
Abstract
Acromegaly is a rare disease (updated estimated prevalence: 80–130 cases per million inhabitants) that is due to excessive production and exposure to growth hormone (GH), generally driven by a pituitary GH-secreting adenoma. Acromegaly is characterized by slowly progressive somatic disfigurement (mainly involving the face and extremities) and multiorgan/system impairment. The rheumatologic, cardiovascular, respiratory, metabolic, and tumoral consequences are prevalent and generally determine the prognosis. The diagnosis is confirmed by elevated and nonsuppressible serum GH concentrations and by increased levels of age- and sex-adjusted circulating insulin-like growth factor-I (IGF-I). Treatment aims at reducing clinical manifestations, normalizing GH and IGF-I levels and correcting (or preventing) pituitary tumor compression of surrounding tissues and preventing the incidence/worsening of disease-related comorbidities. Transsphenoidal resection of the pituitary adenoma is the treatment of choice. If surgery fails to correct GH/IGF-I hypersecretion, medical treatment using three drug classes (somatostatin receptor ligands, dopamine agonists, or the GH-receptor antagonist pegvisomant), alone or in combination, is successful in most cases. Radiotherapy is currently considered a third-line treatment. This multistep therapeutic strategy currently allows clinical/hormonal control in most patients. The effectiveness of the current approach is probably the reason why the life expectancy of patients with acromegaly currently seems to approach that of the general population. Despite better disease control, persistent sequelae may nonetheless persist and consistently impair the quality of life of patients affected by acromegaly.
The chapter has been endorsed by Prof. Vera Popovic, , University of Belgrade, Belgrade, Serbia
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
5.1 Introduction
Acromegaly is a typical rare multisystemic disease associated with progressive enlargement of some parts of the body, featuring somatic disfigurement mainly involving the face and extremities. The archetypal body changes and gigantism are renowned features (Figs. 5.1 and 5.2). Apart from body changes and disproportion, a number of multiorgan comorbidities are frequently associated with acromegaly. Most clinical repercussions originate from growth hormone (GH) and insulin-like growth factor-1 (IGF-I)-dependent organ overgrowth. In most cases, GH excess is due to a pituitary adenoma. Acromegaly itself and the related comorbidities might lead to premature death if not adequately treated.

Schematic representation of signs and symptoms of acromegaly. Adapted from [1], with permission

Disease evolution in a woman with acromegaly. Series of photographs taken over time showing the progressive changes in facial appearance. It is possible to presume that the first signs appeared between 1988 and 1990, 22 years before the diagnosis. In 1990, the patient had to enlarge her ring she bought 2 years before because it was too narrow (adapted from [2], with permission)
5.2 Epidemiology
The prevalence of acromegaly has recently been estimated to be approximately 28 to 137 cases per million inhabitants [3, 4], challenging the historical figure of 40 to 70 cases per million [5]. A prevalence of approximately 1000 per million inhabitants was found in a German study based on screening with IGF-I measurement in the general population [6]. Raappana estimated the annual incidence of acromegaly at 3.4 cases per million in Finland [7]. A recent survey analyzing all the studies providing epidemiological data refined the incidence to a range between 0.2 and 1.1 cases/100,000 [8].
Age at diagnosis typically falls within the fourth decade of life, following a quasi-Gaussian curve [9, 10]. However, although more rarely, acromegaly might be found in children and in the elderly.
The sex ratio has been found to be more or less constant across studies. A female to male ratio of 1.26 has been calculated by analyzing data concerning more than 16,000 patients across national registries [11]. Age at diagnosis is typically earlier in males than in females, and a clearly distinguishable sex-related dimorphic Gaussian curve is observed [9, 12, 13].
Owing to the insidious clinical onset and slow progression, acromegaly is often diagnosed late. Older series, in the 1980s, suggested a mean diagnostic latency of 3–10 years after onset, at an average age of approximately 40 years [8, 14,15,16,17].
Studies focusing on disease latency seem to show that diagnostic delay appears to be more or less constant throughout decades, without any improvement over time with an earlier diagnosis [9, 18].
5.3 Pathogenesis
In more than 95% of cases, acromegaly is secondary to GH hypersecretion by a benign monoclonal pituitary adenoma that develops from somatotroph cells [19,20,21,22]. Pituitary somatotroph adenomas are mostly isolated (sporadic). Rarely, they may develop in the frame of a genetic predisposing disease (Fig. 5.3a).

Prevalence of somatotroph tumor forms and associated clinical genetic diseases. Panel a: Schematic representing the relative prevalence of sporadic pure or mixed somatotroph adenomas and associated mosaic/germline genetic diseases. Panel b: Characteristic clinical features of a patient with McCune Albright syndrome. Please note the typical café-au lait skin spots and X-rays and MRI hallmarks illustrating fibrous bone dysplasia. Panel c: Characteristic clinical features in a patient with acromegaly with multiple endocrine neoplasia type-1. Please note the pituitary tumor on a MRI T1W coronal post-gadolinium view, a pancreatic tumor revealed by echo-endoscopy, and parathyroid hyperplasia/adenoma found on 99mTc -123I subtraction parathyroid scintigraphy. Panel d: Characteristics of a patient with Carney complex, from [23], with permission. Panel e: Growth curve of a patient affected by X-LAG acro-gigantism
5.3.1 Somatotroph Pituitary Adenoma
Pure somatotroph pituitary adenomas (60%) are constituted by eosinophilic cells containing either densely or sparsely granulated (secretory granules) cell elements after immunolabeling [19]. In some adenomas, immunostaining reveals colocalization of free alpha-subunits [24]. Silent somatotroph adenomas do not determine clinical acromegaly. Diagnosis is almost exclusively made by tumor immunostaining. Nonetheless, some patients bearing these tumors may have supranormal circulating GH levels without overt clinical signs [25].
Most human somatotroph adenomas seem to be associated with the clonal expansion [26] of cells carrying a specific somatic mutation. However, as in other types of pituitary adenoma, it has proven difficult to isolate a single causative factor explaining pituitary tumorigenesis [27,28,29]. Mutations in stimulating G-protein [30, 295] have been identified in 35–55% of somatotroph adenomas, according to some series [31,32,32]. gsp mutations are able to inhibit GTPase activity and to lead to constitutive adenyl-cyclase activation [33]. Cell cycle disruption also seems to play an important role, as demonstrated in MEN1 or in patients with CDKN1B mutations (coding the cyclin-dependent kinase inhibitor p27KIP1, a key regulator of the cell cycle) [29]. A number of other genes have been implicated in somatotroph tumorigenesis [29]. The function of the disrupted proteins deriving from mutated genes spans from oncogene/tumor suppression to cyclin/cell proliferation inhibition. The general belief is that a preexisting mutation within somatotrophs should be a predisposing factor for further cell proliferation and GH secretion. Premature senescence likely explains the persistence of a benign phenotype and the rarity of progression to carcinoma. Epigenetic mechanisms may also contribute to cell proliferation by silencing genes such as CDKN2A, encoding p16, a cell proliferation inhibitor. The sequence of events leading to somatotroph cell clonal expansion seems to be multifactorial [34, 35].
Cytogenetic studies show that somatotroph pituitary adenomas display substantial intertumor and intratumor DNA copy-number heterogeneity. Intriguingly, somatic GNAS-mutated adenomas have low copy number variations, whereas a higher heterogeneity is observed in GNAS-intact tumors [36].
5.3.2 Mixed Somatotroph Adenoma
The most frequent mixed adenomas coexpress GH and prolactin (PRL), accounting for 25% of cases. Histopathologists often distinguish between true mixed adenomas, containing either somatotroph or lactotroph cell types, and lactosomatotroph stem cells consisting of more mature monomorphic cells coexpressing GH and PRL [19]. Mixed GH- and TSH-secreting adenomas are rarer and are associated with acromegaly associated with hyperthyroidism by inappropriate secretion of thyroid-stimulating hormone [37, 38]. ACTH cosecretion in somatotroph adenomas is extremely rare.
5.3.3 Genetic Syndromes Associated with Acromegaly
Various genetic syndromes and diseases include acromegaly in a wider spectrum of clinical features. Despite representing a rare cause, the burden of related morbidities requires not neglecting them (Fig. 5.3a–e). This paragraph summarizes the main features of these diseases along with some peculiarities in acromegaly manifestation. In general terms, a germinal genetic disorder should be suspected in patients with early-onset acromegaly bearing other organ involvement. These diseases have also been reviewed in recent articles and book chapters [20, 40,41,42,42].
McCune-Albright syndrome (MAS) is a rare genetic disease associated with multiple fibrous bone dysplastic lesions, precocious puberty, “café-au-lait” spots, and multiorgan and soft-tissue tumors (Fig. 5.3b). Pathological features are related to postzygotic somatic mutations leading to constitutive activation of the Gs protein alpha subunit [44,45,45]. It is of note that this gene is also responsible for most sporadic pure GH adenomas, underlying the importance of stimulating G-protein in the somatotroph environment. In the case of diffuse germinal mutations (MAS), acromegaly is found in approximately 20% of patients [43, 44, 46]. A peculiarity of this form is a relative resistance to somatostatin analogs (approximately 30% of responders). The therapeutic approach is also markedly influenced by the coexistence of skull base dysplasia, making any neurosurgical approach challenging [45].
Acromegaly can also be associated with hyperparathyroidism, neuroendocrine tumors (e.g., gastrinoma, insulinoma, or a nonfunctional pancreatic tumor), adrenal and other endocrine or nonendocrine tumors in the frame of multiple endocrine neoplasia (MEN) type 1, which is related to menin (MEN1) germline mutations (Fig. 5.3c) [47, 48]. Pituitary adenomas are not enriched in the somatotroph lineage in MEN1 patients. Few cases of GHRH-secreting neuroendocrine tumors have been reported in patients with MEN1 (see the “Extrapituitary acromegaly” paragraph) [49, 50].
Mutations in the CDKN1B gene are responsible for a rarer and newer MEN syndrome, multiple endocrine neoplasia type 4 (MEN4, initially known as MEN-X), which combines hyperparathyroidism, pituitary adenomas (including acromegaly), and other endocrine or nonendocrine tumors [51, 52]. Twenty cases have been published to date. Hyperparathyroidism seems to be the most prevalent disease. Nonetheless, acromegaly by a somatotroph adenoma is found in up to 20% of patients harboring a deleterious CDKN1B mutation. Furthermore, contrary to MEN1, pituitary adenomas in MEN4 seem to be more enriched in somatotroph adenomas.
When acromegaly is associated with bilateral pigmented micronodular adrenal hyperplasia (causing ACTH-independent hypercortisolism) or with typical cutaneous pigmentations or cardiac myxomas, the patient should be screened for the Carney complex (Fig. 5.3D). This genetic disease is related to a germline mutation of the regulatory subunit of protein kinase A (PRKAR1A), whose signaling cascade is also located downstream of the stimulating G-protein [53, 54]. A simplified schematic of the cyclic AMP-dependent signaling pathway with relevant targets for somatotroph tumorigenesis is provided in Fig. 5.4.

The cyclic AMP-dependent signaling pathway in pituitary somatotroph cells as a model to understand different disease forms. GHRH induces a conformational change in the class II G protein-coupled receptor GHRHR. The Gs-α subunit exchanges GDP for GTP, which activates adenylyl cyclase (AC), converting ATP to cAMP. Elevated cAMP levels activate protein kinase A (PKA). PKA consists of a tetramer of two homo- or heterodimer regulatory subunits (R) and two catalytic subunits (C) responsible for the phosphorylation of several enzymes and transcription factors downstream [e.g., cAMP-response element-binding protein (CREB)]. MCAS McCune-Albright syndrome, LOH loss of heterozygosity, MEN1 multiple endocrine neoplasia type-1, α, β, γ Gs-protein subunits, AC adenyl cyclase, cAMP cyclic AMP, CREB cyclic AMP response element-binding protein, a transcription factor, PKA protein kinase A, R the regulatory subunits of protein kinase A, C the catalytic subunit of protein kinase A
Acromegaly is also one of the features described in familial isolated pituitary adenoma, partly related to AIP germline mutations (aryl hydrocarbon receptor interacting protein) [56,57,57]. These mutations can also, albeit rarely, be found in some apparently sporadic cases of acromegaly, particularly in young patients [59,60,61,62,62].
GPR101 was the latest gene to be discovered in association with a very early-onset form of X-linked gigantism or X-LAG syndrome [63]. Affected patients develop large or giant adenomas at a very young age. The pathophysiology is related to Xq26.3 microduplication involving the orphan G-protein-coupled receptor GPR101. However, germline GPR101 mutations are very rare in patients with sporadic pituitary adenomas, particularly in patients with gigantism or acromegaly [64]. Little is known about the pathophysiology and sequence of events leading to somatotroph tumorigenesis (Fig. 5.3E).
5.3.4 GH-Secreting Carcinomas
Somatotroph carcinomas are exceptional (fewer than 20 published cases). The presence of distant metastases is required to support the diagnosis of malignancy [39].
5.3.5 Extrapituitary Acromegaly
Extrapituitary acromegaly refers to growth hormone releasing hormone (GHRH) or GH hypersecretion other than from a pituitary adenoma [39].
GHRH hypersecretion could originate either from hypothalamic tumors, such as gangliocytomas, hamartomas, choristomas, and gliomas, or from the periphery. More often, GHRH hypersecretion comes from ectopic sources. The GHRH peptide was indeed originally identified and cloned from large pancreatic tumors [65]. GHRH is expressed by several tissues. However, very large amounts are needed to induce pituitary somatotroph hyperplasia and clinical acromegaly. GHRH hypersecretion may derive from pancreatic cell tumors, small-cell lung cancers, bronchial and other-site carcinoids, adrenal adenomas, and pheochromocytomas. The delivery of high GHRH concentrations stimulates normal pituitary somatotrophs to become hyperplastic and to hypersecrete GH to produce acromegaly [50, 66, 67]. The diagnosis is established by measuring plasma GHRH and by identifying the source (a GHRH-staining neuroendocrine tumor) [68]. The prognosis largely depends on the characteristics of the underlying tumor [50].
GH can also be directly secreted by an ectopic somatotroph adenoma (located near the sella turcica, for example, in the sphenoidal sinus, petrous temporal bone, nasopharyngeal cavity) or, in exceptional cases, by a peripheral tumor (pancreatic islet tumor or lymphoma) [69, 70].
5.4 Clinical Presentation
Acromegaly is generally suspected based on clinical signs and symptoms, which are important to recognize (Fig. 5.1) [2, 16, 17, 72,73,74,74].
5.4.1 The Dysmorphic Syndrome
In typical forms, patients present broadened extremities (hands and feet), widened, thickened and stubby fingers, and soft tissue thickening. When specifically asked, affected patients describe enlarged rings over the last years or the need to change shoe size. The facial aspect is somehow characteristic and includes a widened and thickened nose, prominent cheekbones, bulged forehead, thick lips, and marked facial lines (Figs. 5.1 and 5.2). The forehead and the overlying skin are thickened, sometimes leading to frontal bossing. The lower face is also affected, with several degrees of prognathism, maxillary widening, teeth separation, and, more rarely, jaw occlusion impairment. A useful step is to analyze comparatively ancient photographs. This could show a slow, insidious demarcation of the acrofacial syndrome spreading over several years (Fig. 5.2). Because of this slow progression, relatives and physicians may be unaware, and acromegaly may be diagnosed very late. Typically, after variable latency, the diagnosis is raised by a physician who has not seen the patient before [16, 17, 75]. A recent multicenter survey investigating medical practices of more than 3000 patients with acromegaly across several European countries reported that most diagnoses were reportedly made by an endocrinologist (45%), followed by general practitioner (17.5%), internist (13.2%), orthopedist (3.6%), neurologist (3.3%), ophthalmologist (2.3%), and the patient him or herself or one of their relatives (2.3%) [10]. It is therefore not uncommon that a patient could make the diagnosis him or herself by searching the Internet. In most cases, diagnosis is made owing to changes in the face or extremities; in some other cases, acromegaly is diagnosed not for its signs and symptoms but during the biochemical exploration of a pituitary adenoma.
5.4.2 Symptoms
Acromegaly can cause a broad variety of symptoms [16].
5.4.2.1 Skin Changes
Nearly 70% of patients have sweaty and oily skin. Skin thickening is due to glycosaminoglycan deposition and to increased collagen production by connective tissue. These changes may lead to hyperhidrosis and malodorous sweating. Facial wrinkles, nasolabial folds, and heel pads are increased in thickness, and body hair may become coarsened. Skin tags are frequent and may be a marker of colonic polyps. Raynaud’s disease is present in one-third of cases. In some cases, patients describe night-time malodorous sweating.
5.4.2.2 Bone Changes
In response to both GH and IGF-I, new periosteal bone formation leads to an increase in skeletal growth, especially at the level of the mandible (prognathism). Jaw thickening, tooth separation, frontal bossing, malocclusion, and nasal bone hypertrophy are the standard facial bony deformities in acromegaly.
Radiography shows a thickening of the cranial vault and protuberances, frontal internal hyperostosis, and condensation of the sella turcica walls with clinoid hypertrophy. Hypertrophy of the sinuses, especially the frontal sinuses, is also clearly visible. This, along with laryngeal hypertrophy, may explain why the voice tends to become deeper and acquires a sonorous resonance [76].
