
Abstract
Non-thermal plasma (NTP) driven catalytic decomposition of gas-phase benzene under oxygen plasma was carried out at room temperature over silver on titanium dioxide catalysts. Three preparation methods were investigated as well as the effect of silver loading amount. The catalysts were prepared using impregnation method (IP), deposition–precipitation method with NaOH (DPN), and deposition–precipitation method with H2O2 (DPH) with 1, 3 and 5 % silver loading by weight. The catalysts were characterized using XRD, XPS, SEM–EDS and N2 adsorption, and the catalytic activity was tested on 200 ppm benzene. Low energy treatment by NTP alone only showed insignificant change in benzene concentration but upon the addition of catalysts, IP catalysts were able to decompose the pollutant up to 20 % and with DPN and DPH catalysts up to 100 %. DPN catalysts were also able to decompose the ozone produced in the reactor. The 3 % Ag/TiO2 prepared by DPN method showed the highest energy efficiency capable of decomposing benzene at 392 g/kWh.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Recently, raising environmental awareness and promoting environmental protection is becoming an issue of growing concern as the world is becoming more polluted. Industrialization has led to the emission of various substances in our atmosphere that pose significant threats to both human and ecological life.
Emissions by industrial, commercial and agricultural processes are major sources of air pollution and are a concern for human health and the environment. One major group of these pollutants are volatile organic compounds (VOCs). Depending on their structure and concentration, VOCs together with nitrogen oxides and sulfur containing compounds reacts in a series of complex chemical reactions once released in the troposphere. These lead to the formation of secondary pollutants such as photochemical smog, secondary aerosol, suspended particulate matter, tropospheric ozone, acid deposition, and formation of organosulfuric compounds [1–3].
Conventional methods dealing with VOCs can be classified as either recovery or destruction of the pollutant. Recovery includes the use of technologies such as absorption, adsorption, condensation, and separation and destruction, this usually involves thermal and catalytic oxidation, and bio-filtration. However, the problem with these methods is that they are only cost effective for mid-to-high concentrations and are usually energy intensive [2].
Benzene was chosen as a representative VOC due to its wide use in many industrial processes primarily as a raw material, additive in gasoline and as a solvent. Industrial processes, its presence in gasoline, and its use in the production of ethylbenzene and styrene are major emission sources into waste streams and to the atmosphere. Exposure to benzene can cause drowsiness, dizziness, confusion and unconsciousness. Long-term exposure can lead to adverse effects such as anemia, leukemia, blood diseases, and cancer [4]. Also, compared to other aromatic compounds, benzene has low reactivity. The reaction rate coefficients of aromatic compounds to OH radicals (in 10−12 cm3 molecule−1 s−1) are in the following order: benzene (1.2) < toluene (5.96) < ethylbenzene (7.1) < o-xylene (13.7) < p-xylene (14.3) < m-xylene (23.6) at 298 K [5].
Non-thermal plasma (NTP), such as those produced in dielectric barrier discharge (DBD), corona discharge, surface discharge, and packed-bed plasma reactor, defined by the non-equilibrium characteristic between electrons and particles including ions, radicals, and neutrals, has recently been investigated for VOC abatement [1–3]. The most widely used method for generating NTP is by the application of an external electric field between two electrodes surrounded by a volume of gas and this is achieved by application of a sufficiently high voltage across the electrodes. The breakdown voltage defines the minimum voltage required to break down a gas to form a plasma discharge. At the point where the breakdown voltage is reached, the current flow will increase sharply due to an intensive avalanche of electrons in the discharge gap between the electrodes. These highly energetic electrons will collide with gas molecules leading to the formation of new active plasma species which includes excited molecules and atoms, radicals, ions and new stable gas molecules [6].
NTP has promising applications due to its low power requirement and its capacity to induce physical and chemical reactions with gases at low temperatures. Compared with high-temperature technologies such as combustion and catalytic oxidation, NTP can be operated at room temperature thus removing the need for warm-up time. NTP can be applied to a wide range of VOC concentrations up to 10,000 ppm, and is especially effective for concentrations lower than 100 ppm, in which conventional methods are no longer appropriate [2, 7]. The electrons within the discharge can reach temperatures ranging from 10,000 K to 100,000 K, while the bulk gas temperatures can remain as low as room temperature. It is the mixture of high temperature electrons, highly excited atoms and molecules, ions, radicals, and photons that are generated in NTP that is used in VOC decomposition [8–10].
