
Abstract
Pure MoO3 undergoes dehydration under helium stream between room temperature and 220 °C before being transformed into MoO2.87 and lost by evaporation at 650 °C. However, in the presence of NH4+–ZSM-5 zeolite (Mo/Al = 1), the dehydration of the mixture and the deammonization of the zeolite take place below 440 °C. In this water–rich atmosphere, a fraction of MoO3 is rehydrated and transformed into gaseous MoO2(OH)2 at 220–440 °C instead of being reduced. Despite the significant Mo weight loss, MoO2(OH)2 (g) reacts with ZSM-5 zeolite and is transformed into polymolybdate species upon 4 h of isothermal exchange at 500 °C. However, when the reaction time is prolonged from 4 to 8 h, the residual MoO3 undergoes an agglomeration before being transformed into mono- and dimolybdate species following 12 h of exchange. The determination of band gap energy by means of the diffuse reflectance spectroscopy allowed the identification of Mo species and the quantification of residual MoO3 (55% of the total Mo), which occupied the zeolite surface (46% of residual MoO3) and the internal cavities (9% of residual MoO3).
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Molybdenum (42Mo) exchanged zeolites represent one of the technologically most important category of solid catalysts. In fact, they have been reported to catalyze phenanthrene hydrogenation [1], ethylene carbonylation [2], methyl–hydroxypentadecanoate macrolactonization [3], methane dehydroaromatization [4, 5], C2H x (x = 4 and 6) ammoxidation [6], partial oxidation of i-butene [7], methanol conversion into gasoline [8], bitumen methanotreating [9], etc.
The most widely used method for the introduction of Mo into zeolites is the impregnation of the host structure with an ammonium heptamolybdate tetrahydrate solution [10, 11] followed by thermal treatments performed at different temperatures and extended for different time durations [12]. However, the introduction of molybdenum into zeolites by solid–state ion exchange (SSIE) is still a promising method [6, 11, 13]. Unfortunately, some authors [14, 15] pointed out that the SSIE of chlorinated Mo salts (e.g. MoOCl4 and MoCl5) into zeolites results in a substantial loss in crystallinity due to the attack by gaseous hydrogen chloride. Moreover, following the decomposition of ammonium heptamolybdate tetrahydrate under air [10], the diffusion of MoO3 crystallites into the ZSM-5 zeolite channels requires high temperatures (~ 500 °C), while the stabilization of Mo species with the framework oxygen occurs at 700 °C [10]. Such a stern thermal treatment would result in the extraction of Al ions from the framework with the subsequent formation of water and the disappearance of two Brönsted acid sites [16, 17]. Nonetheless, the solid–state ion exchange of organic precursors, e.g. Mo acetylacetonate, led to the formation of carbon at the surface of ZSM-5 zeolite, as covered by ourselves in a concise work [18], which shall not be reiterated here.
In the solid state, pure MoO3 is a suitable candidate for the introduction of Mo into ZSM-5 zeolite, and the studies dealing with the mechanistic aspects of solid–state ion exchange continue till date [19, 20]. Within the literature, MoO3 precursor is initially deposited on the external ZSM-5 surface and transformed to molybdenum oxo species, which are redistributed between the external surface and the internal channel structure [21]. Using Raman and X–ray absorption spectroscopies, Iglesia et al. [17, 22, 23] pointed out that the thermal activation of a physical mixture of MoO3 and ZSM-5 (Si/Al = 14.3) under dry air results in the formation of (MoO2(OH))+ species, which can condense with another identical moiety to form H2O and (Mo2O5)2+ dimer, which bridges two Al framework sites. Moreover, (MoO2(OH))+ species can also react with a zeolite OH group to form a (MoO2)2+ cation bridging two acid sites and water. In 2006, Li et al. [24] mentioned the formation of Mo5O126+ clusters upon the thermal treatment of ZSM-5 (Si/Al = 19) and MoO3 under nitrogen stream between room temperature and 500 °C. Besides these clusters, located at the ZSM-5 channel intersections, the X–ray diffraction (XRD) patterns revealed the presence of small MoO3 oxide particles on the external zeolite surface after calcination at 500 °C for 2 h [24]. Mosqueira et al. [25,26,27] placed particular emphasis on the thermal treatment of MoO3 in the presence of γ–alumina [25, 26], Y (Si/Al = 2.7) [25,26,27] and ZSM-5 (Si/Al = 37) [25, 26] zeolites at 450 °C under static air for different time durations (from 5 min to 24 h). These authors [25,26,27] have concluded that the migration of MoO x species onto Y and ZSM-5 zeolites stabilized a significant fraction of tetrahedral Mo species (monomers and dimers) at the detriment of octahedral Mo heptamers. Specifically, the UV/visible diffuse reflectance spectroscopy (DRS) analysis revealed that the heptameric species were provided from the polymerization of Mo monomers over ZSM-5, while dimeric species were directly formed from MoO3 [26]. However, the thermal treatment of MoO3 in the presence of Y zeolite [27] led to the formation of MoO2(OH)2 volatile species which favoured the formation of monomers, firstly, then dimeric species, while Mo heptamers originated from the condensation of dimers.
The controversy in this literature overview is clear and the exact structure of Mo species stabilized on the zeolite is yet to be resolved. This work would contribute to the enlightenment and the enrichment of the points described in previous studies, dealing with the thermal activation of MoO3 and zeolite in the solid state. We have placed particular prominence on the interaction between MoO3 and NH4+–ZSM-5 (Si/Al = 26) under inert atmosphere using numerous characterization techniques.