These changes are not only due to soft tissue hypertrophy and excessive growth of bone and cartilage but also to bone deformation. Indeed, radiographic findings are abnormal in half of these patients, showing distal tufting of the phalanges, widening of the base of the phalanges with osteophyte formation, enthesopathy (mineralization of ligamentous insertions), widening of the cortical bone diaphyses, and widening of joint spaces due to cartilage hypertrophy. Deformations can also affect the rest of the skeleton, and dorsal kyphosis with distortion of the rib cage may be observed in severe chronic forms, leading to the paradigmatic “punchinello” aspect, especially when GH hypersecretion begins prior to epiphysis closure.
Bony deformations also affect the spine, with upper dorsal kyphosis and compensatory lumbar hyperlordosis. Vertebral enlargement, widened intervertebral spaces, and osteophyte formation were also observed. The thorax is deformed by protuberance of the lower portion of the sternum and by elongation and divergence of the ribs due to overgrowth of the chondrocostal joints.
Imaging studies show diaphyseal cortical thickening of the long bones and widened joint spaces, sometimes with osteophytes.
Concerning mineral changes, bone remodeling is increased in acromegaly [77, 78]. Cortical bone thickens (as measured by the metacarpal index and histomorphometric parameters) and its porosity is diminished. Trabecular bone mass may be decreased, normal or increased. Measurement of spinal bone mass can give contradictory results, probably because acromegaly is often associated with other endocrine disorders interfering with bone mass. In general, bone mass is normal in the lumbar spine in patients with isolated acromegaly but could be decreased in patients having other related endocrinopathies impacting bone metabolism, such as hypogonadism or hyperparathyroidism. Despite similar bone mineral density values, a lower lumbar spine assessed by the trabecular bone score (TBS) technique was demonstrated in patients with acromegaly compared to controls, especially in hypogonadal patients and women [79]. In a study [80] independent of BMD, the prevalence of vertebral fractures was found to be higher in patients with acromegaly (57.5% vs 22.6%). Fractures were associated with higher serum IGF-I values, a longer duration of active disease, and a longer history of untreated hypogonadism. This higher prevalence of vertebral fractures persists despite biochemical control of acromegaly [81].
5.4.2.3 Rheumatologic Comorbidity
5.4.2.3.1 Peripheral Osteoarthritis
The topic has been reviewed in detail in [82, 83].
Peripheral joint symptoms are very frequent. Arthralgia and myalgia occur in 30–70% of patients. Among the sites, large joints such as the knees, shoulders, hands, wrists, and hips seem to be more affected. Acromegalic osteoarthritis develops within an average of 10 years after diagnosis.
Osteoarthritis develops in two stages. In the initial stage, the growth of joint cartilage and periarticular ligaments is stimulated, leading to enlargement and congestion of the interarticular space that limits mobility and induces joint pain; in the second stage, more degenerative changes of the joint geometry are observed: intra-articular microtrauma and exuberant repair reactions induce scars and subchondral resorption and osteophytotic development, leading to progressive joint deterioration. The pain is thus mainly mechanical, degenerative, and noninflammatory and often persists after treatment of acromegaly. However, rarely, some patients may present symptoms and signs of pseudoinflammatory osteoarthritis, which are dramatically relieved by the treatment of acromegaly. Joint mobility (especially of the shoulders) can be limited in the later stages of the disease. Joint effusion is rare, and synovial aspiration shows a generally degenerative picture with no evidence of inflammation; it may also reveal calcium microcrystals (associated chrondrocalcinosis).
Physical examination of the joints often provides little information. The abnormalities are generally minor despite subjective functional discomfort. The shoulders and hips may show a loss of mobility and function. In contrast, some patients have joint hyperlaxity. There was no correlation between the presence or severity of osteoarthropathy and the age of onset of acromegaly or the mean GH or IGF-I concentration at baseline or during follow-up. Osteoarthritis appears to be more frequent after 45 years of age.
Radiological studies show a widening of the joint spaces, reflecting hypertrophy of the hyaline cartilage, as well as the presence of osteophytes, bone proliferation at the attachment sites of tendons and ligaments, periarticular calcium deposition, and exostosis of the bone. The joint space subsequently diminishes due to destructive arthritis. Sonography shows a thickening of the cartilage in the shoulder, wrist, and knee joints, which is improved with treatment for acromegaly.
Osteoarthritis inexorably progresses in advanced stages of the disease. It is not influenced by successful treatment of acromegaly, with the exception of diffuse articular symptoms and some sites of pain [84]. Acromegalic osteoarthritis considerably impairs patients’ quality of life [86,87,87].
5.4.2.3.2 Spinal Involvement
The estimated prevalence of spinal involvement is approximately 40–50% [88]. Backache is more frequent at the level of the lumbar spine than the cervical or dorsal segments. The pain is mainly mechanical in nature, but inflammatory features can occur in later stages (16%). Spinal involvement may be accompanied by nerve compression. Occasionally, bilateral intermittent claudication reveals lumbar spinal stenosis. Pain may also be related to an increased prevalence of vertebral fractures despite normal BMD [80, 89].
Radiological examination shows typical features, including ossification of the anterior and lateral surfaces of the vertebral bodies, contributing to enlargement of their anteroposterior diameter, as well as a biconcave vertebral aspect and scalloping of the vertebral bodies (exaggerated concavity of the posterior vertebral wall). The mechanism of these morphometric changes is poorly understood but may involve hypertrophy of the intraspinal soft tissues (ligamentous hypertrophy, epidural lipomatosis) or bone. In more severe cases, ossification of the anterior surface of the vertebral bodies can bridge the disk space and give an aspect of diffuse idiopathic skeletal hyperostosis. An increased number of vertebral fractures with wedge deformity and thoracic kyphosis is also more prevalent in patients with acromegaly than in the general population [90].
5.4.2.4 Neuropathies
Symptomatic carpal tunnel syndrome is frequent. Nerve conduction studies have shown that the vast majority of patients with acromegaly have subclinical abnormalities of nerve conduction. Magnetic resonance imaging (MRI) shows a higher amplitude and intensity of the median nerve signal in patients with symptomatic carpal tunnel syndrome compared to asymptomatic patients [91]. The mechanism appears to involve median nerve edema more than extrinsic compression due to an excess of connective tissue, bone or synovial hypertrophy or an increase in extracellular fluid within the carpal tunnel itself with Schwann cell demyelination. Nerve edema, which can also easily be evaluated with ultrasonography [92], improves when GH and IGF-I levels fall, suggesting that hormonal control is a key prerequisite for improving these patients’ neurological status. Sometimes, however, carpal tunnel syndrome may persist.
Ulnar nerve neuropathy at the cubital tunnel is also frequent in patients with acromegaly [93] and improves with treatment of acromegaly.
Apart from mechanical/compressive effects on nerves, autonomic nervous system dysfunction is present in patients with acromegaly, as shown by assessing the heart rate variability indices (mean sinus heart rate, RR intervals) and reverses after effective treatment [94].
5.4.2.5 Psychologic Consequences
Self-esteem may diminish along with progressive facial and bodily disfigurement. Patients with acromegaly further exhibit impairment in body image distortion, disruption in interpersonal relations, and social withdrawal anxiety [95].
Patients reported more negative illness perceptions than patients with acute illness but more positive illness perceptions than patients with other chronic diseases [96].
Nonetheless, direct unstructured interviews reveal an association between the diagnostic delay and the doctor–patient encounter and the experience of this disease, which is often described as catastrophic, both before and after the diagnosis [97].
It is unclear whether reported depression, mood swings, and apathy result solely from these physical changes or whether they are intrinsic high GH exposure central effects.
Acromegaly carries a significant lifelong burden for the affected patient. When evaluating health-related quality of life by means of dedicated questionnaires (such as the ACROQoL), it is clear that values barely normalize despite disease remission or cure [98]. All the biological, environmental, and biopsychosocial aspects of this burden have been extensively covered in a recent review by Biermasz [99].
5.4.2.6 Cardiovascular Manifestations
5.4.2.6.1 Arterial Hypertension
Hypertension occurs in 20–50% of patients. Its prevalence increases with time after the onset of acromegaly, as well as with GH level and age. Several concomitant factors are likely to play a role in the pathogenesis of hypertension in acromegaly. The chronic expansion of extracellular fluid volume (hypervolemia) leads to fluid retention, with plasma volume being 10–40% above normal. At the kidney level, increased renal sodium reabsorption at the distal tubule level is generally observed [100]. Body fluid expansion is related to enhanced epithelial sodium channel (ENaC) activity [1, 101, 102]. Hypertension can also result from endothelial dysfunction [103]. Neither renin-angiotensin aldosterone nor the sympathetic system appears to be involved in the pathogenesis of hypertension in this setting. Other contributors to the onset and maintenance of hypertension in acromegaly are also the increase in peripheral vascular resistance, insulin resistance and diabetes, and the development of obstructive sleep apnea [105,106,106].
5.4.2.6.2 Cardiomyopathy
Cardiac disorders are a consistent feature. Many lines of evidence, especially from experimental studies, point to the existence of specific cardiac disorders in acromegaly that are completely independent of coronary involvement (currently found in only a minority of patients) or valve disorders, diabetes, or hypertension [107, 108].
The first step of acromegaly-related cardiomyopathy mainly consists of myocardial hypertrophy of the interventricular septum and left ventricular posterior wall. This condition is initially asymptomatic, at least at rest. The assessment of initial cardiomyopathy is generally performed by means of cardiac ultrasound examination or magnetic resonance imaging (MRI).
Generally, left ventricle parameters are normal (concentric hypertrophy). Myocardial hypertrophy can occur in the absence of hypertension and even in young patients (<30 years), reflecting the impact of GH excess itself on the myocardium. Its prevalence is likely to be overestimated by echocardiography compared to MRI [109]. Hypertension further aggravates cardiac hypertrophy. Echocardiography and isotope studies show altered diastolic function (abnormal left and right ventricle filling) related to abnormal relaxation: parietal stiffness is, at least in part, probably linked to edematous infiltration of the ventricular wall [110] and perhaps also to a certain degree of fibrosis. Clinical symptoms such as dyspnea during exercise may be observed in patients who are asymptomatic at rest. Systolic function is normal if assessed by conventional methods. However, novel techniques such as two- or three-dimensional speckle-tracking echocardiography reveal, even at an earlier stage, an increased frequency of subclinical systolic impairment in active acromegaly [111, 112]. At later stages, hyperkinetic syndrome (increased cardiac index) is frequent.
Arrhythmias and/or conduction disorders may occur at any stage of acromegalic cardiomyopathy [113]. Their prevalence has long been underestimated in these patients. Ventricular premature complexes have been shown to frequently occur in patients with acromegaly. In one study, systematic 24 h Holter ECG recordings showed complex ventricular arrhythmias in 48% of patients compared to only 12% of controls [114]. Most of these arrhythmias are subclinical and persist despite successful treatment of acromegaly. Myocardial remodeling, hypertrophy, and fibrosis are all likely to play a role in their onset. However, recent studies did not confirm the high prevalence of dysrhythmias [115].
Congestive heart failure can occur if the cardiac disorders progress (if GH hypersecretion persists and, probably, if other risk factors such as diabetes, hypertension, and sleep apnea are also present). Functional signs first appear on effort before becoming permanent. At this stage, echocardiography shows variable degrees of cavity dilation. Fortunately, these severe forms are now far less frequent (prevalence 3%) [116].
A number of cardiovascular parameters improve during effective treatment of acromegaly, even if some changes appear to be irreversible in certain patients. In general, younger patients and patients with a relatively short history of acromegaly show better “recovery” (from diastolic disorders, myocardial hypertrophy, or systolic dysfunction) [71, 117]. In contrast, when dilated congestive heart failure occurs, cardiac function (especially systolic function) may show a short-term improvement [100], allowing some patients to survive or to avoid heart transplantation, but the longer-term prognosis is worse than that of patients with heart failure due to other causes (5-year mortality rate 37%) [116].
There is controversy surrounding the cardiovascular (ischemic) risk carried by patients with acromegaly [118]. An increased prevalence of hypertension, a history of diabetes mellitus and decreased levels of high-density lipoprotein, low-density lipoprotein, and total cholesterol were found in patients with acromegaly, leading to significantly higher Framingham risk scores than in controls [119]. Biomarkers of cardiovascular disease were also found to be altered in another study [120]. However, carotid atherosclerosis and carotid internal media thickening are not more extensive in patients with acromegaly than in nonacromegalic subjects [121, 122]. Importantly, no increase in the prevalence of coronary artery disease (assessed by different means, such as cardiovascular events, calcium scores, or myocardial scintigraphy) is found in patients with newly diagnosed acromegaly compared to the general population [124,125,126,127,127]. The reason for this apparent discordance between observed and expected coronary events is currently unclear. It has been suggested that the known atherogenic effects of hypertension, insulin resistance, and diabetes induced by GH excess are counterbalanced by some other cardioprotective factors, such as decreased endothelial and systemic inflammation [129,130,130].
5.4.2.6.3 Cardiac Valve Disease
Cardiac valve disorders are highly prevalent in patients with acromegaly and can, along with other cardiac abnormalities, also contribute to the onset or aggravation of heart disease in patients with acromegaly [131]. The risk of valve disease increases with time since onset [132]. Acromegaly-related cardiac valve abnormalities, which may be related to fibrotic changes, seem to persist after effective treatment of acromegaly [133]. Furthermore, no cabergoline-induced cardiac valve remodeling was observed.
5.4.2.7 Metabolic Complications
Physiologically, GH increases blood glucose levels, exerts a lipolytic effect, and promotes triglyceride hydrolysis into free fatty acids and glycerol.
GH excess leads to insulin resistance at the level of the liver or in the periphery, leading to fasting and stimulated hyperinsulinemia. The prevalence of type-2 diabetes mellitus (T2DM) in acromegalic patients is more or less constant across studies and ranges from 20% to 56%, depending on the series [71]. The weighted mean T2DM prevalence in individuals with acromegaly is approximately 27% when comparing data from 14 national registries [11]. As long as the compensatory increase in insulin secretion by pancreatic ß cells counterbalances the reduction in insulin sensitivity, glucose tolerance remains normal. Impaired glucose tolerance occurs when insulin secretion is altered, followed by the onset of diabetes [134].
Acromegaly is associated with a decrease in fat mass (both visceral and subcutaneous) but an increase in intermuscular fat mass (which may contribute to insulin resistance) and lean body mass [135, 136]. A recent study found an increase in exercise-induced myokine irisin circulating levels in patients with acromegaly [137]. This increase was independent of the disease status. The consequences on either glucose metabolism or thermogenesis of these findings still need to be demonstrated.
Alterations in lipid metabolism are reported in 30–40% of patients with acromegaly. In uncontrolled disease, a typical lipid profile is found, characterized by increased levels of lipoprotein(a) and triglycerides and decreased levels of HDL cholesterol [138]. The course of lipid parameters (and other cardiovascular risk factors) may vary with the treatment modality after therapeutic control of acromegaly [139].
Hypercalciuria is frequent in patients with acromegaly and may be associated with an increased incidence of nephrolithiasis. It is related to an IGF-I-mediated and PTH-independent increase in calcitriol synthesis, which is responsible for both absorptive hypercalciuria and increased fasting plasma calcium linked to enhanced distal tubular calcium resorption [1, 140]. An increased prevalence of hyperparathyroidism is also observed in patients with acromegaly, either in the context of multiple endocrine neoplasia (see the above “5.3.3 Genetic Syndromes Associated with Acromegaly” section) or independently (phenocopy) as a usual sporadic hyperparathyroidism.
5.4.2.8 Respiratory Complications
Sleep apnea affects 60–80% of all patients with acromegaly at the diagnosis of acromegaly. Men seem to be affected more than women [141]. Sleep apnea is more likely to be sought in patients who snore (reported by 78% of patients with acromegaly) and in those with daytime sleepiness (51%) or morning fatigue and morning headache (16%). Sleep apnea may be a contributory factor in hypertension, cardiovascular disease, and even cognitive decline. In most cases, apnea is obstructive, but one-third of patients have central apnea. Obstructive apnea is linked to anatomical changes due to mandibular and maxillary growth, soft-tissue thickening (especially of the palate and uvula), and changes in the angles of the different bone segments, leading to hypercollapsibility of the posterior and lateral hypopharyngeal walls. Hypertrophy of the tongue also plays a role [142], as does hypertrophy of the submaxillary glands.
Changes in respiratory function are frequent but less well documented. Anatomical modifications of thoracic bones and cartilage (leading to profound changes in the geometry of the rib cage) and mechanical changes in thoracic elasticity and the inspiratory muscles can lead to ventilatory disorders. Respiratory muscle strength is also abnormal. Altered mechanical and energetic properties of some upper airway dilator muscles have recently been demonstrated [143]. The inspiratory time is shorter, and the breathing frequency may increase.
Patients with acromegaly often have an increase in their total lung capacity (81% of men and 56% of women), owing to an increase in alveolar volume. An obstruction is found in 20–30% of patients (small airway or upper airway narrowing). Subclinical hypoxemia may be present. No ventilation-perfusion mismatching has been demonstrated.