However, energy efficiency (EE) still needs to be improved for further industrial application. To address these problems, NTP catalysis is being investigated. This combination of NTP and catalysis combines the advantages from these technologies, the fast ignition and response from the NTP and the high selectivity from catalysis [11–15].
Non-thermal plasma driven catalysis (PDC) has been researched as an alternative technology and has shown great potential in decomposing VOCs. This has been studied on various types of catalysts such as zeolite, noble metal, supported metal and metal oxide catalysts. Silver on titanium dioxide has repeatedly shown great activity toward VOC decomposition at a wide range of energy densities raging from several hundreds to a thousand. The addition of small amounts of silver on titanium dioxide significantly increased the decomposition efficiency as well as the selectivity towards carbon dioxide [1, 2, 10].
In PDC, active chemical species are generated from the excitation of electrons and various molecules that form short- and long-lived radical species, and in the case of oxygen, atomic oxygen and ozone. These species promote the oxidation of pollutants over the catalyst surface at room temperature where normally oxidation would not occur. Active oxygen species from catalytic decomposition of O3 are considered to be mainly responsible for the promotion effects of VOC oxidation in PDC [16, 17].
As NTP driven catalysis systems are mainly concerned with gas-phase and surface-phase reactions, this study aims to investigate the effect of different preparation methods of Ag/TiO2 with varying Ag loading on the decomposition of gas-phase benzene in a PDC system operated at low energy range under oxygen plasma. Three catalyst preparation methods for Ag/TiO2 at 1, 3 and 5 % silver loading, catalyst properties, conversion of benzene, ozone generation and decomposition, and EE were investigated.
Experimental
Catalyst preparation
The Ag/TiO2 catalysts were prepared by three methods: impregnation (IP), deposition–precipitation with sodium hydroxide (DPN), and deposition–precipitation with hydrogen peroxide (DPH) with varying silver loading of 1, 3 and 5 % by weight. Anatase type TiO2 pellets were obtained commercially from Sakai Chemical Industry Co., Ltd., Japan and were used as the base catalyst. The pellets were sieved through various mesh sizes and pellets with diameters between 1.4 and 1.7 mm were used. All other reagents used were analytical grade and used without further purification.
In the IP method, silver nitrate (99.8 % AgNO3, Wako Ltd.) was dissolved in pure water and was added to a beaker with TiO2 and aged for 24 h under room temperature before drying in an oven at 100 °C. The dried catalyst was then calcined at 500 °C for 5 h under air flow. In the DPN method, the same aqueous precursor solution was used and was added to TiO2 in a beaker, heated to 80 °C then 0.5 M NaOH was added dropwise to the mixture under vigorous stirring until a pH = 9 was reached. The mixture was continuously stirred for 2 h at 80 °C and then repeatedly filtered and washed until a pH of 7 was obtained before drying in an oven at 100 °C. In the DPH method, silver chloride (99 % AgCl, Sigma Aldrich) was dissolved in ammonia solution to form a silver diammine complex and this was added to a beaker containing TiO2. The mixture was stirred for 30 min and then 2 % by weight H2O2 was added dropwise under vigorous stirring to facilitate silver precipitation. The mixture was stirred for another 30 min before repeated filtering and washing until a pH of 7 was reached and finally, dried in an oven at 100 °C.
Catalyst characterization
The catalysts were characterized by powder X-ray diffraction (XRD) using a Rigaku diffractometer with Cu Kα radiation. X-ray photoelectron spectra (XPS) were recorded using a ULVAC-PHI 1600 fitted with a monochromated Al Kα source at 1486.6 eV. The anode was operated at 300 W and the analyzer at a constant pass energy of 23.5 eV with a resolution of 0.025 eV. The binding energy shifts due to surface charging were corrected using the C 1 s level at 284.8 eV from adventitious carbon. The XPS peaks were resolved assuming Lorentzian–Gaussian line profiles and a Shirley function was applied for background correction. Catalyst surface morphology and element distribution were scanned by a JEOL JSM-5310LV scanning electron microscope with energy dispersive X-ray spectroscopy (SEM–EDS). Catalyst surface area, pore volume and diameter were determined by BET method from N2 adsorption isotherm at 77 K using Autosorb-1 gas sorption analyzer.