Experimental
Preparation of MoO3/NH4 +–ZSM-5 mixtures
The MoO3/zeolite mixture was prepared by manually mixing the zeolite powder (NH4+–ZSM-5, Si/Al = 26, from Zeolyst) with the desired quantity (theoretical molar ratio Mo/Al = 1, i.e. 6 wt% of Mo and 9 wt% of MoO3) of MoO3 (M = 143.94 g mol−1, from Merck). The prepared mixture was divided into three equal fractions. Each fraction was treated under helium stream from room temperature to 500 °C (2 °C min−1) then kept isothermally at 500 °C for a particular length of time (this period was denoted as x and expressed in hours). The samples were labelled as Ox–Z (x), i.e. Ox–Z (4 h), Ox–Z (8 h) and Ox–Z (12 h), where Z stands for the zeolite, while x is the duration of isothermal exchange at 500 °C (expressed in hours).
A reference sample was prepared by mixing NH4+–ZSM-5 with the desired quantity of MoO3 (6 wt% of Mo, i.e. 9 wt% of MoO3) and stored without undergoing thermal treatment. This reference material was labelled as Ox/Z (m.m.), where m.m. stands for mechanical mixture.
Characterization
Thermal analysis curves were recorded under nitrogen flow (Air Liquide 99.998%, 30 mL min−1) from 30 to 800 °C (from 30 to 700 °C in the case of NH4+–ZSM-5) with a 2 °C min−1 heating rate using an SDT Q 600 apparatus. The outlet gases were simultaneously analyzed by a quadruple mass spectrometer (Pfeiffer Vacuum, OmniStarTM GSD 301). The chemical analysis was performed by inductively coupled plasma–optical emission spectrometry (ICP–OES) on an Optima 3300 DV Perkin Elmer spectrometer. The morphology of the samples was studied by scanning electron microscopy (SEM) using a HITACHI 4800 S electron microscope, while the chemical composition was determined by energy dispersive X–ray (EDX). Textural analysis was performed by nitrogen physisorption at − 196 °C on an automatic ASAP 2020 apparatus from Micromeritics. XRD measurements were performed on a Siemens D–500 diffractometer with CuK α radiation (λ = 1.54060 Å), generator setting of 40 kV and 40 mA, a scanning speed of 0.2° min−1 and a scanning region of 2–70°. X–ray photoelectron spectroscopy (XPS) spectra were recorded with an Omicron photoelectron spectrometer (MgK α = 1256 eV, 150 W, check supplementary material, page 7). Temperature programmed reduction under hydrogen (H2–TPR) was performed with a Micromeritics Autochem 2920 analyzer after a pretreatment under air flow (30 mL min−1) at 450 °C for 1 h. The sample was cooled and flushed with helium at room temperature before being reduced from 50 to 850 °C (or 750 °C) at a heating rate of 5 °C min−1 with H2 (3%)/Ar flow (30 mL min−1). Similarly to our previous works [6, 28,29,30], the TPR profiles were decomposed manually into Gaussian peaks using Origin 8.0 (Microcal Software, Inc., U.S.). Temperature programmed desorption of ammonia (NH3–TPD) analysis was performed with the same H2–TPR equipment. The sample was first exposed to air flow (30 mL min−1) at 450 °C for 1 h then saturated with flowing ammonia at 100 °C and flushed with helium at the same temperature. Finally, the temperature was ramped to 700 °C (10 °C min−1). UV–visible DRS spectra were recorded at room temperature on a Perkin Elmer Lambda 45 spectrophotometer. More details on the experimental protocol have been previously reported [6, 31].
Results
Thermal analyses of NH4 +–ZSM-5, MoO3 and Ox/Z (m.m.) samples
Thermal analysis of NH4 +–ZSM-5
Derivative thermogravimetric (DTG) and the mass spectrometry (MS) curves obtained during the thermal treatment of NH4+–ZSM-5 zeolite (Si/Al = 26) under helium stream were previously presented in Ref. [18] and are reproduced in Fig. S1. The DTG curve (Fig. S1A) reveals the occurrence of different weight losses ascribed specifically to the dehydration and/or deammonization of the zeolite [18]. Interestingly, we noticed in Fig. S1B that the evolution of the signal intensity of H2O+ MS fragment (blue–coloured curve, with a maximum at 30 °C) does not overlap with the weight loss interval situated between 30 and 170 °C in the DTG curve (Fig. S1A). According to Prilipko et al. [32], the shift of MS curve (relative to H2O+) to the lower temperature region (in comparison with the DTG curve) is due to the fact that the reaction in Eq. 1 is displaced to the right.
The plots compiled in Fig. S1B indicate that gaseous ammonia (evacuated during the zeolite deammonization) is oxidized into N2 and H2O at 420 °C as was reported earlier [15, 18, 33]. However, the moderate weight loss situated above 520 °C (yellow grid in Fig. S1A, inset) is attributed to the partial dehydroxylation of the zeolite [18].
Thermal analyses of MoO3 and Ox/Z (m.m.) samples
TG–DTG and derivative differential scanning calorimetry (deriv. DSC) curves recorded during the thermal treatment of pure MoO3(s) and MoO3/NH4+–ZSM-5 mechanical mixture (i.e. Ox/Z (m.m.) sample) under helium stream are illustrated in Fig. 1. On the other hand, Figs. S2 and S3 represent the corresponding MS study results.

Thermal analysis curves of A pure MoO3 (from Merck), and B MoO3/NH4+–ZSM-5 mechanical mixture (6 wt% of Mo) samples treated under helium stream (30 mL min−1, 2 °C min−1)
According to Fig. 1A, pure MoO3 oxide exhibits a dehydration process between room temperature and ~ 220 °C with a weight loss of 5% (see the evolution of the signal intensity of H2O+ MS fragment in Fig. S2A). In this context, Murugan et al. [34] investigated the thermal analysis of hydrated Mo oxide and noticed a deviation in the baselines at around 390 °C in both DTA and DSC thermograms. These authors [34] ascribed this deviation to the transformations between the polymorphic phases of MoO3 (Eq. 2).
