87.
Kreutzer J, Vance ML, Lopes MBS, Laws ER Jr. Surgical management of GH-Secreting pituitary adenomas: an outcome study using modern remission criteria. J Clin Endocrinol Metab 2001;86(9):4072–7.
Clayton. How many surgeons to operate on acromegalic patients. Clin Endocrinol 1999;50(5):557–9.
Fahlbusch R, Honegger J, Buchfelder M. Surgical management of acromegaly. Endocrinol Metab Clin North Am 1992;21(3):669–92.
Barkan AL, Halasz I, Dornfeld KJ, Jaffe CA, Friberg RD, Chandler WF, et al. Pituitary irradiation is ineffective in normalizing plasma insulin-like growth factor i in patients with acromegaly. J Clin Endocrinol Metab 1997;82(10):3187–91.
Biermasz NR, van Dulken H, Roelfsema F. Long-term follow-up results of postoperative radiotherapy in 36 patients with acromegaly. J Clin Endocrinol Metab 2000;85(7):2476–82.
Powell JS, Wardlaw SL, Post KD, Freda PU. Outcome of radiotherapy for acromegaly using normalization of insulin-like growth factor I to define cure. J Clin Endocrinol Metab 2000;85(5):2068–71.
Landolt A, Haller D, Lomax N, Scheib S, Schubiger O, Siegfried J, et al. Stereotactic radiosurgery for recurrent surgically treated acromegaly: comparison with fractionated radiotherapy. J Neurosurg 1998;88(6):1002–8.
Castinetti F, Taieb D, Kuhn JM, Chanson P, Tamura M, Jaquet P, et al. Outcome of gamma knife radiosurgery in 82 patients with acromegaly: correlation with initial hypersecretion. J Clin Endocrinol Metab 2005;90(8):4483–8.
Bevan JS. The antitumoral effects of somatostatin analog therapy in acromegaly. J Clin Endocrinol Metab 2005;90(3):1856–63.
Ferjoux G, Bousquet C, Cordelier P, Bali NL, et al. Signal transduction of somatostatin receptors negatively controlling cell proliferation. J Physiol Paris 2000;94:205–10.
Buscail L, Esteve J, Saint-Laurent N, Bertrand V, Reisine T, O’Carroll A, et al. Inhibition of cell proliferation by the somatostatin analogue RC-160 is mediated by somatostatin receptor subtypes SSTR2 and SSTR5 through different mechanisms. Proc Nat Acad Sci USA 1995;92(5):1580–4.
Cordelier P, Esteve JP, Bousquet C, Delesque N, O’Carroll AM, Schally AV, et al. Characterization of the antiproliferative signal mediated by the somatostatin receptor subtypeásst5. Proc Nat Acad Sci USA 1997;94(17):9343–8.
Lamberts SWJ, van der Lely A-J, de Herder WW, Hofland LJ. Octreotide. N Engl J Med 1996;334(4):246–54.
Koizumi M, Onda M, Tanaka N, Seya T, Yamada T, Takahashi Y. Antiangiogenic effect of octreotide inhibits the growth of human rectal neuroendocrine carcinoma. Digestion 2002;659(4):200–6.
Battershill P, Clissold S. Octreotide. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in conditions associated with excessive peptide secretion. Drugs 1989;38(5):658–702.
Lancranjan I, Bruns C, Grass P, Jaquet P, Jervell J, Kendall-Taylor P, et al. Sandostatin(R) LAR(R): A promising therapeutic tool in the management of acromegalic patients. Metabolism 1996;45 Supplement 1:67–71.
Cozzi R, Attanasio R, Lodrini S, Lasio G. Cabergoline addition to depot somatostatin analogues in resistant acromegalic patients: efficacy and lack of predictive value of prolactin status. Clin Endocrinol 2004;61(2):209–15.
Bruns C, Lewis I, Briner U, Meno-Tetang G, Weckbecker G. SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol 2002;146(5):707–16.
Anthony L. Long-acting formulations of somatostatin analogues. Ital J Gastroenterol Hepatol 1999;31 Suppl 2:S216–8.
Lightman S. Somatuline Autogel: an extended release lanreotide formulation. Hosp Med 2002;63(3):162–5.
Colao A, Ferone D, Marzullo P, Cappabianca P, Cirillo S, Boerlin V, et al. Long-term effects of depot long-acting somatostatin analog octreotide on hormone levels and tumor mass in acromegaly. J Clin Endocrinol Metab 2001;86(6):2779–86.