Experimental set-up and PDC reactor
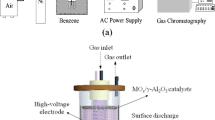
Continuous flow-type experiments were carried out using a plasma-driven catalysis (PDC) system. The experimental set-up used for the activity experiments is shown in Fig. 1. The bubbling method was used to set the benzene concentration in the gas stream and the concentration was controlled either by the flow rate of oxygen gas into liquid benzene or by adjusting the temperature of the water bath. Total feed flow rate was controlled using oxygen as balance gas and was set at 550 ml/min at standard conditions. The resulting gas feed was flowed through a mixing tank before entering into the DBD reactor. The corresponding gas hourly space velocity (GHSV) in the PDC reactor was 26,750 h−1 and was calculated based on the gas residence time in the void of the catalyst bed which was 0.35.

Schematic diagram of experimental setup
Activity tests were performed by varying the voltage applied to the reactor and effluent composition were analyzed using a gas chromatograph-mass spectrometer (GCMS-PQ5050A, Shimadzu Corp.) equipped with an Equity-5 column and an ozone gas detection tube system (Ozone 182SA&B, Kitagawa Komyo Rikagaku Kogyo).
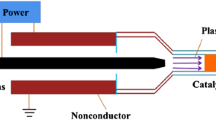
The DBD reactor (Fig. 2) was made of a quartz tube with an outside diameter of 15 mm, length of 200 mm and thickness of 1 mm. A 9 mm diameter stainless steel rod was fixed inside the center of the quartz to function as a high-voltage electrode and silver paste was coated outside the reactor as the ground electrode. The silver paste was coated at a length of 50 mm on the center of the quartz tube to make the effective length of the reactor. The Ag/TiO2 catalysts were packed within the effective length of the reactor during experiments.

Schematic diagram of the DBD reactor
Electrical measurements
AC high voltage was supplied by a neon transformer (LECIP Co. Ltd, 100-A-15HCS) connected to a volt slider and a 50 Hz 100 V AC power source. The applied high voltage, V, was measured with a 1000:1 high voltage probe (Agilent Technologies, N2771B) and the charge, Q (i.e. time-integrated current), was determined by measuring the voltage across a 10 nF capacitor connected in series with the ground line of the reactor. The signals were recorded by an oscilloscope (GW Instek, GDS-1072A-U) by averaging 64 scans. The discharge power was calculated using V-Q Lissajous figure method. The area of the plotted V-Q parallelogram was multiplied by the frequency to get the discharge power and the specific input energy (SIE) by dividing the discharge power with the gas flow rate. Energy efficiency was calculated by dividing the amount of decomposed pollutant by the input energy used and corrected with the necessary conversion units.
Results and discussion
Characterization of Ag/TiO2 catalysts
Bulk crystal structure was analyzed by XRD and Fig. 3 shows the XRD diffractograms for TiO2 and 5 % Ag/TiO2 prepared by different methods. All prepared catalysts retained its anatase crystal structure. Silver or any of its oxides were not seen in the XRD diffractograms due to the small amount loaded on TiO2.

XRD diffractograms of TiO2 and 5 % Ag/TiO2 prepared by IP method, deposition–precipitation method with sodium hydroxide (DPN) and with hydrogen peroxide (DPH)
The chemical state and chemical binding energy of the catalysts were investigated by XPS. Depending on the preparation method, various oxidation states of silver is assumed to be produced. The Ti 2p, Ag 3d, O 1 s and C 1 s core level XPS high-resolution spectra was recorded for TiO2 and the prepared Ag/TiO2 catalysts. The spectra were decomposed assuming Gaussian line profiles and resolution of the Ti 2p spectrum and Ag 3d spectrum were fitted to conform with the differences in binding energies (BEs) of the spin–orbit pairs, 5.7 eV for titanium oxide and 6.0 eV for silver oxide.
Fig. 4a shows the narrow scans of Ti 2p peaks for the different catalysts. The peak for the Ti 2p3/2 state for TiO2 catalyst is narrow and sharp with slight asymmetry and has a binding energy of 458.2 eV with a full width at half maximum (FWHM) of 1.13 and is attributed to the tetravalent Ti4+ state. As silver was incorporated in the catalysts, the Ti 2p peaks became broader for IP and DPN catalysts but retained a similar value for the DPH catalyst. Curve-fitting analysis of the Ti 2p3/2 peaks of the different catalysts revealed two peaks for the DPN catalyst at 458.2 eV and at 457.4 eV, which corresponds to the tetravalent Ti4+ state and trivalent Ti3+ states of Ti, respectively [18, 19]. Although the FWHM of the Ti 2p3/2 of the IP catalyst increased to 1.55, curve-fitting analysis did not show the Ti4+ and Ti3+ states of Ti.