As a matter of fact, the ejected water molecules (Fig. S2A) would be initially present at the surface or inside the layers of Mo oxide and cannot originate from Eq. 2.
Following the dehydration of MoO3, Fig. 1A revealed a weight loss of 1.42% between 220 and ~ 615 °C, while Fig. S2B revealed the exit of oxygen. In their eminent work, Murugan et al. [34] reported that MoO3 is reduced into MoO3–δ above 400 °C (i.e. after the dehydration). Accordingly, δ value was estimated in our study to be 0.13, in agreement with Murugan et al. results (i.e. 0.09 < δ < 0.25 [34]).
The sudden weight loss observed at 650 °C in Fig. 1A would belong to the evaporation of MoO3 [18, 33] and probably MoO2.87 moiety.
In the case of Ox/Z (m.m.), Fig. 1B revealed four distinctive weight loss intervals. The first one, situated between room temperature and 220 °C, is related to the dehydration of both zeolite and salt (check the blue-colored curve in Fig. S3A). The second weight loss between 220 and 440 °C is associated with the desorption of ammonia (view the evolution of NH+ MS fragment in Fig. S3A) during zeolite deammonization. Between 440 and 650 °C, the observed weight loss would be associated with the exit of H2O (Fig. S3A). However, above 650 °C, MoO3 is lost by evaporation as revealed by the drastic decrease in the TG curve intensity (Fig. 1B), while the zeolite dehydroxylation occurs above 700 °C as revealed by the evolution of the signal intensity of H2O+ (pink gird in Fig. S3A).
It is worth to note that, throughout the 220–440 °C temperature range, the removal of water and oxygen is inhibited as confirmed by the evolution of H2O+ and O+ signal intensities in Fig. S3A and Fig. S3B, respectively.
Analysis of Ox–Z (x) mixtures treated isothermally at 500 °C
Chemical analyses
The chemical analyses (ICP and EDX) results associated with NH4+–ZSM-5 zeolite and Ox–Z (12 h) solid are given in Table 1.
Considering the theoretical Mo content (6 wt%), the pronounced weight loss following 12 h of isothermal exchange at 500 °C (38.80%, Table 1) would come from the evaporation of molybdenum, probably in the form of MoO2(OH)2(g). In fact, the sublimation of MoO3 is not envisaged below 650 °C on the basis of our thermal analysis results (Fig. 1A) and based on Mosqueira et al. [26] results, which reported that residual MoO3 is still detected by XRD even after 24 h of thermal treatment at 450 °C.
In our study, despite the significant weight loss, molybdenum is well dispersed at the zeolite surface after 12 h of isothermal exchange as revealed by the EDX study results (average Mo wt% = 6.62, Table 1).
SEM and nitrogen physisorption analyses
The micrographs of NH4+–ZSM-5 and Ox–Z (12 h) samples are illustrated in Fig. S5. According to the SEM images of Ox–Z (12 h) depicted in Fig. S5 (C and D), the shapes and the sizes (1 to 6 μm) of the zeolite crystals are similar to those observed in Fig. S5 (A and B). These results indicate that the solid–state ion exchange of Mo oxide does not affect the morphology of the zeolite crystals, even after 12 h of thermal treatment under helium stream at 500 °C.
The textural analysis results, expressed in terms of specific surface area (SBET), microporous and external areas (S μ and Sext), and (micro)porous volume(s) (V μ and Vp), are compiled in Table 2.
Additionally, the theoretical dispersion capacity (W) of MoO3 [35] inside the micropores of NH4+–ZSM-5 zeolite is included in Table 2. Considering the fact that the dynamic diameter of MoO3 is 5.0 Å [36, 37] and that the dimension of zeolite channel is 6 Å [35], it is therefore reasonable to believe that each MoO3 molecule could diffuse throughout the channels of NH4+–ZSM-5 and reach the internal cavities. Based on this postulation, the surface area occupied by each MoO3 molecule should be calculated by Eq. 3 [35].
Here d stands for the diameter of the zeolite channel, while l represents the distance occupied by MoO3 in the channel and A is the surface area (Å2) occupied by each MoO3 molecule [35].
According to Table 2, the internal surface area of NH4+–ZSM-5 zeolite is 291 m2g−1. Therefore, the total number of MoO3 molecules dispersed inside the micropores of one gram of zeolite should be calculated by Eq. 4.
Here S μ is the internal surface area of NH4+–ZSM-5 zeolite (Table 2) and N refers to the number of MoO3 molecules dispersed inside the micropores.
Taking into consideration Eqs. 3 and 4, the theoretical dispersion capacity (W) of MoO3 inside the micropores of NH4+–ZSM-5 would be calculated by Eq. 5.
In a similar way, W values were calculated in the case of Ox–Z (4 h), Ox–Z (8 h) and Ox–Z (12 h) solids (Table 2).
As reported in Table 2, the external area of NH4+–ZSM-5 zeolite exhibits a significant increase following 4 h of isothermal exchange at 500 °C. Such an increase in Sext value (from 76 to 132 m2 g−1) would be originated from the thermal treatment which might create some cracks, reduce the crystallites size and generate some meso/macroporosity following the exchange. Nevertheless, the SEM analysis results would exclude the massive cracking due to the similarity of the micrographs depicted in Fig. S5 (A and B) and S5 (C and D). Consequently, the change in the zeolite texture is ascribed to the presence of Mo.