Newman CB, Melmed S, George A, Torigian D, Duhaney M, Snyder P, et al. Octreotide as Primary Therapy for Acromegaly. J Clin Endocrinol Metab 1998;83(9):3034–40.
Amato G, Mazziotti G, Rotondi M, Iorio S, Doga M, Sorvillo F, et al. Long-term effects of lanreotide SR and octreotide LARR on tumour shrinkage and GH hypersecretion in patients with previously untreated acromegaly. Clin Endocrinol 2002;56(1):65–71.
Lucas T, Astorga R, Catala M. Preoperative lanreotide treatment for GH-secreting pituitary adenomas: effect on tumour volume and predictive factors of significant tumour shrinkage. Clin Endocrinol 2003;58(4):471–81.
Gutt B, Bidlingmaier M, Kretschmar K, Dieterle C, Steffin B, Schopohl J. Four-year follow-up of acromegalic patients treated with the new long-acting formulation of Lanreotide (Lanreotide Autogel). Exper Clin Endocrinol Diabetes 2005;3:139–44.
Caron P, Beckers A, Cullen DR, Goth MI, Gutt B, Laurberg P, et al. Efficacy of the new long-acting formulation of lanreotide (Lanreotide Autogel) in the management of acromegaly. J Clin Endocrinol Metab 2002;87:99–104.
van Thiel SW, Romijn JA, Biermasz NR, Ballieux BE, Frolich M, Smit JW, et al. Octreotide long-acting repeatable and lanreotide Autogel are equally effective in controlling growth hormone secretion in acromegalic patients. Eur J Endocrinol 2004;150(4):489–95.
Alexopoulou O, Abrams P, Verhelst J, Poppe K, Velkeniers B, Abs R, et al. Efficacy and tolerability of lanreotide Autogel therapy in acromegalic patients previously treated with octreotide LAR. Eur J Endocrinol 2004;151(3):317–24.
Astruc B, Marbach P, Bouterfa H, Denot C, Safari M, Vitaliti A, et al. Long-acting octreotide and prolonged-release lanreotide formulations have different pharmacokinetic profiles. J Clin Pharmacol 2005;45(7):836–44.
Melmed S. Acromegaly. N Engl J Med 2006;355(24):2558–73.
Verhelst JA, Pedroncelli AM, Abs R, Montini M, Vandeweghe MV, Albani G, et al. Slow-release lanreotide in the treatment of acromegaly: a study in 66 patients. Eur J Endocrinol 2000;143(5):577–84.
Livadas S, Hadjidakis J, Argyropoulou M, Stamatelatou M, Kelekis D, Raptis S. Disappearance of a growth hormone secreting macroadenoma during long-term somatostatin analogue administration and recurrence following somatostatin withdrawal. Hormones (Athens) 2006 2007;5(1):57–63.
Kovacs K, Horvath E. Pathology of pituitary tumors. Endocrinol Metab Clin North Am 1987;16(3):529–1.
Colao A, Ferone D, Cappabianca P, del Basso De Caro ML, Marzullo P, Monticelli A, et al. Effect of octreotide pretreatment on surgical outcome in acromegaly. J Clin Endocrinol Metab 1997;82(10):3308–14.
Abs R, Verhelst J, Maiter D, Van Acker K, Nobels F, Coolens JL, et al. Cabergoline in the treatment of acromegaly: A study in 64 patients. J Clin Endocrinol Metab 1998;83(2):374–8.
Chiodini PG, Cozzi R, Dallabonzana D, Oppizzi G, Verde G, Petroncini M, et al. Medical treatment of acromegaly with SMS 201–995, a somatostatin analog: a comparison with bromocriptine. J Clin Endocrinol Metab 1987;64(3):447–53.
Plockinger O, Quabbe H. Evaluation of a repeatable depot-bromocriptine preparation(Parlodel LAR) for the treatment of acromegaly. J Endocrinol Invest 1991;14(11):943–8.
Liuzzi A, Chiodini PG, Botalla L, Cremascoli G, Muller EE, Silvestrini F. Decreased plasma growth hormone (GH) levels in acromegalics following CB 154(2-Br-alpha ergocryptine) administration. J Clin Endocrinol Metab 1974;38(5):910–2.