High resolution XPS spectra of a Ti 2p and b Ag 3d peak profiles for TiO2 and 5 % Ag/TiO2 catalysts
The binding energy of the Ag 3d peaks exhibit a negative shift as oxidation state is increased and these shifts are in the range of a few tenths of an eV [20]. Resolution of the Ag 3d spectra (Fig. 4b) shows peaks at 368.0 eV for Ag0, 367.6 eV for Ag+, 367.3 eV for Ag2+ and 366.5 eV for Ag3+. These results match well with previous studies found in literature [20–22]. The Ag 3d5/2 XPS signal found at 366.5 eV can be attributed to a chemical state of Ag3+, which was first found in doped TiO2, and this could explain the presence of Ti3+ in this sample. Previous works also reported the existence of Ag3+ in the form of Ag2O3, which could easily decompose into Ag2O at room temperature in air [22]. Since the peak for Ag3+ was found for the DPN catalyst even after long storage period, this indicates that Ag in this catalyst exist as both surface Ag oxide (Ag2O3) and substitutional Ag formed by the substitution of Ti sites by Ag atoms in the crystalline lattice.
From these results, it can be inferred that the IP catalyst is composed of metallic Ag while DPN catalyst is composed mainly of Ag2O and a small fraction of Ag2O3, and finally for DPH catalyst, mainly composed of AgO and a small fraction of metallic Ag. Table 1 shows the Ti 2p3/2- and Ag 3d5/2 peak positions for the different chemical species, FWHM and content fractions for the catalysts.
Images of surface morphologies and metal dispersion on the catalyst surface were taken using SEM–EDS. Fig. 5 shows the different images of the 5 % Ag/TiO2 prepared catalysts and in Fig. 6, the corresponding Ag metal dispersion is displayed. IP catalysts showed small aggregate structures on its surface from calcination and was not seen in DPN and DPH catalysts. Digital mapping of Ag by EDS showed that DPN and DPH catalysts had very good Ag dispersion on the catalyst surface as compared to IP catalysts. In the DPN and DPH methods, slow addition of the precipitating agent facilitates the precipitation of silver on the catalyst surface as compared to IP method. Furthermore, the long aging time in the IP method as compared DPN and DPH methods allowed the silver salt to enter into the pores of the catalyst instead of retaining it on the TiO2 surface.

SEM images of the prepared 5 % Ag/TiO2 catalysts

SEM–EDS digital mapping of Ag on the prepared 5 % Ag/TiO2 catalysts
Table 2 shows the characterization results of N2 adsorption by BET method. For IP catalysts, the surface area decreased from 64.95 m2/g of the base TiO2 to 49.00 m2/g with increasing Ag loading, the pore diameter increased from 17.30 to 20.88 nm and the total pore volume slightly decreased from 0.2809 to 0.2557 cm3/g in 5 % Ag/TiO2. This is mainly due to the calcination of the catalysts, which resulted in the widening of its pores and also the presence of the silver within the pores of TiO2. DPN and DPH catalysts showed no significant change in its structural properties.
Benzene decomposition
Benzene decomposition experiments were performed using NTP and a PDC system. The initial benzene concentration was set at 200 ppm and benzene decomposition experiments were performed by varying the voltage applied to the reactor while monitoring effluent composition and concentration. The effect of plasma on benzene concentration was tested by allowing the pollutant to go through the reactor without the catalysts while the plasma was turned on and testing its concentration afterwards. For the PDC system, the catalysts in the reactor were first saturated with gas-phase benzene until adsorption equilibrium was reached. After the adsorption equilibrium was established, activity tests were performed by using the same range of applied voltages to the reactor similar with the plasma alone experiment.