The data compiled in Table 2 also revealed a pronounced decrease (26%) in the internal surface area of the zeolite following 4 h of isothermal exchange (i.e. Ox–Z (4 h) solid). This result indicates that MoO3 oxide diffuses throughout the internal cavities of the zeolite as there are no size restrictions (effectively, d = 6 Å and l = 5 Å). However, in the case of Ox–Z (4 h) and Ox–Z (8 h) samples, the theoretical dispersion capacity values are 0.054 and 0.061 g. Seemingly, the increase in W values would indicate a pronounced diffusion of MoO3 throughout the micropores between 4 and 8 h. Nevertheless, for Ox–Z (4 h) and Ox–Z (12 h) solids, the similarity in W value (~ 0.054 g, which corresponds to the nominal MoO3 quantity (i.e. 0.09 × (100 − 38.80) × 10−2 would discard the sublimation of MoO3 between 8 and 12 h of isothermal exchange.
XRD
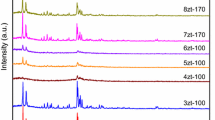
XRD patterns of pure α–MoO3 oxide and Ox–Z (x) samples are illustrated in Fig. 2.

XRD patterns of A MoO3/NH4+–ZSM-5 mechanical mixtures (6 wt% of Mo) treated from 30 to 500 °C (2 °C min−1) then isothermally at 500 °C for 4, 8 and 12 h under helium stream (30 mL min−1), and B pure α–MoO3. *Crystalline α–MoO3
The XRD patterns of MoO3/NH4+–ZSM-5 mixtures, illustrated in Fig. 2A, exhibits clear diffraction peaks at 2θ = 12.87, 25.78 and 39.10°. Based on the XRD analysis performed with pure MoO3 (Fig. 2B), these peaks would correspond to crystalline α–MoO3 (020), (040) and (060) planes [38].
Using the Scherrer formula illustrated in Eq. 6 [39] and the main diffraction peak (Fig. 2B, inset), we were able to determine the size of MoO3 particles.
Here D stands for the crystallite size, λ is the wavelength of the X–ray (0.154056 nm), β is the full width at half maximum (FWHM) of the Bragg peak (in radians) and θ is the diffraction angle. The results are compiled in Table 3 and Fig. S6.
According to Table 3, the D values determined in the case of pure MoO3 oxide and Ox–Z (4 h) solid do not differ significantly. However, D value increases from 68 to 114 nm between 4 and 8 h of isothermal exchange at 500 °C then decreases to 22 nm between 8 and 12 h. In line with these results, the agglomeration of MoO3 particles at the surface (and therefore the increase in D value) would occur between 4 and 8 h of isothermal exchange at 500 °C. Nonetheless, extending the thermal treatment between 8 and 12 h reduces the crystallite size (from 114 to 22 nm) and increases the dispersion state of Mo (EDX results, Table 1).
XPS
The Mo 3d XPS spectra of pure α–MoO3 and Ox–Z (x) solids are illustrated in Figs. S7 and S8, while the quantitative study results are summarized in Table 4.
In the XPS spectrum of pure oxide (Fig. S7A), the core level binding energy peaks corresponding to 3d5/2 and 3d3/2 are observed at 234 and 237.2 eV, respectively [6, 40]. However, the XPS spectra of Ox–Z (x) solids (Fig. S7) undergo a broadening in the Mo 3d line shape, which could be attributed to the presence of more than one type of Mo6+ specie [41]. The peak fitting analysis performed over the spectra of Ox–Z (4 h), Ox–Z (8 h) and Ox–Z (12 h) solids (check Fig. S8) revealed the presence of two additional components at ~ 232 and ~ 235 eV, which could be attributed to octahedral/tetrahedral Mo(VI) species [42]. In this context, Lede et al. [43] reported that molybdenum oxides may adopt different environments over ultrastable zeolite depending on the location of the anchoring site, being the tetrahedral structure, more or less distorted, is the most preserved one.
Interestingly, the XPS peak relative to bulk MoO3 increases in intensity and undergoes a shift from 233.5 to 234 eV between 4 and 8 h of isothermal exchange at 500 °C (check red and magenta curves in Fig. S7C). These results corroborate the increase in the D values from 68 to 114 nm (Table 3) and confirm the agglomeration of MoO3 oxide at the surface of Ox–Z (8 h) solid. However, between 8 and 12 h of isothermal exchange at 500 °C, the peak ascribed to bulk MoO3 decreases in intensity (check wine and magenta curves in Fig. S7C) despite the increase in the Mo concentration at the surface (from 9.60 to 12.08%, Table 4). These results are in line with the decrease in D values between 8 and 12 h and agree with the fact that MoO3 molecules do not diffuse throughout the micropores (check the decrease in W value from 0.061 to 0.056 g between 8 and 12 h of exchange, Table 2).
H2–TPR
The TPR profiles of pure MoO3 and Ox/Z (m.m.) samples are presented in Figs. S9 and S10. The shape of the TPR curves shows, at high temperatures, that the total reduction to Mo metal is not achieved at this stage. It is therefore possible that a small part of molybdenum species has not been reduced in the studied temperature range [44].
The TPR profile of pure MoO3 (Fig. S9A) exhibits two main regions of hydrogen consumption. The first region situated below 783 °C is related to the reduction of MoO3 into MoO2 [45]. However, the hydrogen consumption above 783 °C would correspond to the reduction of MoO2 into metallic molybdenum [45, 46]. It is worth noting that the formation of Mo4O11 intermediate during the reduction of MoO3 into MoO2 [46] is not excluded.