The apnea-hypopnea index improves during effective treatment of acromegaly, along with the obstructive apnea index and oximetry values [141, 142, 144]. However, while apnea can disappear in some patients whose acromegaly is cured, it may persist or even worsen (likely due at least in part to associated obesity [145]) in others who thus require nocturnal positive end expiratory pressure. The reevaluation of sleep apnea is thus useful even if patients are cured or well controlled after acromegaly treatment.
Vocal changes have been described in patients with acromegaly. A deepening of the voice and a low fundamental frequency are observed in the population with acromegaly [76]. Modifications of the laryngeal cords and muscles, as well as upper respiratory tract thickening, may be responsible for these findings. However, the clinical consequences and the phonetic handicap related to these changes are not currently known.
5.4.2.9 Pituitary and Sellar Mass Effects
Headache is a very common symptom. In contrast with nonfunctioning adenoma, headache may be present even in patients bearing a microadenoma, thus reflecting a multifactorial genesis other than a direct adenoma-related compressive/expansive effect. In large tumors (macroadenomas or giant adenomas), low visual acuity and visual field defects may be observed in cases of suprasellar progression. Compression of the normal pituitary may also lead to anterior pituitary deficiency, which must be explored clinically and biochemically. Diabetes insipidus is never associated with acromegaly, except after neurosurgery or in the context of pituitary apoplexy [146].
5.4.2.10 Neoplasia and Acromegaly
Through the GH- and IGF-I-related promotion of cellular proliferation and differentiation, neoplasm and cancer risks have always been a major issue when dealing with acromegaly. In vitro and in vivo studies have shown a direct effect of GH or IGF-I in mediating cell proliferation. Pharmacological blockade of these targets in some cases allowed tumor inhibition in cell and animal models [147]. Despite these data on molecular biology, the link between GH excess and cancer risk in acromegaly is still unclear [147]. Although cancer-related mortality varies across studies, it seems that an excess of cancer and related mortality is present in patients with acromegaly with uncontrolled disease [148]. An overall cancer prevalence of 10% (any type, any site) is found in national registries collecting real-life data [11]. There is also some controversy regarding the incidence of each individual cancer type in patients with acromegaly.
Figures of colorectal cancer relative risk compared with the general population, initially widely overestimated at 10–20, are probably only 2–3 as per novel estimates [150,151,152,153,153]. There are various potential biological mechanisms that could explain the increased risk of colonic cancer in acromegaly: direct effects of GH and/or IGF-I; hyperinsulinemia; increases in IGFBP-3, IGF-II, and IGFBP-2 levels; altered bile acid secretions and local immune response; increased large bowel length; and obesity [150, 153]. Some authors claim that epigenetic alterations predispose patients with acromegaly to cancer development [154]. As colonic cancer may be the consequence of colonic polyp degeneration, many studies have examined the prevalence of colon polyps in patients with acromegaly. Prospective studies show that up to 45% of patients with acromegaly have colonic polyps, which are adenomatous in 24% of cases [155] and can arise in all parts of the colon. The acromegaly-associated colonic lesions seem to exhibit some peculiarities, such as larger, multiple, and more dysplastic adenomatous polyps than in nonacromegaly patients [150]. There is no clear correlation between GH or IGF-I concentrations and the incidence of colonic polyps. Colonoscopy guidelines for patients with acromegaly are controversial. The British Society of Gastroenterology [156] recommends performing a colonoscopy in patients with acromegaly by the age of 40. A subsequent examination should depend on the findings at the original screening and on the disease activity: screening every 3 years in patients with a previous adenoma or with elevated IGF-I and every 5–10 years in those without adenomatous/dysplastic polyps or those with only hyperplastic polyps. Some technical difficulties may be encountered in patients with acromegaly because of the increased colon length [153].
Goiter is found in a large proportion of patients with acromegaly. Thyroid nodules have been found in nearly 60–70% of patients [157]. Multinodular goiter is autonomous in 10–20% of patients, sometimes causing patent thyrotoxicosis. Although thyroid nodules are in most cases benign, the risk of thyroid cancer has been found to be higher than that in the general population (odds ratio, OR = 7.9, relative risk, RR = 7.6), with a prevalence of nearly 4%. These findings were confirmed by a recent Finnish study [158]. Nevertheless, contrary to colorectal cancer, most studies about thyroid cancer contain recruitment biases, and the real incidence of thyroid cancer in acromegaly is still a matter of debate [153, 157, 159]. As is the case for colonic cancer, a relative overestimation of thyroid cancer may arise because of increased physician awareness for these tumors, as well as the large use of ultrasonography during the screening of comorbidities in acromegaly.
Neoplasms of the breast, lung, prostate, skin, soft tissues, brain, bone, and lympho-hematopoietic system, initially described in association with acromegaly, do not seem to be overrepresented in these patients [160]. There is therefore remarkable agreement among all experts and reported guidelines, pointing out that surveillance in relation to these cancer sites should follow the same recommendations as for the general population [161].
It is currently acknowledged that, along with other cancers and neoplasms, the description of cancer occurrence is probably overestimated because of enhanced proactive screening. Modern imaging techniques may detect subclinical lesions and therefore affect the incidence rates. Other benign lesions may be found at a higher prevalence in patients with acromegaly. A higher incidence of meningioma has been found when analyzing encephalic MRI in patients with somatotroph adenomas versus those with other cell-type pituitary adenomas [162].
5.5 Diagnosis of Acromegaly
The diagnosis of acromegaly is suspected on clinical grounds and is confirmed by a typical biochemical profile [2, 74]. Clinical diagnosis is suggested by typical disfigurement due to progressive acral enlargement and modification of the facial appearance. In the case of very low progression or clinical incertitude, it is sometimes useful to assess the evolution by comparing serial photographs over several years (Fig. 5.2). Deep learning approaches are currently going to be tested to assist semiology [163]. It is of note that the regions of interest of these tools using aprioristic algorithms are primarily the same as those used by clinicians [163].
IGF-I (with reference to the age-adjusted normal range), the main GH-dependent growth factor, is the screening test recommended for acromegaly, with the diagnosis being confirmed by a nonsuppressive level of GH after an oral glucose load, OGTT [161].
5.5.1 GH and IGF-I
The introduction of international standards has minimized GH variability, which was mainly due to the use of polyclonal or monoclonal antibodies recognizing a mixture of different molecular forms. Manufacturers were recently advised to calibrate their GH assay kits with the international standard (IS) 98/574 [164].
The latest assays allow the limit of quantification to be as low as 0.05 μg/l with an interassay coefficient of variation (CV) of <20% [164, 165].
In most cases, GH levels are elevated, both at baseline and after OGTT [166]. GH levels in the population with acromegaly are inversely correlated with age, in which the youngest patients have the most elevated serum GH concentrations, and with the maximal tumor diameter [10]. Previous recommendations consider a diagnosis of acromegaly if nadir GH levels are above 1 μg/l [74, 167]. However, a few patients with clear clinical signs of acromegaly and high IGF-I levels could have low GH output and can thus suppress GH levels to less than 1 μg/l during the OGTT. Thus, a more stringent criterion of a nadir GH at 0.4 μg/l after OGTT has been proposed [168] and is now increasingly recognized as the recommended threshold if a sensitive GH assay is used. This is in line with recent normative data in healthy subjects underlining the importance of sex, BMI, and the use of contraceptive (estroprogestative) pills in defining the threshold for GH under OGTT [169]. However, it must be emphasized that the last Endocrine Society guidelines continue to recommend the 1 μg/ml threshold rather than the 0.4 μg/l threshold, considering that in the United States, the use of ultrasensitive GH assays is not yet generalized [161]. A paradoxical increase in GH following OGTT is observed in approximately 10–30% of patients with acromegaly [170, 171].
For the IGF-I assay, the IS 02/254 WHO reference standard has recently become available. It is an ~97%-pure recombinant standard recommended for manufacturers [164, 165]. The IGF-I level increases in parallel to the log of the GH concentration and must be determined by using age-adjusted norms because levels fall with age. A multicenter cohort study comparing six IGF-I immunoassays in 911 healthy individuals showed good agreement at lower but not upper levels [172]. This variability, especially in upper levels, which are more interesting when evaluating the biologic control of disease, leads to a marked variability in each individual’s IGF-I levels. Concordance between assay values in intraindividual patients with acromegaly was on average good (ranging from moderate to excellent) [173]. These differences in assay performances must be considered when evaluating disease control in subjects with acromegaly [173].
Similar to GH, IGF-I levels in patients with acromegaly are inversely correlated with age and with the maximal tumor diameter [10].
High IGF-I concentrations are also systematically found in other physiological states, such as pregnancy, puberty, and the postpubertal period. The concentration of IGFBP-3, the main IGF carrier protein, is usually increased in patients with acromegaly, but this marker offers little further diagnostic information in differential diagnosis.
GH and/or IGF-I measurements are of limited use for diagnosis (or treatment efficacy assessment) in patients with uncontrolled diabetes mellitus, chronic renal failure or pregnancy, and at the time of puberty.
Estradiol increases either basal GH or nadir GH levels after OGTT. This explains why GH is rarely inhibited by OGTT in women taking estrogen-containing pills. GH nadir concentrations are also significantly higher in lean and normal weight compared to overweight or obese subjects [169].
There are some individuals with a typical clinical picture of acromegaly but normal IGF-I and GH concentrations. This situation could correspond to two different situations: (1) spontaneously resolving real acromegaly, probably through necrosis or apoplexy of a previous GH-secreting pituitary adenoma; facial sequelae and disfigurement could have persisted despite the normalization of the somatotroph axis after spontaneous adenoma shrinkage; and (2) acromegaloid features may also be encountered in other diseases, such as severe insulin resistance, severe hypothyroidism, some forms of lipodystrophy, genodermatoses, or rarer overgrowth disease [175,176,176]. An extensive review focusing on various causes of pseudoacromegaly has recently been published [177].
Finally, some adenomas excised for mass effect or upon another surgical indication were revealed to derive from the somatotroph lineage only after histopathological examination [178]. In most of these cases, the somatotroph adenoma is silent, and no clinical signs of acromegaly are found. Nonetheless, subtle abnormalities revealing GH/IGF-I hypersecretion may be encountered [25, 179].
5.5.2 Neuroimaging
MRI is the imaging method of choice to detect a pituitary lesion. T1- and T2-weighted coronal and T1-weighted sagittal sections are routinely performed in diencephalic studies; gadolinium contrast usually shows a retardation in lesion enhancement demarcating the remaining hypophyseal tissue.
The majority of patients clearly have a pituitary macroadenoma (lesion above 10 mm). In patients with a sellar macroadenoma, once the diagnosis is established, before initiating treatment for acromegaly, patients must undergo a thorough work-up focusing on tumor mass effects (headaches, changes in the visual field and acuity, MRI abnormalities) and anterior pituitary function.
Although the majority of somatotroph adenomas are large tumors, in recent decades, the prevalence of microadenomas has seemingly increased in patients with acromegaly. It has to be known whether this trend depends on an improved clinical skill to detect disease (and therefore smaller lesions) or an intrinsic biological characteristic of somatotroph adenomas.
GH-secreting pituitary adenomas can be hypo-, iso-, or hyperintense on T2-weighted MRI sequences. Some authors suggest that hypointense imaging on T2-weighted MRI predicts a better outcome after somatostatin analog treatment either in terms of biochemical profile or tumoral shrinkage [180].
When MRI is contraindicated, a skull base CT scan may still be used. In patients with macroadenoma, this technique may show the presence of the pituitary mass and various extents of enlargement of the sella turcica.
Novel tools such as 11C-methionine positron emission tomography seem to detect small pituitary remnants, especially those with a high metabolic rate and hypersecretion [181]. This technique seems particularly promising in equivocal MRI images [182]. Nevertheless, the accuracy of this technique has yet to be extensively established, and 11C-methionine PET sequences are not routinely indicated in assessing pituitary imaging. Moreover, this technique is available in very few centers where a cyclotron is available on site due to the very short half-life of the radionuclide.
5.5.3 Pituitary Assessment
Associated prolactin hypersecretion is present in up to 30% of cases and may be either functional, secondary to impairment of hypothalamic dopamine production or compression of the pituitary stalk by the tumor, or due to a mixed adenoma.
In patients with microadenoma, no other pituitary defect or sellar mass effect is expected.
5.5.4 Total Body Imaging
If, despite an overt disease, no image is found on MRI, an ectopic GHRH secretion must be suspected and appropriate imaging requested [50].
Some occult neuroendocrine tumors may require total body scans (CT scans, MRI) or functional imaging (Octreoscan®, F-DOPA, or DOTATOC) [183]. Biopsy may help prove the neuroendocrine nature of these neoplasms. Complete excision of the underlying tumor usually cures disease.
5.6 Management and Follow-Up
Management of acromegaly is multimodal and quite consensual across different American and European guidelines and clinical practices [74, 165, 185,186,187,188,188].
The main aim is to relieve symptoms, normalize or decrease GH/IGF-1 excess, remove or reduce pituitary tumors, and improve long-term morbidity and mortality [185, 189]. Recent epidemiological studies have helped to refine the definitions of “cure” and “good disease control”, which are now far more precise: the GH concentration (in a random sample) must return to less than 1 μg/l in the new sensitive assays that are now widely used (if the OGTT is used, the nadir needs to be less than 0.4 μg/l) and the IGF-I level must return to normal according to sex and age [165, 187]. A stepwise therapeutic strategy based on surgery and/or radiotherapy and/or medical treatment allows these goals to be achieved.
5.6.1 Neurosurgery
Surgery is generally the first-line treatment. Tumor excision, usually by the transsphenoidal route, is the most rapid way of reducing GH and IGF-I concentrations in patients with acromegaly. Nevertheless, these levels normalize in only 40–70% of cases after surgery [11, 191,192,193,194,194]. The success rate depends on a range of features, such as the tumor size (microadenomas are more amenable to cure), the preoperative GH concentration (the success rate is higher when GH concentrations are <10 μg/l), and the surgeon’s experience. Endoscopic techniques, now used in the majority of expert centers [195], though not improving the success rate, may attenuate local adverse effects [192].
Postoperative outcome in terms of symptom relief and biological disease control is generally assessed 3 months after surgery. When surgery fails to achieve disease control or when surgery is impossible or contraindicated, patients are offered radiotherapy and/or pharmacological treatments.
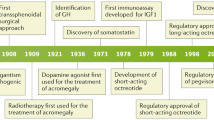
5.6.2 Radiating Techniques
Radiotherapy techniques have evolved over time, refining the techniques and number of sessions. Radiosurgery is a term used to define high-dose radiation delivery. It better applies to small targets and requires a single or few sittings. Fractionated radiotherapy refers to radiation therapy delivered at smaller doses but with multiple treatments (typically 25–30 sittings during 5–6 weeks). In order to minimize the dose to surrounding tissues, stereotactic localization is now used. Stereotactic radiosurgery (SRS) may use different radiating particles, such as photons (gamma knife, Linac, CyberKnife) or charged ions (protons). Stereotactic fractionated radiotherapy (SFR) is a hybrid form combining stereotactic localization with fractionated therapy administered by 3D-conformal radiation therapy, intensity-modulated radiation therapy, or proton radiation therapy [196].
Radiating techniques have consistently evolved over the last decade, from conventional radiotherapy to three-dimensional (3-D) conformal and stereotactic techniques. Technical improvements have been performed in all aspects of radiation treatment, including better imaging and 3-D planning, patient immobilization, sophisticated imaging systems for accurate patient repositioning and a more precise dose delivery, and reduction in normal surrounding brain structures exposed to high radiation doses [197].
In patients with somatotroph adenomas, normalization of GH/IGF-I levels occurs in approximately 40–60% of patients 5–10 years after treatment, with a 50% decline in GH and IGF-1 preradiation levels in approximately 2 and 5 years, respectively [190, 198, 199].
The choice between SFR and SRS, like for any other pituitary adenoma, in part depends on the size of the tumor and on its contiguity with the optic apparatus [200]. It is of note that the baseline GH concentrations predict treatment outcome and the time-to-normalization of patients with high (>3–4-fold ULN) IGF-1 levels requiring up to 10 years to achieve biochemical control of disease [199, 201].
SRS provides more focused irradiation. In a French series of over 80 patients, the efficacy of gamma-knife irradiation was close to that of SFR [202]. In a recent meta-analysis, disease control (without complementary medical treatment) was achieved in 48–53% of cases after a mean follow-up of 4 years. The relatively larger figures by SRS are probably explained by the small size of tumors (2.1 ± 1.2 mL) [203]. Apart from disease control, the different SRS techniques give excellent results in controlling tumor growth, with >95% success according to different series [200, 203,204,204].
A recent review of the literature proved a similar rate of tumor control between stereotactic radiosurgery and SFR for patients with persistent active acromegaly after surgery and/or during medical therapy [205]. Tumors were stable or decreased in 93–100% of patients at 5–10 years, whereas endocrinological remission was achieved in 40–60% of patients at 5 years [205].