Thorner M, Chait A, Aitken M, Benker G, Bloom SR. Bromocriptine treatment of acromegaly. Br Med J 1975;1:299–303.
Sachdev Y, Gopal K, Garg VK. Bromocriptine therapy in acromegaly. A long-term review of 35 cases. Postgrad Med J 1981;57(666):210–6.
Jaffe C, Barkan A. Treatment of acromegaly with dopamine agonists. Endocrinol Metab Clin North Am 1992;21(3):713–35.
Lindholm J, Riishede J, Vestergaard S, Hummer L, Faber O, Hagen C. No effect of bromocriptine in acromegaly: a controlled trial. N Engl J Med 1981;304(24):1450–4.
Nortier J, Croughs R, Thijssen J, Schwarz F. Plasma growth hormone suppressive effect of bromocriptine in acromegaly. Evaluation by plasma GH day profiles and plasma GH concentrations during oral glucose tolerance tests. Clin Endocrinol 1984;20 May (5):565–71.
Quabbe H. Treatment results in 235 patients with acromegaly. A report of the acromegaly study group. Acta Endocrinol (Copenh). 1981;240 Suppl:66.
Yamamoto M, Uesugi T, Nakayama T. Dopamine agonists and cardiac valvulopathy in Parkinson disease: A case-control study. Neurology 2006;67(7):1225–9.
Chen WY, Wight DC, Wagner TE, Kopchick JJ. Expression of a mutated bovine growth hormone gene suppresses growth of transgenic mice. Proc Natl Acad Sci USA 1990;87(13):5061–5.
Fuh G, Cunningham BC, Fukunaga R, Nagata S, Goeddel DV, Wells JA. Rational design of potent antagonists to the human growth hormone receptor. Science 1992;256(5064):1677–80.
Trainer PJ, Drake WM, Katznelson L, Freda PU, Herman-Bonert V, van der Lely AJ, et al. Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant. N Engl J Med 2000;342(16):1171–7.
van der Lely AJ, Hutson RK, Trainer PJ, Besser GM, Barkan AL, Katznelson L, et al. Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet 2001;358(9295):1754–9.
Feenstra J, de Herder WW, ten Have SMTH, van den Beld AW, Feelders RA, Janssen JAMJ, et al. Combined therapy with somatostatin analogues and weekly pegvisomant in active acromegaly. The Lancet 2005;365(9471):1644–6.
Schreiber I, Buchfelder M, Droste M, Forssmann K, Mann K, Saller B, et al. Treatment of acromegaly with the GH receptor antagonist pegvisomant in clinical practice: Safety and efficacy evaluation from the German Pegvisomant Observational Study. Eur J Endocrinol 2007;156(1):75–82.
Neggers SJCM, van Aken MO, Janssen JAMJ, Feelders RA, de Herder WW, van der Lely A-J. Long-term efficacy and safety of combined treatment of somatostatin analogs and pegvisomant in acromegaly. J Clin Endocrinol Metab 2007 (in press).
van der Hoek J, de Herder WW, Feelders RA, van der Lely A-J, Uitterlinden P, Boerlin V, et al. A single-dose comparison of the acute effects between the new somatostatin analog SOM230 and octreotide in acromegalic patients. J Clin Endocrinol Metab 2004;89(2):638–45.
Surya S, Barkan A. GH Receptor antagonist: mechanism of action and clinical utility. Rev Endoc Metab Disord 2005;6(1):5–13.
Bai M. Dimerization of G-protein-coupled receptors: roles in signal transduction. Cell Signal 2004;16(2):175–86.
Bulenger S, Marullo S, Bouvier M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol Sci 2005;26(3):131–7.
Ferone D, Saveanu A, Culler MD, Arvigo M, Rebora A, Gatto F, et al. Novel chimeric somatostatin analogs: facts and perspectives. Eur J Endocrinol 2007;156 suppl_1:S23–8.
Rocheville M, Lange DC, Kumar U, Patel SC, Patel RC, Patel YC. Receptors for dopamine and somatostatin: formation of Hetero-Oligomers with enhanced functional activity. Science 2000;288(5463):154–7.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bush, Z.M., Vance, M.L. Management of acromegaly: Is there a role for primary medical therapy?. Rev Endocr Metab Disord 9, 83–94 (2008). https://doi.org/10.1007/s11154-007-9061-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-007-9061-1