The peak fitting analysis performed over the TPR curve of Ox/Z (m.m.) sample (see Fig. S9B) revealed the presence of three peaks at 550, 585 and 640 °C. Given that a significant fraction of MoO3 crystallites would occupy the external surface of the zeolite following the short TPR pretreatment (1 h at 450 °C), these peaks would correspond to the reduction of MoO3 crystallites dispersed at the surface. However, the peaks situated at 710, 740, 800 and 840 °C in Fig. S9B would belong to the reduction of Mo4O11 into MoO2 or MoO2 into metallic molybdenum [46].
Given that the reduction of the zeolite dehydroxylation–products and the evaporation of MoO2 are not discarded, the deep interpretation of the high–temperature region should be avoided.
The peak fitting analyses performed over the TPR curves of Ox–Z (x) solids are presented in Fig. 3 (see also Fig. S10), while Table 5 compiles the quantitative study results.

H2–TPR profiles of MoO3/NH4+–ZSM-5 mechanical mixtures (6 wt% of Mo) treated from 30 to 500 °C (2 °C min−1) then isothermally at 500 °C for 4, 8 and 12 h under helium stream (30 mL min−1)
The TPR profile of Ox–Z (4 h) solid exhibits four peaks at 480, 520, 540 and 575 °C (dashed lines in Fig. 3, check also Table 5) ascribed to the reduction of MoO3 as will be later confirmed by XPS. However, the peaks at 350 and 440 °C (solid–lines) would correspond to the reduction of easy–reducible surface species other than MoO3 oxide, while the peak at 730 °C (dotted line in Fig. 3) belongs to the reduction of MoO2 into Mo (or Mo4O11 into MoO2 [46]). In the case of Ox–Z (8 h) solid, the easily reduced surface species exhibit three reduction peaks at 220, 310 and 385 °C (Table 5), while MoO3 is reduced at 480 and 590 °C. For Ox–Z (12 h) solid, the reduction of MoO3 occurs at 500 and 555 °C, while the peaks centred at 670, 810 and 840 °C (dotted–lines in Fig. 3) have been previously ascribed to the reduction of MoO2 or Mo4O11 [46]. Nevertheless, the peak centred at 610 °C would belong to the reduction of tetrahedral Mo6+ species [6].
In order to verify the previous attribution of the reduction peaks, we calculated for each sample the percentage of MoO3 using Eq. 7 (see more details in Table S1).
It is worth noting that the dashed–line peaks in Fig. 3 correspond to the reduction of MoO3, while the total area is the sum of the areas under the dashed line and solid line peaks. In Fig. S11, we represented the percentage of MoO3 determined by TPR versus its homologue determined by XPS. The linear plot presented in Fig. S11 corroborates the identification of reduction peaks, previously adopted.
Optical spectroscopy study
In this study, we explored the absorption band gap using the Shuster–Kubelka–Munk function, F(R∞), as given in Eqs. (8–10) [47].
Here R∞ is the diffuse reflectance of an infinitely thick sample, n is an exponent, hν is the photon energy and Eg is the optical energy gap of the material. The exponent n can take the values of 2, 3, 1/2 and 3/2 for indirect allowed, indirectly forbidden, directly allowed and directly forbidden transitions [47]. Plotting [F(R∞) × hν]1/n versus hν and extrapolating to [F(R∞) × hν]1/n = 0 (in Eq. 10) yields the value of Eg.
In the case of MoO3, only n = 1/2 (i.e. direct allowed transition) [26, 31, 48, 49] and n = 2 (i.e. indirect allowed transition) [50,51,52,53] have been reported in the literature.
For pure MoO3 and Ox/Z (m.m.) samples, the plots corresponding to either [F(R∞) × hν]2 or [F(R∞) × hν]0.5 versus hν are presented in Fig. 4. Additional functions, i.e. [F(R∞) × hν]1/3 and [F(R∞) × hν]2/3 versus hν were depicted in Figs. S12 and S13.

Optical absorption spectra of pure MoO3 and MoO3/NH4+–ZSM-5 fresh mechanical mixture (6 wt% of Mo, 9 wt% of MoO3): A [F(R∞) × hν]2 versus hν, and B [F(R∞) × hν]0.5 versus hν curves
In the case of the direct allowed transition (n = 1/2), i.e., the plot representing [F(R∞) × hν]2 vs. hν (Fig. 4A), we determined the area under the curve ascribed to Ox–Z (m.m.) to be 3.24 a.u.. However, the area under the curve attributed to pure MoO3 is 55.62 a.u.. The ratio of these areas (i.e. (3.24/55.62) × 100) does not correspond to the theoretical percentage of MoO3 in the mixture (i.e. 9%). In a similar way, and using the plots in Fig. 4B (representing the indirect allowed transition (n = 2, i.e. [F(R∞) × hν]0.5 vs. hν), the area ratio (i.e. (0.26/2.78) × 100) was calculated to be 9.4%, which is close to the theoretical MoO3 wt% in the mixture. Nevertheless, in the case of forbidden transitions (indirect transition for n = 3 and direct transition for n = 3/2), the area ratios for pure and diluted MoO3 do not correspond to 9% (Figs. S12 and S13). Consequently, it is concluded that the function [F(R∞) × hν]0.5 satisfactorily describes our system.
Figs. 5 and S14 represent the deconvolution of the DRS spectrum of Ox–Z (12 h) sample (see Ref. [31] for more details), while Table 6 summarizes the corresponding quantitative study results.