On the other hand, radiotherapy leads to variable degrees of anterior pituitary insufficiency in 50–100% of patients after 10–15 years, regardless of the technique. Complications such as radionecrosis and optic neuropathy are very rare. In contrast, the risk of stroke and cerebrovascular events may be increased, sometimes many years after irradiation [206]. When compared to patients not exposed to radiotherapy, stroke incidence appears to be increased from 1.7 to 2.8 times [200, 207]. Along with cortisol deficiency and inadequate hormonal substitution, these findings seem to account for the excess mortality in these patients [208]. The question of whether cerebrovascular risk may be lowered by newer radiating techniques is presently still unanswered.
5.6.3 Medical Treatment
5.6.3.1 Dopamine Agonists (DA)
Cabergoline appears to be the most effective among DA agents [209, 210]. In a meta-analysis of all published studies, IGF-I normalization was achieved in up to 34% of cases [211]. Multivariate analysis showed that the efficacy depended on the initial IGF-I concentration, the treatment duration, and the basal concentration of PRL (and, to a lesser degree, the dose of cabergoline) [211]. As in patients with hyperprolactinemia, cardiac valve disease does not seem to be increased in patients with acromegaly treated with cabergoline in the long term [133, 212].
Because of their dual origin, mixed lactotroph/somatotroph tumors are more likely to respond to DA [213]. Half of the patients with GH/PRL-secreting adenomas normalize their IGF-I levels, and 60% of those with macroadenomas display tumor shrinkage [209].
5.6.3.2 Somatostatin Receptor Ligands (SRLs)
SRLs suppress GH secretion by binding to somatostatin receptor subtypes (sst) sst2 and sst5, which are mainly present on somatotroph adenoma cells [214]. These drugs have been demonstrated to exert either antisecretory or antitumoral effects (Fig. 5.5).

Trends of disease control, treatment approach, and medical therapy over time in acromegaly—the example of the French Registry of Acromegaly. Panel a. Evolution of disease status across 4-year follow-up periods in the French acromegaly registry. Histograms indicate the percentages of patients and standard deviations; trt: medical treatment. Panel b. Distribution of treatment approaches in different follow-up periods. MT medical treatment, RT radiotherapy. Data are reported as percentages of patients. Panel c. Evolution of drug therapy in medically treated patients: somatostatin analogs (SSA), dopamine agonists (DA), GH receptor antagonist (GHRA). The histograms report the prescribed classes of treatment per patient according to different follow-up periods as the percentage of prescriptions. Note: Pegvisomant was approved for the treatment of acromegaly in France in late 2003. Adapted from [23], with permission
The first SRL to be marketed, octreotide (Sandostatin®), can be injected subcutaneously (SC), generally by the patient him/herself, at a dose of 100–200 μg two or three times a day [215]. Sustained-release SRLs (lanreotide and octreotide LAR) progressively followed and had an impact on the market due to their comfort. The former requires deep SC injections every 28 days at variable doses (Somatuline® Autogel® 60, 90, or 120 mg). The latter is administered intramuscularly once a month (Octreotide LAR, Sandostatin® LAR 10–20 or 30 mg). The dose and frequency of injections may be initiated and adjusted depending on the GH/IGF-1 concentration.
These SRLs bear similar efficacy [216] in driving GH concentrations below 2 μg/l (60% to 70% of cases) and in normalizing IGF-1 levels (50–80%) [217, 218]. A recent meta-analysis has emphasized that control rates were highly variable from one study to the other. If clinical design characteristics had no statistically significant impact on efficacy determination, then later year of publication, study duration, and prior somatostatin analog use were significant efficacy determinants for acromegaly trial outcomes. In that meta-analysis, overall achieved control rates were 56% for mean GH and 55% for IGF-1 normalization [219].
Several long-term studies have shown that the cure rate may improve over time [221,222,222].
In a handful of good responders, SRL injection frequency may be lengthened or even safely halted with no subsequent increase in GH/IGF-I concentrations [223, 224].
Tumor volume shrinks in a weighted mean of 37–51% of patients [225]. It seems that the reduction in tumor volume is larger when an SRL is used as first-line treatment [226]. When not shrinking, tumor volumes remain at least stable in the vast majority of cases [217].
SRLs may cause gastrointestinal disorders (abdominal bloating, nausea, diarrhea), which are generally transient. SRLs induce the occurrence of gallstones in 10–20% of cases that did not respond to ursodeoxycholic acid [227, 228]. Some practitioners prescribe pancreatic enzymes in SRL-related diarrhea. Changes in glucose metabolism are sometimes observed, including impaired glucose tolerance or even diabetes in patients who are overweight. In other cases, however, glucose tolerance improves following the reduction in insulin resistance due to the lowering of GH concentrations. Overall, according to recent meta-analyses, despite a decrease in fasting plasma insulin levels, no consistent changes in fasting glucose and HbA1c levels have been observed [229, 230].
Pasireotide (SOM230 or Signifor®) is a second-generation SRL compound that binds to sst1, 2, 3, and 5 with high affinity [231], which has been proven to be effective in controlling acromegaly [233,234,234]. When directly compared to octreotide LAR, pasireotide was able to control a higher proportion of patients (36% versus 21%) [232]. In a crossover study, 15% of noncontrolled patients under maximal octreotide doses responded to pasireotide [235].
Concerning the side effects, glucose metabolism abnormalities (diabetes or glucose intolerance) were far more frequent in patients receiving pasireotide than in those administered conventional SRLs [232, 233]. Gastrointestinal symptoms after pasireotide treatment seem to occur at a similar or slightly increased frequency [233].
This drug may be particularly interesting in patients with partial resistance to first-generation SRLs.
Among the factors believed to influence SRL effectiveness, there is the T2-weighted hypointense signal on MRI [236, 237], the presence of specific somatostatin receptor subtypes, and the aspect of densely granulated cells at histological examination [238].
5.6.3.3 GH-Receptor Antagonists
Pegvisomant (Somavert®) acts peripherally, blocking the effects of GH on its target organs by binding to GH receptors and by preventing their dimerization, GH signal transduction, and downstream activity, including IGF-I production [239]. As pegvisomant inhibits the action of GH but not its secretion, GH concentrations cannot be used to evaluate treatment efficacy. IGF-I is used as a surrogate marker, together with clinical parameters. Pegvisomant is administered subcutaneously at a daily dose of 10–20 mg (sometimes more), with the dose being adapted to the hormone response (IGF-I normalization). Pegvisomant is highly effective, as IGF-I levels normalize in more than 90% of patients in the initial trials reported [240, 241]. In routine practice, the pegvisomant efficacy rate seems to be as low as 70% of cases, as shown by observational studies [243,244,245,246,247,247]. This treatment is reserved for patients in whom SRLs fail.
In a series of 304 patients in whom tumor volume was monitored for at least 3 years, an increase in tumor volume occurred in 9 cases within 8 months after commencing pegvisomant. This is likely related to rebound expansion after discontinuation of SRLs and/or to the natural history of aggressively growing pituitary tumors [248]; this latter situation may justify combination with an SRL to reduce tumor volume [249]. Tumor volume must therefore be monitored (by MRI) during this treatment. Available clinical data on pegvisomant concern a relatively small number of patients and relatively short treatment periods. Independent of disease control, pegvisomant improves glucose metabolism [250]. Adverse effects are limited to rare liver enzyme elevations, which are observed in between 2.5% and 3% of patients according to surveillance studies [242, 247]. Liver enzyme elevation generally normalizes either spontaneously or after treatment interruption. Exceptional cases of true hepatitis have been reported [246, 247]. Gilbert disease has been suggested as a risk factor for severe hepatitis [251, 252], but this was not confirmed by a recent Italian study [253].
SRL-pegvisomant combination therapy has also been developed [254]: a slow release formulation of the SRL is given once a month at the highest marketed dose (30 mg octreotide LAR or 120 mg lanreotide Autogel), and pegvisomant is injected once a week at escalating doses until the IGF-I level normalizes. IGF-I normalization was obtained in all patients with a median weekly pegvisomant dose of 60 mg [255]. This decreased dose requirement during combined therapy might be partially explained by an increase of approximately 20% in serum levels of pegvisomant [256]. Biochemical hepatic anomalies were quite frequent (although always transient) with this combination and appeared to be twice as common in patients with acromegaly with diabetes [257]. Compared with octreotide monotherapy, this combination appears to have a greater positive impact on quality of life for a given degree of IGF-I normalization [258]. This has raised the hypothesis of an extrahepatic effect of pegvisomant [259].
Cabergoline-pegvisomant combination therapy has also been proposed. In a multicenter, open-label, prospective clinical trial [260], the combination of cabergoline and low-dose pegvisomant (10 mg/day) was associated with a significant decrease in IGF-I levels compared with cabergoline alone, and 68% of patients achieved normalization. Then, when cabergoline was withdrawn and pegvisomant continued as monotherapy, only 26% of patients maintained normal IGF-I levels. The adjunction of cabergoline may be interesting when pegvisomant alone achieved minimally increased IGF-I levels [261].
5.6.4 Treatment Strategy
By analyzing large caseload series, several studies have evaluated medical practices and the evolution of treatment strategies over time [9, 11, 13]. The advantages, disadvantages, and costs of the different treatment options must be considered [262]. A marked evolution in clinical practice has been observed in recent decades (Fig. 5.5a–c).
An algorithm indicating a putative therapeutic strategy is proposed in Fig. 5.6. A surgical procedure is tried whenever possible. This depends on the availability of an experienced neurosurgeon, on the feasibility, on the fact that there are no anesthesiology constraints, and on the patient’s choice. Surgery has been chosen in nearly 80% of cases, considering the weighted mean of 19 studies across different countries encompassing more than 16,000 patients with acromegaly [11].

Proposed algorithm of the treatment strategy for acromegaly. After confirmation of acromegaly, the first step is to establish the patient’s eligibility for neurosurgery. In the absence of disease remission/cure after surgery, long-acting somatostatin receptor ligands (SRLs) are indicated. SRL doses and frequencies should be adapted and optimized, especially in partial responders (≥50% decrease in growth hormone (GH) and/or insulin-like growth factor 1 (IGF-I)). In the case of mild IGF-I elevation (<2–2.5-fold of the adjusted value for sex and age of the upper limit of normal value (ULN)), the addition of cabergoline can be considered. If disease control is not achieved, patients should be switched to the second-generation SRL pasireotide if there is clinically relevant residual tumor on imaging and/or clinical concern of tumor growth. Patients with impaired glucose tolerance should be switched to the GH antagonist pegvisomant (PegV). Patients with impaired glucose tolerance and tumor concern could be treated with a combination of a first-generation SRL and PegV. Patients who remain uncontrolled despite this second-line medical therapy should be discussed by a multidisciplinary team and considered for a second surgical intervention, a radiating therapy or temozolomide (features of aggressiveness, high Ki-67, tumor progression). *: consider using cabergoline in place of first-generation SRL if IGF-I elevation is <2–2.5-fold the ULN and/or in case of mixed GH/prolactin-secreting tumor; **near-normal IGF-I is considered for IGF-I values <1.3 ULN
In the case of surgery failure to cure acromegaly, medical treatment with SRLs is preferred as an elective option. SRL therapy is not only indicated after surgical failure but can sometimes be used for first-line treatment, especially when severe comorbidities create a risk of perioperative complications. Thus, when heart failure or respiratory problems are associated with acromegaly [71, 141], it is preferable to prepare the patient for surgery by administering SRLs for a few months first. In some cases, when a large tumor extends outside the sella and is not completely extractable by surgery, SRLs can be administered in the hope of controlling GH hypersecretion and tumor growth, thus avoiding the need for surgery [226, 264,265,266,267,267]. First-line SRL treatment before surgery has been chosen in 0–52% of cases, according to different series [11]. In these cases, when clinical conditions improve, neurosurgery could be performed as second-line treatment. According to a meta-analysis, IGF-I more likely normalizes after second-line treatment than after first-line drug therapy [218].
There is some controversy surrounding the ability of preoperative SRL therapy to improve surgical outcome: some studies [269,270,271,272,273,274,274] indicate that, in some patients with isolated somatotroph macroadenomas, surgery provides better control of acromegaly when patients are pretreated with an SRL, while other studies showed no difference [276,277,278,278]. Yang et al., in their latest meta-analysis, showed that preoperative SRL treatment was able to improve short-term (OR 2.07, 95% CI 1.50–2.87, p < 0.00001) but not long-term biochemical control [279].
The overall ability to normalize IGF-I by a first-line full-dose SRL (either octreotide LAR or lanreotide) is approximately 50% in recent series and the latest meta-analyses [280].
In some selected patients, especially in those with moderately increased IGF-I levels (below 2–2.5-fold the upper limit of normal) or in those with elevated concomitant prolactin levels, cabergoline may be tried first. It has indeed been shown that cabergoline is particularly useful in patients with low IGF-I excess [211].
After a first surgical approach, when full-dosed SRL therapy fails to achieve remission, several options may be chosen, mainly according to the patient status, his/her willingness, and the severity of clinical/biochemical disease persistence:
-
(a)
In the case of a large tumor remnant, it may be of utmost interest to repeat surgery. The main aim, in this case, will not be to cure disease but to debulk and decrease the secretory mass before trying medical treatment again [281, 282].
-
(b)
In the case of partial response to SRLs, physicians may choose to further adapt the adjusted doses [283, 284], to combine with other agents such as cabergoline [211], to administer pasireotide, or to initiate pegvisomant. When analyzing current anti-GH/IGF-I medical choices in these cases, a striking similarity is found between Germany and France, two countries where drugs are similarly available [9, 13]. In second-line treatment (after surgery failure), 60% of patients have been treated with SRLs alone, 10% by a combination of SRLs and DA, 10% by pegvisomant, 8.6% by DA alone, 6.8% by SRLs and pegvisomant, 2% by DA and pegvisomant, and 2.7% by tritherapy (SRLs+DA + pegvisomant). Notably, at the time of the survey, pasireotide was not yet available in these countries.
-
(c)
In the case of full persistence or disease that is still clinically and biochemically severe, pegvisomant should be rapidly tried. Increasing doses may be chosen to improve symptoms and to normalize IGF-I levels. If possible, surgical debulking may also be interesting to propose for reducing the dose of pegvisomant necessary to achieve normal IGF-I.
-
(d)
Radiotherapy is far less chosen as the first or second line. Studies evaluating clinical practices over time show a remarkable reduction in radiotherapy use between <2001 and > 2007 (Fig. 5.5b) [9]. However, radiotherapy may be particularly interesting when aiming at avoiding remnant enlargement and controlling GH secretion. In the case of a small remnant, gamma- or CyberKnife could be proposed. In the case of large tumors, fractionated radiotherapy may be proposed to avoid further tumor enlargement. Apart from clinical and biochemical issues, the cost of these long-term medical treatments should be weighed against the risks of radiotherapy. In any event, medical treatment will be necessary while waiting for the benefits of radiotherapy to emerge.
All these treatments must be reassessed on a yearly basis. After radiotherapy, if medical treatment is necessary while waiting for the effects of irradiation, regular withdrawal is necessary for assessing the persistence of active disease.
5.7 Prognosis
Several targeted studies and meta-analyses have been conducted to explore mortality in populations with acromegaly. The overall body of evidence globally shows an increased mortality rate in patients with acromegaly [11, 285, 286].
According to the earliest series published in the 1980s–1990s, approximately 60% of patients die from cardiovascular disease, 25% from respiratory complications, and 15% from cancer. If untreated, patients with acromegaly have been reported to die approximately 10 years earlier than healthy subjects [5]. Several studies have shown that cerebrovascular disorders are a frequent cause of death, especially among women, but these studies involved patients treated in various ways (craniotomy, radiotherapy), many years ago, and a deleterious effect of these treatments (especially radiotherapy) cannot be ruled out [287, 288]. Two recent meta-analyses [286, 289] showed a standardized mortality ratio (SMR), i.e., the ratio of observed mortality in the population with acromegaly to expected mortality in the general population, of 1.72 (IC 95%, 1.62–1.83). However, recent reports describe a reduction in this trend, with mortality rates that seem no longer higher than the general population [290]. Bolfi et al. compared the mortality rates across 26 studies according to the date of publication of the series [285]. They found that the mortality in acromegaly was increased from the 17 studies published before 2008, while no difference from the general population was found from the nine studies published after this date (SMR, 1.35; CI, 0.99–1.85). The posttreatment GH concentration is probably the best predictor of survival for all causes of death, independent of the type of complication. Thus, life expectancy outcomes can be stratified according to the posttreatment GH concentration: if GH secretion is controlled (<2 or 2.5 μg/l, or IGF-I normalization), life expectancy merges with that of the matched general population [287, 290, 291].
In the meta-analysis published by Holdaway and colleagues, it has been shown that the prognosis of acromegaly has improved in the last 20 years. This improvement is probably due to better disease control through a multimodal treatment strategy or to better management of comorbidities through improved awareness by physicians. Concerning the former aspect, a multimodal strategy and/or various anti-GH drug combinations witnessed a more aggressive treatment of the disease [9, 13, 292]. Concerning the latter, it is believed that mortality in acromegaly largely depends on the numerous and severe comorbidities. High GH/IGF-I concentrations, arterial hypertension, and cardiomyopathy are factors of poor prognosis, while the duration of symptoms and other factors (diabetes, lipid disorders, and cancer) are less important. Quality of life is also altered in acromegaly and is improved by effective treatment [293].
Finally, it must be stressed that, with the current therapeutic strategy, the vast majority of patients with acromegaly achieve very good control of GH/IGF-I secretion and have no problems related to tumor growth. Up to two-thirds of patients have normal IGF-I at the last visit, and these numbers seem to have increased in the latest series [11].
Adverse effects are infrequent and minor, even in the very long term. The figure is broadly different from the disease management 20 or more years ago, before the advent of somatostatin analogs. In addition, the use of more stringent criteria to define cure, together with aggressive treatment of comorbidities, has significantly improved the outlook of patients with acromegaly [118, 294]. However, even cured or well-controlled patients may have invalidating sequelae, such as joint pain, deformities, and impaired quality of life [145].
5.8 Conclusion
Acromegaly is a rare disease characterized by excessive GH and IGF-I exposure. In addition to the typical dysmorphic syndrome, a number of highly morbid diseases are associated with acromegaly. The cardiovascular, respiratory, rheumatologic and metabolic consequences and an increased oncologic risk represent the real burden of acromegalic disease and determine the prognosis. A detailed work-up of the various organs potentially involved in these complications is therefore recommended. Treatment is aimed at correcting (or preventing) tumor compression of neighboring tissues by excising the culprit lesion and at reducing GH and IGF-I levels to normal values. Selective pituitary sphenoidal surgery is often the first-line treatment. When surgery fails to correct GH/IGF-I hypersecretion, a multimodal therapeutic strategy is available, including several medical treatment classes (standard and novel somatostatin analogs, dopamine agonists, or the GH receptor antagonist pegvisomant). Radiotherapy is currently proposed as a third-line treatment. The prognosis of acromegaly has improved in recent years: adequate hormonal control is achieved in most cases, providing a life expectancy increasingly similar to that of the general population. It remains to be shown whether the criteria used to define control of the disease in terms of mortality also apply to optimal management of comorbidities.
5.9 Learning Points
-
Acromegaly, even if clinicaly manifest is diagnosed after a long delay which makes the cure of the disease more difficult and aggravates comorbidities.
-
Preoperative GH level (high GH output) is an important predictor of remission in acromegaly.
-
Persistent elevation of biochemical markers (GH and IGF1) following surgery indicates uncontrolled disease.
-
Medical therapy nowadays is an important part of the treatment of patients with acromegaly.
-
Age, size of the tumor, GH levels on presentation, histopathological type, and the somatostatin receptor status of the tumor in acromegaly may predict response to medical therapy.
-
Despite the availability of multiple treatment modalities (surgery, radiotherapy, somatostatin analogs first and second generation, GH receptor antagonists, etc.), acromegaly sometimes is a challenging condition to treat.
References
Kamenicky P, Mazziotti G, Lombes M, Giustina A, Chanson P. Growth hormone, insulin-like growth factor-1, and the kidney: pathophysiological and clinical implications. Endocr Rev. 2014;35(2):234–81.
Chanson P, Salenave S, Kamenicky P. Acromegaly. Handb Clin Neurol. 2014;124:197–219.
Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege. Belgium J Clin Endocrinol Metab. 2006;91(12):4769–75.
Fernandez A, Karavitaki N, Wass JA. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol. 2010;72(3):377–82.
Holdaway IM, Rajasoorya C. Epidemiology of acromegaly. Pituitary. 1999;2(1):29–41.
Schneider HJ, Sievers C, Saller B, Wittchen HU, Stalla GK. High prevalence of biochemical acromegaly in primary care patients with elevated insulin-like growth factor-1 levels. Clin Endocrinol. 2008; in press (available on line)
Raappana A, Koivukangas J, Ebeling T, Pirila T. Incidence of pituitary adenomas in northern Finland in 1992–2007. J Clin Endocrinol Metab. 2010;95(9):4268–75.
Lavrentaki A, Paluzzi A, Wass JA, Karavitaki N. Epidemiology of acromegaly: review of population studies. Pituitary. 2017;20(1):4–9.
Maione L, Brue T, Beckers A, Delemer B, Petrossians P, Borson-Chazot F, et al. Changes in the management and comorbidities of acromegaly over three decades: the French Acromegaly Registry. Eur J Endocrinol. 2017;176(5):645–55.
Petrossians P, Daly AF, Natchev E, Maione L, Blijdorp K, Sahnoun-Fathallah M, et al. Acromegaly at diagnosis in 3173 patients from the Liege acromegaly survey (LAS) database. Endocr Relat Cancer. 2017;24(10):505–18.
Maione L, Chanson P. National acromegaly registries. Best Pract Res Clin Endocrinol Metab. 2019;
Bex M, Abs R, T'Sjoen G, Mockel J, Velkeniers B, Muermans K, et al. AcroBel the Belgian registry on acromegaly: a survey of the 'real-life' outcome in 418 acromegalic subjects. Eur J Endocrinol. 2007;157(4):399–409.
Schofl C, Franz H, Grussendorf M, Honegger J, Jaursch-Hancke C, Mayr B, et al. Long-term outcome in patients with acromegaly: analysis of 1344 patients from the German Acromegaly Register. Eur J Endocrinol. 2013;168(1):39–47.
Ezzat S, Forster MJ, Berchtold P, Redelmeier DA, Boerlin V, Harris AG. Acromegaly. Clinical and biochemical features in 500 patients. Medicine (Baltimore). 1994;73(5):233–40.
Nabarro JD. Acromegaly. Clin Endocrinol. 1987;26(4):481–512.
Nachtigall L, Delgado A, Swearingen B, Lee H, Zerikly R, Klibanski A. Changing patterns in diagnosis and therapy of acromegaly over two decades. J Clin Endocrinol Metab. 2008;93(6):2035–41.
Reid TJ, Post KD, Bruce JN, Nabi Kanibir M, Reyes-Vidal CM, Freda PU. Features at diagnosis of 324 patients with acromegaly did not change from 1981 to 2006: acromegaly remains under-recognized and under-diagnosed. Clin Endocrinol. 2010;72(2):203–8.
Dal J, Feldt-Rasmussen U, Andersen M, Kristensen LO, Laurberg P, Pedersen L, et al. Acromegaly incidence, prevalence, complications and long-term prognosis: a nationwide cohort study. Eur J Endocrinol. 2016;175(3):181–90.
Asa SL, Ezzat S. The pathogenesis of pituitary tumours. Nat Rev Cancer. 2002;2(11):836–49.
Dworakowska D, Korbonits M, Aylwin S, McGregor A, Grossman AB. The pathology of pituitary adenomas from a clinical perspective. Front Biosci (Schol Ed). 2011;3:105–16.
Heaney AP, Melmed S. Molecular targets in pituitary tumours. Nat Rev Cancer. 2004;4(4):285–95.
Melmed S. Pathogenesis of pituitary tumors. Nat Rev Endocrinol. 2011;7(5):257–66.
Ho KKY, On behalf of the 2007 GH Deficiency Consensus Workshop Participants. Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: a statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur J Endocrinol. 2007;157(6):695–700. https://doi.org/10.1530/EJE-07-0631.
Beck-Peccoz P, Bassetti M, Spada A, Medri G, Arosio M, Giannattasio G, et al. Glycoprotein hormone alpha-subunit response to growth hormone (GH)-releasing hormone in patients with active acromegaly. Evidence for alpha-subunit and GH coexistence in the same tumoral cell. J Clin Endocrinol Metab. 1985;61(3):541–6.
Sakharova AA, Dimaraki EV, Chandler WF, Barkan AL. Clinically silent somatotropinomas may be biochemically active. J Clin Endocrinol Metab. 2005;90(4):2117–21.
Herman V, Fagin J, Gonski R, Kovacs K, Melmed S. Clonal origin of pituitary adenomas. J Clin Endocrinol Metab. 1990;71:1427–30.
Horvath A, Stratakis CA. Clinical and molecular genetics of acromegaly: MEN1, Carney complex, McCune-Albright syndrome, familial acromegaly and genetic defects in sporadic tumors. Rev Endocr Metab Disord. 2008;9(1):1–11.
Lytras A, Tolis G. Growth hormone-secreting tumors: genetic aspects and data from animal models. Neuroendocrinology. 2006;83(3–4):166–78.
Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009;119(11):3189–202.
Bi WL, Horowitz P, Greenwald NF, Abedalthagafi M, Agarwalla PK, Gibson WJ, et al. Landscape of genomic alterations in pituitary adenomas. Clin Cancer Res. 2017;23(7):1841–51.
Gadelha MR, Kasuki L, Korbonits M. The genetic background of acromegaly. Pituitary. 2017;20(1):10–21.
Valimaki N, Demir H, Pitkanen E, Kaasinen E, Karppinen A, Kivipelto L, et al. Whole-genome sequencing of growth hormone (GH)-secreting pituitary adenomas. J Clin Endocrinol Metab. 2015;100(10):3918–27.
Vallar L, Spada A, Giannattasio G. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature. 1987;330(6148):566–8.
Asa SL, Ezzat S. The cytogenesis and pathogenesis of pituitary adenomas. Endocr Rev. 1998;19(6):798–827.
Melmed S. Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest. 2003;112(11):1603–18.
Hage M, Viengchareun S, Brunet E, Villa C, Pineau D, Bouligand J, et al. Genomic alterations and complex subclonal architecture in sporadic GH-secreting pituitary adenomas. J Clin Endocrinol Metab. 2018;103(5):1929–39.
Beck-Peccoz P, Brucker-Davis F, Persani L, Smallridge RC, Weintraub BD. Thyrotropin-secreting pituitary adenomas. Endocr Rev. 1996;17(6):610–38.
Socin HV, Chanson P, Delemer B, Tabarin A, Rohmer V, Mockel J, et al. The changing spectrum of TSH-secreting pituitary adenomas: diagnosis and management in 43 patients. Eur J Endocrinol. 2003;148(4):433–42.
Chanson P, Salenave S, Droumaguet C, Cazabat L, Galland F, Young J. Rare causes of acromegaly. In: Wass JA, editor. Acromegaly: a handbook of history, current therapy and future prospects. Bristol: Bioscientifica Ltd; 2009. p. 70–98.
Daly AF, Tichomirowa MA, Beckers A. The epidemiology and genetics of pituitary adenomas. Best Pract Res Clin Endocrinol Metab. 2009;23(5):543–54.
Gadelha MR, Trivellin G, Hernandez Ramirez LC, Korbonits M. Genetics of pituitary adenomas. Front Horm Res. 2013;41:111–40.
Lecoq AL, Kamenicky P, Guiochon-Mantel A, Chanson P. Genetic mutations in sporadic pituitary adenomas—what to screen for? Nat Rev Endocrinol. 2015;11(1):43–54.
Chanson P, Salenave S, Orcel P. McCune-Albright syndrome in adulthood. Pediatr Endocr Rev. 2007;4(Suppl4):453–63.
Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J Rare Dis. 2012;7(Suppl 1):S4.
Salenave S, Boyce AM, Collins MT, Chanson P. Acromegaly and McCune-Albright syndrome. J Clin Endocrinol Metab. 2014;99(6):1955–69.
Vortmeyer AO, Glasker S, Mehta GU, Abu-Asab MS, Smith JH, Zhuang Z, et al. Somatic GNAS mutation causes widespread and diffuse pituitary disease in acromegalic patients with McCune-Albright syndrome. J Clin Endocrinol Metab. 2012;97(7):2404–13.
Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97(9):2990–3011.
Verges B, Boureille F, Goudet P, Murat A, Beckers A, Sassolas G, et al. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab. 2002;87(2):457–65.
Sano T, Yamasaki R, Saito H, Hirose T, Kudo E, Kameyama K, et al. Growth hormone-releasing hormone (GHRH)-secreting pancreatic tumor in a patient with multiple endocrine neoplasia type I. Am J Surg Pathol. 1987;11(10):810–9.
Garby L, Caron P, Claustrat F, Chanson P, Tabarin A, Rohmer V, et al. Clinical characteristics and outcome of acromegaly induced by ectopic secretion of growth hormone-releasing hormone (GHRH): a French nationwide series of 21 cases. J Clin Endocrinol Metab. 2012;97(6):2093–104.
Georgitsi M, Heliovaara E, Paschke R, Kumar AVK, Tischkowitz M, Vierimaa O, et al. Large genomic deletions in AIP in pituitary adenoma predisposition. J Clin Endocrinol Metab. 2008;93(10):4146–51. https://doi.org/10.1210/jc.2008-1003.
Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006;103(42):15558–63.
Bertherat J. Carney complex (CNC). Orphanet J Rare Dis. 2006;1:21.
Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al. Mutations of the gene encoding the protein kinase a type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26(1):89–92.
Beckers A, Aaltonen LA, Daly AF, Karhu A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev. 2013;34(2):239–77.
Chahal HS, Stals K, Unterlander M, Balding DJ, Thomas MG, Kumar AV, et al. AIP mutation in pituitary adenomas in the 18th century and today. N Engl J Med. 2011;364(1):43–50.
Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006;312(5777):1228–30.
Barlier A, Vanbellinghen JF, Daly AF, Silvy M, Jaffrain-Rea ML, Trouillas J, et al. Mutations in the aryl hydrocarbon receptor interacting protein gene are not highly prevalent among subjects with sporadic pituitary adenomas. J Clin Endocrinol Metab. 2007;92(5):1952–5.
Cazabat L, Libe R, Perlemoine K, Rene-Corail F, Burnichon N, Gimenez-Roqueplo AP, et al. Germline inactivating mutations of the aryl hydrocarbon receptor-interacting protein gene in a large cohort of sporadic acromegaly: mutations are found in a subset of young patients with macroadenomas. Eur J Endocrinol. 2007;157(1):1–8.
Cazabat L, Bouligand J, Chanson P. AIP mutation in pituitary adenomas. N Engl J Med. 2011;364(20):1973–4. author reply 4-5
Cazabat L, Bouligand J, Salenave S, Bernier M, Gaillard S, Parker F, et al. Germline AIP mutations in apparently sporadic pituitary adenomas: prevalence in a prospective single-center cohort of 443 patients. J Clin Endocrinol Metab. 2012;97(4):E663–70.
Daly AF, Vanbellinghen JF, Khoo SK, Jaffrain-Rea ML, Naves LA, Guitelman MA, et al. Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: analysis in 73 families. J Clin Endocrinol Metab. 2007;92(5):1891–6.
Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med. 2014;371(25):2363–74.
Kamenicky P, Bouligand J, Chanson P. Gigantism, acromegaly, and GPR101 mutations. N Engl J Med. 2015;372(13):1264.
Guillemin R, Brazeau P, Bohlen P, Esch F, Ling N, Wehrenberg WB. Growth hormone-releasing factor from a human pancreatic tumor that caused acromegaly. Science. 1982;218(4572):585–7.
Biermasz NR, Smit JW, Pereira AM, Frolich M, Romijn JA, Roelfsema F. Acromegaly caused by growth hormone-releasing hormone-producing tumors: long-term observational studies in three patients. Pituitary. 2007;10(3):237–49.
Gola M, Doga M, Bonadonna S, Mazziotti G, Vescovi PP, Giustina A. Neuroendocrine tumors secreting growth hormone-releasing hormone: pathophysiological and clinical aspects. Pituitary. 2006;9:221–9.
Thorner MO, Perryman RL, Cronin MJ, Rogol AD, Draznin M, Johanson A, et al. Somatotroph hyperplasia. Successful treatment of acromegaly by removal of a pancreatic islet tumor secreting a growth hormone-releasing factor. J Clin Invest. 1982;70(5):965–77.
Beuschlein F, Strasburger CJ, Siegerstetter V, Moradpour D, Lichter P, Bidlingmaier M, et al. Acromegaly caused by secretion of growth hormone by a non-Hodgkin's lymphoma. N Engl J Med. 2000;342(25):1871–6.
Melmed S, Ezrin C, Kovacs K, Goodman RS, Frohman LA. Acromegaly due to secretion of growth hormone by an ectopic pancreatic islet-cell tumor. N Engl J Med. 1985;312(1):9–17.
Colao A, Ferone D, Marzullo P, Lombardi G. Systemic complications of acromegaly: epidemiology, pathogenesis, and management. Endocr Rev. 2004;25(1):102–52.
Gadelha MR, Kasuki L, Lim DST, Fleseriu M. Systemic complications of acromegaly and the impact of the current treatment landscape: an update. Endocr Rev. 2019;40(1):268–332.
Molitch ME. Clinical manifestations of acromegaly. Endocrinol Metab Clin N Am. 1992;21(3):597–614.
Ribeiro-Oliveira A Jr, Barkan A. The changing face of acromegaly—advances in diagnosis and treatment. Nat Rev Endocrinol. 2012;8(10):605–11.
Danzig J. Acromegaly. BMJ. 2007;335(7624):824–5.
Bogazzi F, Nacci A, Campomori A, La Vela R, Rossi G, Lombardi M, et al. Analysis of voice in patients with untreated active acromegaly. J Endocrinol Investig. 2010;33(3):178–85.