UV absorption edges of MoO3/NH4+–ZSM-5 mechanical mixture (6 wt% of Mo, 9 wt% of MoO3) treated from 30 to 500 °C (2 °C min−1) then isothermally at 500 °C for 12 h under helium stream (30 mL min−1)
Based on the band gap analysis and the deconvolution of the spectrum relative to Ox–Z (12 h) solid, four O2− → Mo6+ charge transfer bands could be distinguished (Fig. 5). These ligand to metal charge transfer bands were assigned to MoO3 (Eg = 2.94 eV), [Mo7O24]6− (Eg = 3.65 eV), [Mo2O7]2− (Eg = 4.70 eV) and [MoO4]2− (Eg = 5.56 eV) [48]. However, despite the significant Mo weight loss (38.80%, Table 1) following 12 h of isothermal exchange at 500 °C, the area below the four bands in Fig. 5 (3.40 a.u.) exceeds the value obtained with pure and diluted MoO3 (2.78 and 0.26 a.u., Fig. 4B). This is predictable due to the fact that monomolybdate [MoO4]2−, dimolybdate [Mo2O7]2− and heptamolybdate [Mo7O24]6− species would exhibit different absorption coefficients (similarly to bare Co2+ ions [54] stabilized in BEA zeolite) and this is the reason for which we quantify only the MoO3 component.
Mosqueira et al. [26] identified different surface molybdate species over ZSM-5 zeolite by using DR measurements in the UV–visible region and carried out a band gap analysis of the deconvoluted spectra. Nevertheless, Mosqueira’s study [26] only provides a semi–quantitative measure of the Mo oxo–species surface concentrations. Herein, we extrapolated our DRS measurements to a fully quantitative study by plotting the appropriate function (i.e. [F(R∞) × hν]0.5).
Discussion
The thermogravimetric analysis results (Figs. 1A, S2A and S4) revealed that unsupported MoO3 is dehydrated under helium stream between room temperature and 220 °C (weight loss = 5%). Similarly to Murugan’s study [34], the desorbed water molecules do not evolve from the dehydration of MoO3 polymorphs (i.e. monoclinic MoO3·2H2O, monoclinic and triclinic monohydrates (MoO3·H2O), monoclinic hemihydrate (MoO3·0.5H2O) and orthorhombic MoO3· 1/3 H2O [55]) given in Eq. (2) since we were not able to detect baseline deviations in our thermal analysis curves.
Following the dehydration, MoO3 losses oxygen atoms (Fig. S2B) between 220 and 615 °C (weight loss = 1.42%) and transforms into MoO3–δ (δ = 0.13). However, at 650 °C, unreduced MoO3 evaporates (Δ sub H = 276.56 kJ mol−1, Δ sub S = 300 J K−1mol−1, Δ sub G = – 3000 J mol−1 [18]) as revealed by the sudden weight loss in Fig. 1A and by the derivative DSC curve in Fig. S4.
In the case of the mixture, the thermogravimetric analysis coupled to mass spectrometry (Figs. 1B and S3A) revealed that the dehydration takes place below 220 °C. However, gaseous ammonia evolved between 220 and 440 °C as revealed by the evolution of the signal intensity of NH+ MS fragment (red curve in Fig. S3A). It is worth noting that the weight loss between 220 and 440 °C (0.84%, i.e. 0.5 mmol of NH3 per gram, determined by NH3–TPD) is in agreement with the Al wt% determined by ICP–OES (Table 1). Therefore, we concluded that the dehydration of the mixture was inhibited between 220 and 440 °C (check also the blue curve in Fig. S3A).
Between 220 and 440 °C, H2O and MoO3 were transformed into gaseous MoO2(OH)2 according to Eq. 11.
It is a noticeable fact that, in this water–rich atmosphere, solid MoO3 is not reduced into MoO3–δ’ (δ ≠ δ’) following the dehydration since the MS study results (Fig. S3B) excluded the formation of gaseous oxygen.
Between 440 and 600 °C, the non–isothermal ion exchange of the volatile MoO2(OH)2 into H+–ZSM-5 is marked by the exit of H2O (Fig. S3A, grey gird) during the subsequent steps illustrated in Scheme S1. However, the exchange process, which involves the formation of H2O [18], is achieved at 500 °C as indicated by the decrease in both H2O+ signal intensity (see the yellow arrow in Fig. S3A) and DSC signal in Figs. 1B and S4.
The partial dehydroxylation of the zeolite occurs at 520 °C (yellow gird in Fig. S1A inset), while MoO3 evaporates at 650 °C. Accordingly, we performed the isothermal solid–state exchange of MoO3 into ZSM-5 at 500 °C in order to avoid the zeolite dealumination and the loss of Mo.
Following 4 h of isothermal exchange at 500 °C (i.e. Ox–Z (4 h) solid), the theoretical dispersion capacity of MoO3 (W) inside the zeolite micropores has been estimated to be 0.054 g (Table 2, last column). This theoretical value stands for the quantity of MoO3 remaining after the weight loss (i.e. 0.09 g MoO3 × (100 − 38.8) × 10−2). As a matter of fact, MoO2(OH)2 is lost by evaporation between 220 and 440 °C since MoO3 sublimates at 650 °C.
Between 4 and 8 h of isothermal exchange, the increase in W value (from 0.054 to 0.061 g) would ostensibly indicate the diffusion of MoO3 inside the zeolite micropores. However, the similar W values for Ox–Z (4 h) and Ox–Z (12 h) solids would exclude the sublimation of MoO3 between 8 and 12 h.