Giustina A, Mazziotti G, Canalis E. Growth hormone, insulin-like growth factors, and the skeleton. Endocr Rev. 2008;29(5):535–59. https://doi.org/10.1210/er.2007-0036.
Ueland T, Fougner SL, Godang K, Schreiner T, Bollerslev J. Serum GH and IGF-I are significant determinants of bone turnover but not bone mineral density in active acromegaly: a prospective study of more than 70 consecutive patients. Eur J Endocrinol. 2006;155(5):709–15.
Hong AR, Kim JH, Kim SW, Kim SY, Shin CS. Trabecular bone score as a skeletal fragility index in acromegaly patients. Osteoporos Int. 2016;27(3):1123–9.
Mazziotti G, Bianchi A, Bonadonna S, Cimino V, Patelli I, Fusco A, et al. Prevalence of vertebral fractures in men with acromegaly. J Clin Endocrinol Metab. 2008;93(12):4649–55.
Claessen KM, Kroon HM, Pereira AM, Appelman-Dijkstra NM, Verstegen MJ, Kloppenburg M, et al. Progression of vertebral fractures despite long-term biochemical control of acromegaly: a prospective follow-up study. J Clin Endocrinol Metab. 2013;98(12):4808–15.
Kropf LL, Madeira M, Vieira Neto L, Gadelha MR, de Farias ML. Functional evaluation of the joints in acromegalic patients and associated factors. Clin Rheumatol. 2013;32(7):991–8.
Liote F, Orcel P. Osteoarticular disorders of endocrine origin. Baillieres Best Pract Res Clin Rheumatol. 2000;14(2):251–76.
Wassenaar MJ, Biermasz NR, van Duinen N, van der Klaauw AA, Pereira AM, Roelfsema F, et al. High prevalence of arthropathy, according to the definitions of radiological and clinical osteoarthritis, in patients with long-term cure of acromegaly: a case-control study. Eur J Endocrinol. 2009;160(3):357–65.
Claessen KM, Ramautar SR, Pereira AM, Romijn JA, Kroon HM, Kloppenburg M, et al. Increased clinical symptoms of acromegalic arthropathy in patients with long-term disease control: a prospective follow-up study. Pituitary. 2013;
Miller A, Doll H, David J, Wass J. Impact of musculoskeletal disease on quality of life in long-standing acromegaly. Eur J Endocrinol. 2008;158(5):587–93.
Wassenaar MJ, Biermasz NR, Kloppenburg M, van der Klaauw AA, Tiemensma J, Smit JW, et al. Clinical osteoarthritis predicts physical and psychological QoL in acromegaly patients. Growth Hormon IGF Res. 2010;20(3):226–33.
Scarpa R, De Brasi D, Pivonello R, Marzullo P, Manguso F, Sodano A, et al. Acromegalic axial arthropathy: a clinical case-control study. J Clin Endocrinol Metab. 2004;89(2):598–603.
Wassenaar MJ, Biermasz NR, Hamdy NA, Zillikens MC, van Meurs JB, Rivadeneira F, et al. High prevalence of vertebral fractures despite normal bone mineral density in patients with long-term controlled acromegaly. Eur J Endocrinol. 2011;164(4):475–83.
de Azevedo Oliveira B, Araujo B, Dos Santos TM, Ongaratti BR, Rech C, Ferreira NP, et al. The acromegalic spine: fractures, deformities and spinopelvic balance. Pituitary. 2019;22(6):601–6.
Jenkins PJ, Sohaib SA, Akker S, Phillips RR, Spillane K, Wass JA, et al. The pathology of median neuropathy in acromegaly. Ann Intern Med. 2000;133(3):197–201.
Tagliafico A, Resmini E, Nizzo R, Bianchi F, Minuto F, Ferone D, et al. Ultrasound measurement of median and ulnar nerve cross-sectional area in acromegaly. J Clin Endocrinol Metab. 2008;93(3):905–9.
Tagliafico A, Resmini E, Nizzo R, Derchi LE, Minuto F, Giusti M, et al. The pathology of the ulnar nerve in acromegaly. Eur J Endocrinol. 2008;159(4):369–73.
Chemla D, Attal P, Maione L, Veyer AS, Mroue G, Baud D, et al. Impact of successful treatment of acromegaly on overnight heart rate variability and sleep apnea. J Clin Endocrinol Metab. 2014;99(8):2925–31.
Furman K, Ezzat S. Psychological features of acromegaly. Psychother Psychosom. 1998;67(3):147–53.
Tiemensma J, Kaptein AA, Pereira AM, Smit JW, Romijn JA, Biermasz NR. Affected illness perceptions and the association with impaired quality of life in patients with long-term remission of acromegaly. J Clin Endocrinol Metab. 2011;96(11):3550–8.
Sibeoni J, Manolios E, Verneuil L, Chanson P, Revah-Levy A. Patients' perspectives on acromegaly diagnostic delay: a qualitative study. Eur J Endocrinol. 2019;180(6):339–52.
Andela CD, Niemeijer ND, Scharloo M, Tiemensma J, Kanagasabapathy S, Pereira AM, et al. Towards a better quality of life (QoL) for patients with pituitary diseases: results from a focus group study exploring QoL. Pituitary. 2015;18(1):86–100.
Biermasz NR. The burden of disease for pituitary patients. Best Pract Res Clin Endocrinol Metab. 2019;33(2):101309.
Chanson P, Timsit J, Masquet C, Warnet A, Guillausseau PJ, Birman P, et al. Cardiovascular effects of the somatostatin analog octreotide in acromegaly. Ann Intern Med. 1990;113(12):921–5.
Kamenicky P, Viengchareun S, Blanchard A, Meduri G, Zizzari P, Imbert-Teboul M, et al. Epithelial sodium channel is a key mediator of growth hormone-induced sodium retention in acromegaly. Endocrinology. 2008;149(7):3294–305.
Kamenicky P, Blanchard A, Frank M, Salenave S, Letierce A, Azizi M, et al. Body fluid expansion in acromegaly is related to enhanced epithelial sodium channel (ENaC) activity. J Clin Endocrinol Metab. 2011;96(7):2127–35.
Maison P, Demolis P, Young J, Schaison G, Giudicelli JF, Chanson P. Vascular reactivity in acromegalic patients: preliminary evidence for regional endothelial dysfunction and increased sympathetic vasoconstriction. Clin Endocrinol. 2000;53(4):445–51.
Colao A, Baldelli R, Marzullo P, Ferretti E, Ferone D, Gargiulo P, et al. Systemic hypertension and impaired glucose tolerance are independently correlated to the severity of the acromegalic cardiomyopathy. J Clin Endocrinol Metab. 2000;85(1):193–9.
Jaffrain-Rea ML, Moroni C, Baldelli R, Battista C, Maffei P, Terzolo M, et al. Relationship between blood pressure and glucose tolerance in acromegaly. Clin Endocrinol. 2001;54(2):189–95.
Puglisi S, Terzolo M. Hypertension and acromegaly. Endocrinol Metab Clin N Am. 2019;48(4):779–93.
Clayton RN. Cardiovascular function in acromegaly. Endocr Rev. 2003;24(3):272–7.
Sacca L, Cittadini A, Fazio S. Growth hormone and the heart. Endocr Rev. 1994;15(5):555–73.
Dos Santos Silva CM, Lima GA, Volschan IC, Gottlieb I, Kasuki L, Neto LV, et al. Low risk of coronary artery disease in patients with acromegaly. Endocrine. 2015;50(3):749–55.
Gouya H, Vignaux O, Le Roux P, Chanson P, Bertherat J, Bertagna X, et al. Rapidly reversible myocardial edema in patients with acromegaly: assessment with ultrafast T2 mapping in a single-breath-hold MRI sequence. AJR Am J Roentgenol. 2008;190(6):1576–82.
Kormanyos A, Domsik P, Kalapos A, Valkusz Z, Lengyel C, Forster T, et al. Three-dimensional speckle tracking echocardiography-derived left atrial deformation analysis in acromegaly (results from the MAGYAR-Path Study). Echocardiography. 2018;35(7):975–84.
Popielarz-Grygalewicz A, Gasior JS, Konwicka A, Grygalewicz P, Stelmachowska-Banas M, Zgliczynski W, et al. Heart in acromegaly: the echocardiographic characteristics of patients diagnosed with acromegaly in various stages of the disease. Int J Endocrinol. 2018;2018:6935054.
Sharma MD, Nguyen AV, Brown S, Robbins RJ. Cardiovascular disease in acromegaly. Methodist Debakey Cardiovasc J. 2017;13(2):64–7.
Kahaly G, Olshausen KV, Mohr-Kahaly S, Erbel R, Boor S, Beyer J, et al. Arrhythmia profile in acromegaly. Eur Heart J. 1992;13(1):51–6.
Warszawski L, Kasuki L, Sa R, Dos Santos Silva CM, Volschan I, Gottlieb I, et al. Low frequency of cardniac arrhythmias and lack of structural heart disease in medically-naive acromegaly patients: a prospective study at baseline and after 1 year of somatostatin analogs treatment. Pituitary. 2016;19(6):582–9.
Bihan H, Espinosa C, Valdes-Socin H, Salenave S, Young J, Levasseur S, et al. Long-term outcome of patients with acromegaly and congestive heart failure. J Clin Endocrinol Metab. 2004;89(11):5308–13.
Maison P, Tropeano AI, Macquin-Mavier I, Giustina A, Chanson P. Impact of somatostatin analogs on the heart in acromegaly: a metaanalysis. J Clin Endocrinol Metab. 2007;92(5):1743–7.
Maison P, Chanson P. Less is more risky? Growth hormone and insulin-like growth factor 1 levels and cardiovascular risk. Nat Clin Pract Endocrinol Metab. 2006;2(12):650–1.
Berg C, Petersenn S, Lahner H, Herrmann BL, Buchfelder M, Droste M, et al. Cardiovascular risk factors in patients with uncontrolled and long-term acromegaly: comparison with matched data from the general population and the effect of disease control. J Clin Endocrinol Metab. 2010;95(8):3648–56.
Boero L, Manavela M, Gomez Rosso L, Insua C, Berardi V, Fornari MC, et al. Alterations in biomarkers of cardiovascular disease (CVD) in active acromegaly. Clin Endocrinol. 2009;70(1):88–95.
Otsuki M, Kasayama S, Yamamoto H, Saito H, Sumitani S, Kouhara H, et al. Characterization of premature atherosclerosis of carotid arteries in acromegalic patients. Clin Endocrinol. 2001;54(6):791–6.
Paisley AN, Banerjee M, Rezai M, Schofield RE, Balakrishnannair S, Herbert A, et al. Changes in arterial stiffness but not carotid intimal thickness in acromegaly. J Clin Endocrinol Metab. 2011;96(5):1486–92.
Akutsu H, Kreutzer J, Wasmeier G, Ropers D, Rost C, Mohlig M, et al. Acromegaly per se does not increase the risk for coronary artery disease. Eur J Endocrinol. 2010;162(5):879–86.
Bogazzi F, Battolla L, Spinelli C, Rossi G, Gavioli S, Di Bello V, et al. Risk factors for development of coronary heart disease in patients with acromegaly: a five-year prospective study. J Clin Endocrinol Metab. 2007;92(11):4271–7.
Cannavo S, Almoto B, Cavalli G, Squadrito S, Romanello G, Vigo MT, et al. Acromegaly and coronary disease: an integrated evaluation of conventional coronary risk factors and coronary calcifications detected by computed tomography. J Clin Endocrinol Metab. 2006;91(10):3766–72.
Sardella C, Cappellani D, Urbani C, Manetti L, Marconcini G, Tomisti L, et al. Disease activity and lifestyle influence comorbidities and cardiovascular events in patients with acromegaly. Eur J Endocrinol. 2016;175(5):443–53.
Schofl C, Petroff D, Tonjes A, Grussendorf M, Droste M, Stalla G, et al. Incidence of myocardial infarction and stroke in acromegaly patients: results from the German Acromegaly Registry. Pituitary. 2017;20(6):635–42.
Parkinson C, Renehan AG, Ryder WD, O'Dwyer ST, Shalet SM, Trainer PJ. Gender and age influence the relationship between serum GH and IGF-I in patients with acromegaly. Clin Endocrinol. 2002;57(1):59–64.
Sesmilo G, Fairfield WP, Katznelson L, Pulaski K, Freda PU, Bonert V, et al. Cardiovascular risk factors in acromegaly before and after normalization of serum IGF-I levels with the GH antagonist pegvisomant. J Clin Endocrinol Metab. 2002;87(4):1692–9.
Verhelst J, Velkeniers B, Maiter D, Haentjens P, T'Sjoen G, Rietzschel E, et al. Active acromegaly is associated with decreased hs-CRP and NT-proBNP serum levels: insights from the Belgian registry of acromegaly. Eur J Endocrinol. 2013;168(2):177–84.
Colao A, Spinelli L, Marzullo P, Pivonello R, Petretta M, Di Somma C, et al. High prevalence of cardiac valve disease in acromegaly: an observational, analytical, case-control study. J Clin Endocrinol Metab. 2003;88(7):3196–201.
Pereira AM, van Thiel SW, Lindner JR, Roelfsema F, van der Wall EE, Morreau H, et al. Increased prevalence of regurgitant valvular heart disease in acromegaly. J Clin Endocrinol Metab. 2004;89(1):71–5.
Maione L, Garcia C, Bouchachi A, Kallel N, Maison P, Salenave S, et al. No evidence of a detrimental effect of cabergoline therapy on cardiac valves in patients with acromegaly. J Clin Endocrinol Metab. 2012;97(9):E1714–9.
Alexopoulou O, Bex M, Kamenicky P, Mvoula AB, Chanson P, Maiter D. Prevalence and risk factors of impaired glucose tolerance and diabetes mellitus at diagnosis of acromegaly: a study in 148 patients. Pituitary. 2014;17(1):81–9.
Freda PU, Shen W, Heymsfield SB, Reyes-Vidal CM, Geer EB, Bruce JN, et al. Lower visceral and subcutaneous but higher intermuscular adipose tissue depots in patients with growth hormone and insulin-like growth factor I excess due to acromegaly. J Clin Endocrinol Metab. 2008;93(6):2334–43.
Katznelson L. Alterations in body composition in acromegaly. Pituitary. 2009;12:136–42.
Calan M, Demirpence M. Increased circulating levels of irisin are associated with cardiovascular risk factors in subjects with acromegaly. Hormones (Athens). 2019;18:435.
Mercado M, Ramirez-Renteria C. Metabolic complications of acromegaly. Front Horm Res. 2018;49:20–8.
Briet C, Ilie MD, Kuhn E, Maione L, Brailly-Tabard S, Salenave S, et al. Changes in metabolic parameters and cardiovascular risk factors after therapeutic control of acromegaly vary with the treatment modality. Data from the Bicetre cohort, and review of the literature. Endocrine. 2019;63(2):348–60.
Kamenicky P, Blanchard A, Gauci C, Salenave S, Letierce A, Lombes M, et al. Pathophysiology of renal calcium handling in acromegaly: what lies behind hypercalciuria? J Clin Endocrinol Metab. 2012;97(6):2124–33.
Attal P, Chanson P. Endocrine aspects of obstructive sleep apnea. J Clin Endocrinol Metab. 2010;95(2):483–95.
Herrmann BL, Wessendorf TE, Ajaj W, Kahlke S, Teschler H, Mann K. Effects of octreotide on sleep apnoea and tongue volume (magnetic resonance imaging) in patients with acromegaly. Eur J Endocrinol. 2004;151(3):309–15.
Attal P, Claes V, Bobin S, Chanson P, Kamenicky P, Zizzari P, et al. Growth hormone excess and sternohyoid muscle mechanics in rats. Eur Respir J. 2009;34(4):967–74.
Ip MSM, Tan KCB, Peh WCG, Lam KSL. Effect of Sandostatin® LAR® on sleep apneoa in acromegaly: correlation with computerized tomographic cephalometry and hormonal activity. Clin Endocrinol. 2001;55:477–83.
Annamalai AK, Webb A, Kandasamy N, Elkhawad M, Moir S, Khan F, et al. A comprehensive study of clinical, biochemical, radiological, vascular, cardiac, and sleep parameters in an unselected cohort of patients with acromegaly undergoing presurgical somatostatin receptor ligand therapy. J Clin Endocrinol Metab. 2013;98(3):1040–50.
Briet C, Salenave S, Bonneville JF, Laws ER, Chanson P. Pituitary apoplexy. Endocr Rev. 2015;36(6):622–45.
Boguszewski CL, Boguszewski M. Growth Hormone's links to cancer. Endocr Rev. 2019;40(2):558–74.
Dal J, Leisner MZ, Hermansen K, Farkas DK, Bengtsen M, Kistorp C, et al. Cancer incidence in patients with acromegaly: a cohort study and meta-analysis of the literature. J Clin Endocrinol Metab. 2018;103(6):2182–8.
Jenkins PJ, Besser M. Clinical perspective: acromegaly and cancer: a problem. J Clin Endocrinol Metab. 2001;86(7):2935–41.
Lois K, Bukowczan J, Perros P, Jones S, Gunn M, James RA. The role of colonoscopic screening in acromegaly revisited: review of current literature and practice guidelines. Pituitary. 2015;18(4):568–74.
Melmed S. Acromegaly and cancer: not a problem? J Clin Endocrinol Metab. 2001;86(7):2929–34.
Renehan AG, O'Connell J, O'Halloran D, Shanahan F, Potten CS, O'Dwyer ST, et al. Acromegaly and colorectal cancer: a comprehensive review of epidemiology, biological mechanisms, and clinical implications. Horm Metab Res. 2003;35(11–12):712–25.
Renehan AG, Brennan BM. Acromegaly, growth hormone and cancer risk. Best Pract Res Clin Endocrinol Metab. 2008;22(4):639–57.
Loeper S, Ezzat S. Acromegaly: re-thinking the cancer risk. Rev Endocr Metab Disord. 2008;9(1):41–58.
Delhougne B, Deneux C, Abs R, Chanson P, Fierens H, Laurent-Puig P, et al. The prevalence of colonic polyps in acromegaly : a prospective colonoscopic and pathological study in 103 patients. J Clin Endocrinol Metab. 1995;80:3223–6.
Cairns SR, Scholefield JH, Steele RJ, Dunlop MG, Thomas HJ, Evans GD, et al. Guidelines for colorectal cancer screening and surveillance in moderate and high risk groups (update from 2002). Gut. 2010;59(5):666–89.
Wolinski K, Czarnywojtek A, Ruchala M. Risk of thyroid nodular disease and thyroid cancer in patients with acromegaly—meta-analysis and systematic review. PLoS One. 2014;9(2):e88787.
Kauppinen-Makelin R, Sane T, Valimaki MJ, Markkanen H, Niskanen L, Ebeling T, et al. Increased cancer incidence in acromegaly—a nationwide survey. Clin Endocrinol. 2010;72(2):278–9.
dos Santos MC, Nascimento GC, Nascimento AG, Carvalho VC, Lopes MH, Montenegro R, et al. Thyroid cancer in patients with acromegaly: a case-control study. Pituitary. 2013;16(1):109–14.
Boguszewski CL, Ayuk J. Management of endocrine disease: acromegaly and cancer: an old debate revisited. Eur J Endocrinol. 2016;175(4):R147–56.
Katznelson L, Laws ER Jr, Melmed S, Molitch ME, Murad MH, Utz A, et al. Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(11):3933–51.
Engelhardt J, Nunes ML, Pouchieu C, Ferriere A, San-Galli F, Gimbert E, et al. Increased incidence of intracranial meningiomas in patients with acromegaly. Neurosurgery. 2019;87:639.
Wei R, Jiang C, Gao J, Xu P, Zhang D, Sun Z, et al. Deep-learning approach to automatic identification of facial anomalies in endocrine disorders. Neuroendocrinology. 2019;110:328.
Clemmons DR. Consensus statement on the standardization and evaluation of growth hormone and insulin-like growth factor assays. Clin Chem. 2011;57(4):555–9.
Giustina A, Chanson P, Bronstein MD, Klibanski A, Lamberts S, Casanueva FF, et al. A consensus on criteria for cure of acromegaly. J Clin Endocrinol Metab. 2010;95(7):3141–8.
Carmichael JD, Bonert VS, Mirocha JM, Melmed S. The utility of oral glucose tolerance testing for diagnosis and assessment of treatment outcomes in 166 patients with acromegaly. J Clin Endocrinol Metab. 2009;94(2):523–7.
Giustina A, Barkan A, Casanueva FF, Cavagnini F, Frohman L, Ho K, et al. Criteria for cure of acromegaly: a consensus statement. J Clin Endocrinol Metab. 2000;85(2):526–9.
Trainer PJ. Editorial: acromegaly–consensus, what consensus? J Clin Endocrinol Metab. 2002;87(8):3534–6.
Schilbach K, Gar C, Lechner A, Nicolay SS, Schwerdt L, Haenelt M, et al. Determinants of the growth hormone nadir during oral glucose tolerance test in adults. Eur J Endocrinol. 2019;181(1):55–67.
Scaroni C, Albiger N, Daniele A, Dassie F, Romualdi C, Vazza G, et al. Paradoxical GH increase during OGTT is associated with first-generation somatostatin analog responsiveness in acromegaly. J Clin Endocrinol Metab. 2019;104(3):856–62.
Hage M, Kamenicky P, Chanson P. Growth hormone response to Oral glucose load: from normal to pathological conditions. Neuroendocrinology. 2019;108(3):244–55.
Chanson P, Arnoux A, Mavromati M, Brailly-Tabard S, Massart C, Young J, et al. Reference values for IGF-I serum concentrations: comparison of six immunoassays. J Clin Endocrinol Metab. 2016;101(9):3450–8.
Mavromati M, Kuhn E, Agostini H, Brailly-Tabard S, Massart C, Piketty ML, et al. Classification of patients with GH disorders may vary according to the IGF-I assay. J Clin Endocrinol Metab. 2017;102(8):2844–52.
Chakraborty PP, Bhattacharjee R, Mukhopadhyay S, Chowdhury S. Pseudoacromegaly in pachydermoperiostosis. BMJ Case Rep. 2016;2016
Chakraborty PP, Datta S, Mukhopadhyay S, Chowdhury S. Pseudoacromegaly in congenital generalised lipodystrophy (Berardinelli-Seip syndrome). BMJ Case Rep. 2016;2016 https://doi.org/10.1136/bcr-2016-214493.
Dahlqvist P, Spencer R, Marques P, Dang MN, Glad CAM, Johannsson G, et al. Pseudoacromegaly: a differential diagnostic problem for acromegaly with a genetic solution. J Endocr Soc. 2017;1(8):1104–9.
Marques P, Korbonits M. Pseudoacromegaly. Front Neuroendocrinol. 2019;52:113–43.
Kovacs K, Lloyd R, Horvath E, Asa SL, Stefaneanu L, Killinger DW, et al. Silent somatotroph adenomas of the human pituitary. A morphologic study of three cases including immunocytochemistry, electron microscopy, in vitro examination, and in situ hybridization. Am J Pathol. 1989;134(2):345–53.
Dimaraki EV, Jaffe CA, DeMott-Friberg R, Chandler WF, Barkan AL. Acromegaly with apparently normal GH secretion: implications for diagnosis and follow-up. J Clin Endocrinol Metab. 2002;87(8):3537–42.
Potorac I, Petrossians P, Daly AF, Alexopoulou O, Borot S, Sahnoun-Fathallah M, et al. T2-weighted MRI signal predicts hormone and tumor responses to somatostatin analogs in acromegaly. Endocr Relat Cancer. 2016;23(11):871–81.
Rodriguez-Barcelo S, Gutierrez-Cardo A, Dominguez-Paez M, Medina-Imbroda J, Romero-Moreno L, Arraez-Sanchez M. Clinical usefulness of coregistered 11C-methionine positron emission tomography/3-T magnetic resonance imaging at the follow-up of acromegaly. World Neurosurg. 2014;82(3–4):468–73.
Feng Z, He D, Mao Z, Wang Z, Zhu Y, Zhang X, et al. Utility of 11C-methionine and 18F-FDG PET/CT in patients with functioning pituitary adenomas. Clin Nucl Med. 2016;41(3):e130–4.
Putzer D, Gabriel M, Kendler D, Henninger B, Knoflach M, Kroiss A, et al. Comparison of (68)Ga-DOTA-Tyr(3)-octreotide and (18)F-fluoro-L-dihydroxyphenylalanine positron emission tomography in neuroendocrine tumor patients. Q J Nucl Med Mol Imaging. 2010;54(1):68–75.
Chanson P, Kamenicky P. Treatment of acromegaly: a critical analysis of the last ten years. Ann Endocrinol (Paris). 2012;73(2):99–106.
Colao A, Grasso LFS, Giustina A, Melmed S, Chanson P, Pereira AM, et al. Acromegaly. Nat Rev Dis Primers. 2019;5(1):20.
Melmed S, Colao A, Barkan A, Molitch M, Grossman AB, Kleinberg D, et al. Guidelines for acromegaly management: an update. J Clin Endocrinol Metab. 2009;94(5):1509–17.
Melmed S, Bronstein MD, Chanson P, Klibanski A, Casanueva FF, Wass JAH, et al. A consensus statement on acromegaly therapeutic outcomes. Nat Rev Endocrinol. 2018;14(9):552–61.
Sherlock M, Woods C, Sheppard MC. Medical therapy in acromegaly. Nat Rev Endocrinol. 2011;7(5):291–300.
Melmed S, Casanueva FF, Cavagnini F, Chanson P, Frohman L, Grossman A, et al. Guidelines for acromegaly management. J Clin Endocrinol Metab. 2002;87(9):4054–8.
Biermasz NR, van Dulken H, Roelfsema F. Long-term follow-up results of postoperative radiotherapy in 36 patients with acromegaly. J Clin Endocrinol Metab. 2000;85(7):2476–82.
Fahlbusch R, Buchfelder M, Nomikos P. Pituitary surgery. In: Melmed S, editor. The pituitary. 2nd ed. Malden, MA: Blackwell Science Inc.; 2002. p. 405–18.
Jane JA Jr, Starke RM, Elzoghby MA, Reames DL, Payne SC, Thorner MO, et al. Endoscopic transsphenoidal surgery for acromegaly: remission using modern criteria, complications, and predictors of outcome. J Clin Endocrinol Metab. 2011;96(9):2732–40.
Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical 'cure'. Eur J Endocrinol. 2005;152(3):379–87.
Swearingen B, Barker FG, Katznelson L, Biller BM, Grinspoon S, Klibanski A, et al. Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegaly. J Clin Endocrinol Metab. 1998;83(10):3419–26.
Cappabianca P, Cavallo LM, de Divitiis E. Endoscopic endonasal transsphenoidal surgery. Neurosurgery. 2004;55(4):933–40. discussion 40-1
Shih HA, Loeffler JS. Radiation therapy in acromegaly. Rev Endocr Metab Disord. 2008;9(1):59–65.
Minniti G, Flickinger J. The risk/benefit ratio of radiotherapy in pituitary tumors. Best Pract Res Clin Endocrinol Metab. 2019;33(2):101269.
Barrande G, Pittino-Lungo M, Coste J, Ponvert D, Bertagna X, Luton JP, et al. Hormonal and metabolic effects of radiotherapy in acromegaly: long-term results in 128 patients followed in a single center [in process citation]. J Clin Endocrinol Metab. 2000;85(10):3779–85.
Minniti G, Jaffrain-Rea ML, Osti M, Esposito V, Santoro A, Solda F, et al. The long-term efficacy of conventional radiotherapy in patients with GH-secreting pituitary adenomas. Clin Endocrinol. 2005;62(2):210–6.
Loeffler JS, Shih HA. Radiation therapy in the management of pituitary adenomas. J Clin Endocrinol Metab. 2011;96(7):1992–2003.
Jenkins PJ, Bates P, Carson MN, Stewart PM, Wass JA. Conventional pituitary irradiation is effective in lowering serum growth hormone and insulin-like growth factor-I in patients with acromegaly. J Clin Endocrinol Metab. 2006;91(4):1239–45.
Castinetti F, Taieb D, Kuhn JM, Chanson P, Tamura M, Jaquet P, et al. Outcome of gamma knife radiosurgery in 82 patients with acromegaly: correlation with initial hypersecretion. J Clin Endocrinol Metab. 2005;90(8):4483–8.
Yang I, Kim W, De Salles A, Bergsneider M. A systematic analysis of disease control in acromegaly treated with radiosurgery. Neurosurg Focus. 2010;29(4):E13.
Castinetti F, Nagai M, Morange I, Dufour H, Caron P, Chanson P, et al. Long-term results of stereotactic radiosurgery in secretory pituitary adenomas. J Clin Endocrinol Metab. 2009;94(9):3400–7.
Gheorghiu ML. Updates in outcomes of stereotactic radiation therapy in acromegaly. Pituitary. 2017;20(1):154–68.
Brada M, Burchell L, Ashley S, Traish D. The incidence of cerebrovascular accidents in patients with pituitary adenoma. Int J Radiat Oncol Biol Phys. 1999;45(3):693–8.
Minniti G, Scaringi C, Maurizi ER. Radiation techniques for acromegaly. Radiat Oncol. 2011;6(1):167.
Sherlock M, Ayuk J, Tomlinson JW, Toogood AA, Aragon-Alonso A, Sheppard MC, et al. Mortality in patients with pituitary disease. Endocr Rev. 2010;31(3):301–42.
Abs R, Verhelst J, Maiter D, Van Acker K, Nobels F, Coolens JL, et al. Cabergoline in the treatment of acromegaly: a study in 64 patients. J Clin Endocrinol Metab. 1998;83(2):374–8.
Newman CB. Medical therapy for acromegaly. Endocrinol Metab Clin N Am. 1999;28(1):171–90.
Sandret L, Maison P, Chanson P. Place of cabergoline in acromegaly: a meta-analysis. J Clin Endocrinol Metab. 2011;96(5):1327–35.
Valassi E, Klibanski A, Biller BM. Clinical review#: potential cardiac valve effects of dopamine agonists in hyperprolactinemia. J Clin Endocrinol Metab. 2010;95(3):1025–33.
Kuhn E, Chanson P. Cabergoline in acromegaly. Pituitary. 2017;20(1):121–8.
Schaer JC, Waser B, Mengod G, Reubi JC. Somatostatin receptor subtypes sst1, sst2, sst3 and sst5 expression in human pituitary, gastroentero-pancreatic and mammary tumors: comparison of mRNA analysis with receptor autoradiography. Int J Cancer. 1997;70(5):530–7.
Lamberts SW, van der Lely AJ, de Herder WW, Hofland LJ. Octreotide. N Engl J Med. 1996;334(4):246–54.
Murray RD, Melmed S. A critical analysis of clinically available somatostatin analog formulations for therapy of acromegaly. J Clin Endocrinol Metab. 2008;93(8):2957–68.
Bevan JS. Clinical review: the antitumoral effects of somatostatin analog therapy in acromegaly. J Clin Endocrinol Metab. 2005;90(3):1856–63.
Freda PU, Katznelson L, van der Lely AJ, Reyes CM, Zhao S, Rabinowitz D. Long-acting somatostatin analog therapy of acromegaly: a meta-analysis. J Clin Endocrinol Metab. 2005;90(8):4465–73.
Carmichael JD, Bonert VS, Nuno M, Ly D, Melmed S. Acromegaly clinical trial methodology impact on reported biochemical efficacy rates of somatostatin receptor ligand treatments: a meta-analysis. J Clin Endocrinol Metab. 2014;99(5):1825–33.
Ayuk J, Stewart SE, Stewart PM, Sheppard MC. Long-term safety and efficacy of depot long-acting somatostatin analogs for the treatment of acromegaly. J Clin Endocrinol Metab. 2002;87(9):4142–6.
Cozzi R, Attanasio R, Montini M, Pagani G, Lasio G, Lodrini S, et al. Four-year treatment with octreotide-long-acting repeatable in 110 acromegalic patients: predictive value of short-term results? J Clin Endocrinol Metab. 2003;88(7):3090–8.
Maiza JC, Vezzosi D, Matta M, Donadille F, Loubes-Lacroix F, Cournot M, et al. Long-term (up to 18 years) effects on GH/IGF-1 hypersecretion and tumour size of primary somatostatin analogue (SSTa) therapy in patients with GH-secreting pituitary adenoma responsive to SSTa. Clin Endocrinol. 2007;67(2):282–9.
Ramirez C, Vargas G, Gonzalez B, Grossman A, Rabago J, Sosa E, et al. Discontinuation of octreotide LAR after long term, successful treatment of patients with acromegaly: is it worth trying? Eur J Endocrinol. 2012;166(1):21–6.
Ronchi CL, Rizzo E, Lania AG, Pivonello R, Grottoli S, Colao A, et al. Preliminary data on biochemical remission of acromegaly after somatostatin analogs withdrawal. Eur J Endocrinol. 2008;158(1):19–25.
Giustina A, Mazziotti G, Torri V, Spinello M, Floriani I, Melmed S. Meta-analysis on the effects of octreotide on tumor mass in acromegaly. PLoS One. 2012;7(5):e36411.
Colao A, Ferone D, Marzullo P, Cappabianca P, Cirillo S, Boerlin V, et al. Long-term effects of depot long-acting somatostatin analog octreotide on hormone levels and tumor mass in acromegaly. J Clin Endocrinol Metab. 2001;86(6):2779–86.
Attanasio R, Mainolfi A, Grimaldi F, Cozzi R, Montini M, Carzaniga C, et al. Somatostatin analogs and gallstones: a retrospective survey on a large series of acromegalic patients. J Endocrinol Investig. 2008;31(8):704–10.
Chanson P, Bertherat J, Beckers A, Bihan H, Brue T, Caron P, et al. French consensus on the management of acromegaly. Ann Endocrinol (Paris). 2009;70(2):92–106.