During the isothermal exchange between 4 and 8 h, we noticed the formation of MoO3 agglomerates inside the zeolite bulk (revealed by the increase in D values from 68 to 114 nm, Table 3) and at the surface (the increase in the bulk MoO3 percentage from 68 to 79%, Table 4, and in the MoO3 XPS peak intensity, Fig. S7C). However, between 8 and 12 h, the size of MoO3 crystallites decreased from 114 to 22 nm. In this context, we were able to exclude, on one hand, the diffusion of MoO3 inside the zeolite channels between 8 and 12 h (since W value decrease from 0.061 to 0.056 g) and the sublimation of MoO3 (similar W values for Ox–Z (4 h) and Ox–Z (12 h) solids) on the other hand. Seemingly, within this period (8–12 h), MoO3 is transformed into octahedral and tetrahedral Mo(VI) species at the surface, leading to the increase in Mo surface concentration (from 9.60 to 12.08%, Table 4) and the decrease in the MoO3 XPS peak intensity (Fig. S7C).
The identification of octahedral and tetrahedral Mo(VI) species was carried out by H2–TPR. In effect, during the isothermal exchange at 500 °C for up to 4 h, gaseous MoO2(OH)2(g) species were transformed into mono– and dimeric species according to the processes described in Scheme S1. However, these tetrahedral species, not yet stabilized at this stage, were transformed into heptamolybdates. According to the XPS study results, the concentration of heptamolybdate species at the surface of Ox–Z(4 h) solid is 32% (Table 4), and they were reduced under hydrogen at 350 and 440 °C (Fig. 3 and Table 5).
Between 4 and 8 h, mono– and dimolybdate species were not yet stabilized. Essentially, the H2–TPR analysis of Ox–Z (8 h) solid (Fig. 3 and Table 5) revealed, on the one hand, the reduction of MoO3 agglomerates (D = 144 nm) at 480 and 590 °C and heptaolybdate species at 220, 310 and 385 °C, on the other hand. Moreover, the decrease in the concentration of surface species (from 32 to 21%, Table 4) is due to the fact that a fraction of MoO3 diffuses inside the zeolite channels (increase in W value, Table 2) and is transformed into internal heptamolybdate species, inaccessible to the XPS beam.
Between 8 and 12 h, the concentration of MoO3 at the surface decreases from 79 to 46 at.%. At this stage, mono–and dimolybdates species were stabilized as revealed by the band gap analysis performed over Ox–Z (12 h) solid (Figs. 5 and S14). On the other hand, the H2–TPR results evidenced the reduction of the monomolybdates at 610 °C (Fig. 3, Table 5). Concerning MoO3, the percentages of residual oxide determined by DRS (Table 6) and XPS (Table 5) are similar (46%), while the value determined by TPR–H2 is 55% (see Table S1). This result indicates that 9% of the total MoO3 occupied the internal channels, unable to be reached by the X–ray and UV beams.
In summary, the results of this study would exclude the formation of Mo5O126+ clusters [24] (at least when using NH4+–ZSM-5, Si/Al = 26). However, until MoO2(OH)2 is not yet formed, the direct transformation of MoO3 into dimeric Mo is not envisaged. Seemingly, MoO2(OH)2 is condensed into polymolybdates in the first place and then into dimolybdates, while monomolybdates occupy, finally, the available sites.
Conclusion
Unsupported α–MoO3 is dehydrated then reduced into MoO3−δ (δ = 0.13) between room temperature and 615 °C before being lost by sublimation at 650 °C. However, in the presence of the hydrated NH4+–ZSM-5 zeolite (Si/Al = 26), the water molecules transformed a fraction of MoO3 into gaseous MoO2(OH)2 between 220 and 440 °C. During 4 h of isothermal exchange at 500 °C, gaseous MoO2(OH)2 species reacted with the zeolite (already in its protonated form), leading to the formation of unstable mono– and dimeric species. The formed species were then transformed into surface heptamolybdates. However, between 4 and 8 h, residual MoO3 is first agglomerated at the surface and then diffused throughout the internal cavities. The stabilization of monomolybdates and dimeric species in the cationic sites takes place within 8–12 h at 500 °C. Following 12 h of exchange we noticed that 55% of the total Mo occupied the zeolite surface and the micropores instead of being transformed into mono–, di– and polymeric species. In this study, the calculation of MoO3 theoretical dispersion capacity improved the comprehension of the solid–state exchange of MoO3 into ZSM-5 zeolite, while the DRS experiments were extrapolated to a fully–quantitative study using the [F(R∞) × hν]0.5 function and a bunch of reference materials.
References
Fu W, Zhang L, Wu D, Xiang M, Zhuo Q, Huang K, Tao Z, Tang T (2015) J Catal 330:423–433
Yang CC, Yacob S, Kilos BA, Barton DG, Weitz E, Notestein JM (2016) J Catal 338:313–320
Lai F, Liu X, Li W, Shen F (2010) React Kinet Mech Catal 100:407–415
Toosi MR, Sabour B, Hamuleh T, Peyrovi MH (2010) React Kinet Mech Catal 101:221–226
Kosinov N, Coumans FJAG, Uslamin EA, Wijpkema ASG, Mezari B, Hensen EJM (2017) ACS Catal 7:520–529
Mannei E, Ayari F, Petitto C, Asedegbega-Nieto E, Guerrero-Ruiz AR, Delahay G, Mhamdi M, Ghorbel A (2017) Microporous Mesoporous Mater 241:246–257
Corma A, Cortés Corberàn V, Fornés V (1986) React Kinet Catal Lett 32:191–197
Zhu JH (1997) React Kinet Catal Lett 62:39–46
Zhao L, He P, Jarvis J, Song H (2016) Energy Fuels 30:10330–10340
Xu Y, Liu S, Guo X, Wang L, Xie M (1995) Catal Lett 30:135–149
Weckhuysen BM, Wang D, Rosynek MP, Lunsford JH (1998) J Catal 175:338–346
Ismagilov ZR, Matus EV, Tsikoza LT (2008) Energy Environ Sci 1:526–541
Li S, Zhang C, Kan Q, Wang D, Wu T, Lin L (1999) Appl Catal A 187:199–206
Dai PSE, Lunsford JH (1980) J Catal 64:173–183
Ayari F, Mannei E, Asedegbega-Nieto E, Mhamdi M, Guerrero-Ruiz AR, Delahay G, Ghorbel A (2017) Thermochim Acta 655:269–277
Liu W, Xu Y, Wong ST, Wang L, Qiu J, Yang N (1997) J Mol Catal A 120:257–265
Borry RW III, Kim YH, Huffsmith A, Reimer JA, Iglesia E (1999) J Phys Chem B 103:5787–5796
Mannei E, Ayari F, Asedegbega-Nieto E, Mhamdi M, Guerrero-Ruiz AR, Delahay G, Ghorbel A (2017) Thermochim Acta 652:150–159
Zhang Z, Zhang Q, Jia L, Wang W, Gao X, Gu Y, Gao X, Han Y, Tan Y (2016) Chem Sel 1:6127–6135
Kosinov N, Coumans FJAG, Li G, Uslamin E, Mezari B, Wijpkema ASG, Pidko EA, Hensen EJM (2017) J Catal 346:125–133
Burns S, Hargreaves JSJ, Stockenhuber M, Wells RPK (2007) Microporous Mesoporous Mater 104:97–102
Li W, Meitzner D, Borry RW III, Iglesia E (2000) J Catal 191:373–383
Lacheen HS, Iglesia E (2005) Phys Chem Chem Phys 7:538–547
Li B, Li S, Li N, Chen H, Zhang W, Bao X, Lin B (2006) Microporous Mesoporous Mater 88:244–253
Mosqueira L, Fuentes GA (2002) Mol Phys 100:3055–3057
Mosqueira L, Gómez SA, Fuentes GA (2004) J Phys 16:S2319–S2327
Mosqueira L, Angeles-Chavez C, Torres-García E (2011) Mater Chem Phys 126:930–937
Ayari F, Mhamdi M, Delahay G, Ghorbel A (2009) J Sol Gel Sci Technol 49:170–179
Ayari F, Mhamdi M, Debecker DP, Gaigneaux EM, Álvarez-Rodríguez J, Guerrero-Ruiz AR, Delahay G, Ghorbel A (2011) J Mol Catal A 339:8–16
Áyari F, Mhamdi M, Alvarez-Rodríguez J, Guerrero-Ruiz AR, Delahay G, Ghorbel A (2013) Appl Catal B 134–135:367–380
Mannei E, Ayari F, Mhamdi M, Almohalla M, Guerrero Ruiz AR, Delahay G, Ghorbel A (2016) Microporous Mesoporous Mater 219:77–86
Prilipko AI, Il’in VG, Turutina NV, Nazarenko VA, Mel’nichenko GN (1990) Theor Exp Chem 25:467–470
Ayari F, Mannei E, Asedegbega-Nieto E, Mhamdi M, Guerrero-Ruiz AR, Delahay G, Ghorbel A (2017) J Therm Anal Calorim. https://doi.org/10.1007/s10973-017-6545-4
Murugan R, Ghule A, Bhongale C, Chang H (2000) J Mater Chem 10:2157–2162
Xiao FS, Zheng S, Sun J, Yu R, Qiu S, Xu R (1998) J Catal 176:474–487
Wells AF (1975) Structural inorganic chemistry, 4th edn. Oxford University Press, London
Shriver DF, Atkins PW, Langford CH (1990) Inorganic chemistry. Freeman, New York
JCPDS data file No 05–0508
Patterson AL (1939) Phys Rev 56:978–982
Hussain OH, Rao KS (2003) Mater Chem Phys 80:638–646
Zingg DS, Makovsky LE, Tischer RE, Brown FR, Hercules DM (1980) J Phys Chem 84:2898–2913
Naumkin AV, Vass AK, Gaarenstroom SW, Powell CJ (2012) XPS database, NIST Standard Reference Database 20, Version 4.1, 2012
Lede EJ, Requejo FG, Pawelec B, Fierro JLG (2002) J Phys Chem B 106:7824–7831
Arnoldy P, Franken MC, Scheffer B, Moulijn JA (1985) J Catal 96:381–395
Arnoldy P, de Jonge JCM, Moulijn JA (1985) J Phys Chem 89:4517–4526
Bhaskar T, Reddy KR, Kumar CP, Murthy MRVS, Chary KVR (2001) Appl Catal A 211:189–201
Khan GA, Hogarth CA (1991) J Mater Sci 26:412–416
Weber RS (1995) J Catal 151:470–474
Braun S, Appel LG, Schmal M (2002) Appl Surf Sci 201:227–235
Anwar M, Hogarth CA (1988) Phys Stat Sol A 109:469–478
Donnadieu A, Davazoglou D, Abdellaoui A (1988) Thin Solid Films 164:333–338
Anwar M, Hogarth CA (1989) J Mater Sci 24:3673–3678
Chithambararaj A, Sanjini NS, Velmathi S, Chandra Bose A (2013) Phys Chem Chem Phys 15:14761–14769
Essid S, Ayari F, Bulánek R, Vaculík J, Mhamdi M, Delahay G, Ghorbel A (2017) Catal Today. https://doi.org/10.1016/j.cattod.2017.08.027
Seguin L, Figlarz M, Cavagnat R, Lassègues JC (1995) Spectrochim Acta Part A 51:1323–1344
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ayari, F., Mannei, E., Asedegbega-Nieto, E. et al. More insight on the isothermal spreading of solid MoO3 into ZSM-5 zeolite. Reac Kinet Mech Cat 124, 419–436 (2018). https://doi.org/10.1007/s11144-018-1357-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-018-1357-5