Cozzolino A, Feola T, Simonelli I, Puliani G, Pozza C, Giannetta E, et al. Somatostatin analogs and glucose metabolism in acromegaly: a meta-analysis of prospective interventional studies. J Clin Endocrinol Metab. 2018;
Mazziotti G, Floriani I, Bonadonna S, Torri V, Chanson P, Giustina A. Effects of somatostatin analogs on glucose homeostasis: a metaanalysis of acromegaly studies. J Clin Endocrinol Metab. 2009;94(5):1500–8.
Bruns C, Lewis I, Briner U, Meno-Tetang G, Weckbecker G. SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol. 2002;146(5):707–16.
Colao A, Bronstein MD, Freda P, Gu F, Shen CC, Gadelha M, et al. Pasireotide versus octreotide in acromegaly: a head-to-head superiority study. J Clin Endocrinol Metab. 2014;99(3):791–9.
Gadelha MR, Bronstein MD, Brue T, Coculescu M, Fleseriu M, Guitelman M, et al. Pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA): a randomised, phase 3 trial. Lancet Diabetes Endocrinol. 2014;2(11):875–84.
Petersenn S, Schopohl J, Barkan A, Mohideen P, Colao A, Abs R, et al. Pasireotide (SOM230) demonstrates efficacy and safety in patients with acromegaly: a randomized, multicenter, phase II trial. J Clin Endocrinol Metab. 2010;95(6):2781–9.
Bronstein MD, Fleseriu M, Neggers S, Colao A, Sheppard M, Gu F, et al. Switching patients with acromegaly from octreotide to pasireotide improves biochemical control: crossover extension to a randomized, double-blind, Phase III study. BMC Endocr Disord. 2016;16:16.
Heck A, Ringstad G, Fougner SL, Casar-Borota O, Nome T, Ramm-Pettersen J, et al. Intensity of pituitary adenoma on T2-weighted magnetic resonance imaging predicts the response to octreotide treatment in newly diagnosed acromegaly. Clin Endocrinol. 2012;77(1):72–8.
Puig-Domingo M, Resmini E, Gomez-Anson B, Nicolau J, Mora M, Palomera E, et al. Magnetic resonance imaging as a predictor of response to somatostatin analogs in acromegaly after surgical failure. J Clin Endocrinol Metab. 2010;95(11):4973–8.
Gatto F, Feelders RA, van der Pas R, Kros JM, Waaijers M, Sprij-Mooij D, et al. Immunoreactivity score using an anti-sst2A receptor monoclonal antibody strongly predicts the biochemical response to adjuvant treatment with somatostatin analogs in acromegaly. J Clin Endocrinol Metab. 2013;98(1):E66–71.
Kopchick JJ, Parkinson C, Stevens EC, Trainer PJ. Growth hormone receptor antagonists: discovery, development, and use in patients with acromegaly. Endocr Rev. 2002;23(5):623–46.
Trainer PJ, Drake WM, Katznelson L, Freda PU, Herman-Bonert V, van der Lely AJ, et al. Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant. N Engl J Med. 2000;342(16):1171–7.
van der Lely AJ, Hutson RK, Trainer PJ, Besser GM, Barkan AL, Katznelson L, et al. Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet. 2001;358(9295):1754–9.
Buchfelder M, van der Lely AJ, Biller BMK, Webb SM, Brue T, Strasburger CJ, et al. Long-term treatment with pegvisomant: observations from 2090 acromegaly patients in ACROSTUDY. Eur J Endocrinol. 2018;179(6):419–27.
Colao A, Pivonello R, Auriemma RS, De Martino MC, Bidlingmaier M, Briganti F, et al. Efficacy of 12-month treatment with the GH receptor antagonist pegvisomant in patients with acromegaly resistant to long-term, high-dose somatostatin analog treatment: effect on IGF-I levels, tumor mass, hypertension and glucose tolerance. Eur J Endocrinol. 2006;154(3):467–77.
Marazuela M, Lucas T, Alvarez-Escola C, Puig-Domingo M, de la Torre NG, de Miguel-Novoa P, et al. Long-term treatment of acromegalic patients resistant to somatostatin analogues with the GH receptor antagonist pegvisomant: its efficacy in relation to gender and previous radiotherapy. Eur J Endocrinol. 2009;160(4):535–42.
Moore DJ, Adi Y, Connock MJ, Bayliss S. Clinical effectiveness and cost-effectiveness of pegvisomant for the treatment of acromegaly: a systematic review and economic evaluation. BMC Endocr Disord. 2009;9:20.
Schreiber I, Buchfelder M, Droste M, Forssmann K, Mann K, Saller B, et al. Treatment of acromegaly with the GH receptor antagonist pegvisomant in clinical practice: safety and efficacy evaluation from the German Pegvisomant observational study. Eur J Endocrinol. 2007;156(1):75–82.
van der Lely AJ, Biller BM, Brue T, Buchfelder M, Ghigo E, Gomez R, et al. Long-term safety of pegvisomant in patients with acromegaly: comprehensive review of 1288 subjects in ACROSTUDY. J Clin Endocrinol Metab. 2012;97(5):1589–97.
Jimenez C, Burman P, Abs R, Clemmons DR, Drake WM, Hutson KR, et al. Follow-up of pituitary tumor volume in patients with acromegaly treated with pegvisomant in clinical trials. Eur J Endocrinol. 2008;159(5):517–23.
van der Lely AJ, Muller A, Janssen JA, Davis RJ, Zib KA, Scarlett JA, et al. Control of tumor size and disease activity during cotreatment with octreotide and the growth hormone receptor antagonist pegvisomant in an acromegalic patient. J Clin Endocrinol Metab. 2001;86(2):478–81.
Feola T, Cozzolino A, Simonelli I, Sbardella E, Pozza C, Giannetta E, et al. Pegvisomant improves glucose metabolism in acromegaly: a meta-analysis of prospective interventional studies. J Clin Endocrinol Metab. 2019;104(7):2892–902.
Bernabeu I, Cameselle-Teijeiro J, Casanueva FF, Marazuela M. Pegvisomant-induced cholestatic hepatitis with jaundice in a patient with Gilbert's syndrome. Eur J Endocrinol. 2009;160(5):869–72.
Bernabeu I, Marazuela M, Lucas T, Loidi L, Alvarez-Escola C, Luque-Ramirez M, et al. Pegvisomant-induced liver injury is related to the UGT1A1*28 polymorphism of Gilbert's syndrome. J Clin Endocrinol Metab. 2010;95(5):2147–54.
Filopanti M, Barbieri AM, Mantovani G, Corbetta S, Gasco V, Ragonese M, et al. Role of UGT1A1 and ADH gene polymorphisms in pegvisomant-induced liver toxicity in acromegalic patients. Eur J Endocrinol. 2014;170(2):249–56.
Feenstra J, de Herder WW, ten Have SM, van den Beld AW, Feelders RA, Janssen JA, et al. Combined therapy with somatostatin analogues and weekly pegvisomant in active acromegaly. Lancet. 2005;365(9471):1644–6.
Neggers SJ, van Aken MO, Janssen JA, Feelders RA, de Herder WW, van der Lely AJ. Long-term efficacy and safety of combined treatment of somatostatin analogs and pegvisomant in acromegaly. J Clin Endocrinol Metab. 2007;92(12):4598–601.
Jorgensen JO, Feldt-Rasmussen U, Frystyk J, Chen JW, Kristensen LO, Hagen C, et al. Cotreatment of acromegaly with a somatostatin analog and a growth hormone receptor antagonist. J Clin Endocrinol Metab. 2005;90(10):5627–31.
Neggers SJ, de Herder WW, Janssen JA, Feelders RA, van der Lely AJ. Combined treatment for acromegaly with long-acting somatostatin analogs and pegvisomant: long-term safety for up to 4.5 years (median 2.2 years) of follow-up in 86 patients. Eur J Endocrinol. 2009;160(4):529–33.
Neggers SJCMM, van Aken MO, de Herder WW, Feelders RA, Janssen JAMJL, Badia X, et al. Quality of life in acromegalic patients during long-term somatostatin analog treatment with and without pegvisomant. J Clin Endocrinol Metab. 2008;93(10):3853–9.
Neggers SJ, van der Lely AJ. Somatostatin analog and pegvisomant combination therapy for acromegaly. Nat Rev Endocrinol. 2009;5(10):546–52.
Higham CE, Atkinson AB, Aylwin S, Bidlingmaier M, Drake WM, Lewis A, et al. Effective combination treatment with cabergoline and low-dose pegvisomant in active acromegaly: a prospective clinical trial. J Clin Endocrinol Metab. 2012;97(4):1187–93.
Bernabeu I, Alvarez-Escola C, Paniagua AE, Lucas T, Pavon I, Cabezas-Agricola JM, et al. Pegvisomant and cabergoline combination therapy in acromegaly. Pituitary. 2013;16(1):101–8.
Leonart LP, Ferreira VL, Tonin FS, Fernandez-Llimos F, Pontarolo R. Medical treatments for acromegaly: a systematic review and network meta-analysis. Value Health. 2018;21(7):874–80.
Attanasio R, Baldelli R, Pivonello R, Grottoli S, Bocca L, Gasco V, et al. Lanreotide 60 mg, a new long-acting formulation: effectiveness in the chronic treatment of acromegaly. J Clin Endocrinol Metab. 2003;88(11):5258–65.
Bevan JS, Atkin SL, Atkinson AB, Bouloux PM, Hanna F, Harris PE, et al. Primary medical therapy for acromegaly: an open, prospective, multicenter study of the effects of subcutaneous and intramuscular slow- release octreotide on growth hormone, insulin-like growth factor-I, and tumor size. J Clin Endocrinol Metab. 2002;87(10):4554–63.
Caron PJ, Bevan JS, Petersenn S, Flanagan D, Tabarin A, Prevost G, et al. Tumor shrinkage with lanreotide Autogel 120 mg as primary therapy in acromegaly: results of a prospective multicenter clinical trial. J Clin Endocrinol Metab. 2014;99(4):1282–90.
Newman CB, Melmed S, George A, Torigian D, Duhaney M, Snyder P, et al. Octreotide as primary therapy for acromegaly. J Clin Endocrinol Metab. 1998;83(9):3034–40.
Sheppard MC. Primary medical therapy for acromegaly. Clin Endocrinol. 2003;58(4):387–99.
Abe T, Ludecke DK. Effects of preoperative octreotide treatment on different subtypes of 90 GH-secreting pituitary adenomas and outcome in one surgical centre. Eur J Endocrinol. 2001;145(2):137–45.
Barkan AL, Lloyd RV, Chandler WF, Hatfield MK, Gebarski SS, Kelch RP, et al. Preoperative treatment of acromegaly with long-acting somatostatin analog SMS 201-995: shrinkage of invasive pituitary macroadenomas and improved surgical remission rate. J Clin Endocrinol Metab. 1988;67(5):1040–8.
Carlsen SM, Lund-Johansen M, Schreiner T, Aanderud S, Johannesen O, Svartberg J, et al. Preoperative octreotide treatment in newly diagnosed acromegalic patients with macroadenomas increases cure short-term postoperative rates: a prospective, randomized trial. J Clin Endocrinol Metab. 2008;93(8):2984–90.
Colao A, Merola B, Ferone D, Lombardi G. Acromegaly. J Clin Endocrinol Metab. 1997;82(9):2777–81.
Lucas T, Astorga R, Catala M. Preoperative lanreotide treatment for GH-secreting pituitary adenomas: effect on tumour volume and predictive factors of significant tumour shrinkage. Clin Endocrinol. 2003;58(4):471–81.
Mao ZG, Zhu YH, Tang HL, Wang DY, Zhou J, He DS, et al. Preoperative lanreotide treatment in acromegalic patients with macroadenomas increases short-term postoperative cure rates: a prospective, randomised trial. Eur J Endocrinol. 2010;162(4):661–6.
Stevenaert A, Harris AG, Kovacs K, Beckers A. Presurgical octreotide treatment in acromegaly. Metabolism. 1992;41(9 Suppl 2):51–8.
Biermasz NR, van Dulken H, Roelfsema F. Direct postoperative and follow-up results of transsphenoidal surgery in 19 acromegalic patients pretreated with octreotide compared to those in untreated matched controls. J Clin Endocrinol Metab. 1999;84(10):3551–5.
Kristof RA, Stoffet-Wagner B, Klingmüller D, Schramm J. Does Octréotide treatment improve the surgical results of macroadenomas in acromegaly ? A randomised study. Acta Neurochir. 1999;141:399–405.
Losa M, Mortini P, Urbaz L, Ribotto P, Castrignano T, Giovanelli M. Presurgical treatment with somatostatin analogs in patients with acromegaly: effects on the remission and complication rates. J Neurosurg. 2006;104(6):899–906.
Pita-Gutierrez F, Pertega-Diaz S, Pita-Fernandez S, Pena L, Lugo G, Sangiao-Alvarellos S, et al. Place of preoperative treatment of acromegaly with somatostatin analog on surgical outcome: a systematic review and meta-analysis. PLoS One. 2013;8(4):e61523.
Yang C, Li G, Jiang S, Bao X, Wang R. Preoperative somatostatin analogues in patients with newly-diagnosed acromegaly: a systematic review and meta-analysis of comparative studies. Sci Rep. 2019;9(1):14070.
Carmichael JD, Broder MS, Cherepanov D, Chang E, Mamelak A, Said Q, et al. Long-term treatment outcomes of acromegaly patients presenting biochemically-uncontrolled at a tertiary pituitary center. BMC Endocr Disord. 2017;17(1):49.
Karavitaki N, Turner HE, Adams CB, Cudlip S, Byrne JV, Fazal-Sanderson V, et al. Surgical debulking of pituitary macroadenomas causing acromegaly improves control by lanreotide. Clin Endocrinol. 2008;68(6):970–5.
Petrossians P, Borges-Martins L, Espinoza C, Daly A, Betea D, Valdes-Socin H, et al. Gross total resection or debulking of pituitary adenomas improves hormonal control of acromegaly by somatostatin analogs. Eur J Endocrinol. 2005;152(1):61–6.
Giustina A, Bonadonna S, Bugari G, Colao A, Cozzi R, Cannavo S, et al. High-dose intramuscular octreotide in patients with acromegaly inadequately controlled on conventional somatostatin analogue therapy: a randomised controlled trial. Eur J Endocrinol. 2009;161(2):331–8.
Giustina A, Mazziotti G, Cannavo S, Castello R, Arnaldi G, Bugari G, et al. High-dose and high-frequency lanreotide autogel in acromegaly: a randomized, multicenter study. J Clin Endocrinol Metab. 2017;102(7):2454–64.
Bolfi F, Neves AF, Boguszewski CL, Nunes-Nogueira VS. Mortality in acromegaly decreased in the last decade: a systematic review and meta-analysis. Eur J Endocrinol. 2018;179(1):59–71.
Dekkers OM, Biermasz NR, Pereira AM, Romijn JA, Vandenbroucke JP. Mortality in acromegaly: a metaanalysis. J Clin Endocrinol Metab. 2008;93(1):61–7.
Ayuk J, McGregor EJ, Mitchell RD, Gittoes NJ. Acute management of pituitary apoplexy—surgery or conservative management? Clin Endocrinol. 2004;61(6):747–52.
Kauppinen-Makelin R, Sane T, Reunanen A, Valimaki MJ, Niskanen L, Markkanen H, et al. A nationwide survey of mortality in acromegaly. J Clin Endocrinol Metab. 2005;90(7):4081–6.
Holdaway IM, Bolland MJ, Gamble GD. A meta-analysis of the effect of lowering serum levels of GH and IGF-I on mortality in acromegaly. Eur J Endocrinol. 2008;159(2):89–95.
Mercado M, Gonzalez B, Vargas G, Ramirez C, de los Monteros AL, Sosa E, et al. Successful mortality reduction and control of comorbidities in patients with acromegaly followed at a highly specialized multidisciplinary clinic. J Clin Endocrinol Metab. 2014;99(12):4438–46.
Holdaway IM, Rajasoorya RC, Gamble GD. Factors influencing mortality in acromegaly. J Clin Endocrinol Metab. 2004;89(2):667–74.
Chanson P, Maison P. Does attainment of target levels of growth hormone and insulin-like growth factor I improve acromegaly prognosis? Nat Clin Pract Endocrinol Metab. 2009;5(2):70–1.
Biermasz NR, van Thiel SW, Pereira AM, Hoftijzer HC, van Hemert AM, Smit JW, et al. Decreased quality of life in patients with acromegaly despite long-term cure of growth hormone excess. J Clin Endocrinol Metab. 2004;89(11):5369–76.
Arosio M, Reimondo G, Malchiodi E, Berchialla P, Borraccino A, De Marinis L, et al. Predictors of morbidity and mortality in acromegaly: an Italian survey. Eur J Endocrinol. 2012;167(2):189–98.
Claudia A, Landis Susan B, Masters Anna, Spada Ann M, Pace Henry R, Bourne Lucia, Vallar GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 1989;340(6236):692–96 10.1038/340692a0.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Maione, L., Chanson, P. (2022). Acromegaly. In: Tamagno, G., Gahete, M.D. (eds) Pituitary Adenomas. Springer, Cham. https://doi.org/10.1007/978-3-030-90475-3_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-90475-3_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-90474-6
Online ISBN: 978-3-030-90475-3
eBook Packages: MedicineMedicine (R0)