Results for the plasma alone treatment and PDC treatment are presented in Fig. 7. The results showed that oxygen plasma alone was not able to decompose benzene significantly but the addition of catalysts greatly improved the decomposition of benzene. IP catalysts decomposed benzene up to 20 % while DPN and DPH catalysts were able to fully decompose benzene even at lower SIE values. The differences in the catalytic activity of the catalysts are attributed to the decrease in the surface area of the IP catalysts as well as the improved metal dispersion on the catalyst surface of the DPN and DPH catalysts. The decrease in the surface area of the IP catalysts reduced the probability of the adsorption of the pollutant on the catalyst surface, thus showed lower decomposition efficiencies than DPN and DPH catalysts. On the other hand, the improved metal dispersion on the DPN and DPH catalysts also increased the active metal surface concentration which allowed more of the pollutant to react on the catalyst surface promoting pollutant decomposition.

Effect of catalysts on the decomposition of benzene using non-thermal plasma driven catalysis system. Reaction conditions standard atmospheric conditions; GHSV, 26,750 h−1; benzene initial concentration, 200 ppm; catalyst weight, 2.0 g
In PDC, the mechanisms are rather complex due to the simultaneous action of plasma and catalysis and their interaction with each other. Many publications report the synergetic effect in plasma-catalysis decomposition of VOCs even at low temperatures [11–15], where thermal catalysis does not occur. Although the detailed mechanism is still unclear, the synergetic effect can be explained by the involvement of catalyst surface activated by the plasma. These include ozone, UV, local heating, changes in the work function of the catalyst, activation of lattice oxygen, plasma-induced adsorption/desorption, generation of electron–hole pairs and their subsequent chemical reactions, direct interaction of gas-phased radicals with the catalyst and the adsorbed molecules and so on [1]. Two important modes for explaining the plausible mechanisms for the catalytic reactions are the Langmuir–Hinshelwood (L–H) model and the Eley–Rideal (E–R) model. In the L–H model, both reactants need to be on the adsorbed surface, and then followed by migration to the active site. In the E–R model, only one reactant is adsorbed on the surface and the other exists in the gas-phase [23].
To help explain the effect of the different catalysts on benzene decomposition, ozone generation efficiency during the decomposition of benzene was investigated. In the next section, the effect of catalysts on ozone concentration and the role of metal oxidation state on ozone generation efficiency and its contribution to benzene decomposition are discussed.
Ozone generation and decomposition
Ozone generation efficiency is often used as a good indicator of DBD reactor performance. Ozone formation proceeds via two-step process involving atomic oxygen formation (Eq. 3) and then recombination of this atomic oxygen with an oxygen molecule to form ozone (Eq. 4). Despite ozone being a powerful oxidant that can be used for VOC oxidation, tropospheric ozone is a hazardous by-product resulting from gas discharge. The decomposition of ozone to dioxygen is a thermodynamically favored process and is thermally stable up to 523 K [24]. Ozone can also be catalytically decomposed into active oxygen species (Eqs. 5 and 6), which is beneficial for PDC as it showed higher activity towards hydrocarbons than ozone [16, 25, 26].
Initial ozone concentrations at specified applied voltages were determined by allowing the feed gas to pass through the plasma reactor without benzene and the catalysts. Ozone concentrations during the PDC treatment of benzene were also monitored. The results showed that ozone concentration increased linearly with SIE obtaining a maximum of 1940 ppm at the highest voltage setting. During PDC treatment, a similar linear trend was observed but with lower ozone concentrations.
The ozone generation efficiency was then calculated from the slope of the linearized data from ozone concentration and SIE. Fig. 8 shows the comparison of the ozone generation efficiency and the effect of catalysts on ozone concentration during benzene decomposition. The ozone generation efficiency decreased from 43.81 to 28.70 ppm/SIE during plasma alone treatment which could indicate that ozone was utilized in benzene decomposition. However, based on the results presented in Fig. 7, there was only a slight decrease in benzene concentration. This may be due to the competing reactions between oxygen and benzene with the energized electrons formed in the discharge, thus, resulting in lower ozone concentrations.

Comparison of ozone generation efficiency during PDC decomposition of benzene. Reaction conditions standard atmospheric conditions; GHSV, 26,750 h−1; benzene initial concentration, 200 ppm; catalyst weight, 2.0 g
Upon the addition of catalysts, ozone generation efficiency further decreased. The results showed that with increasing silver loading, ozone generation efficiency decreased and the degree of reduction observed in ozone generation based on the preparation method of the catalysts were as follows: DPN > DPH > IP. The DPN3 and DPN5 catalysts were also able to fully utilize the ozone produced in the reactor as ozone was no longer detected in the effluent during the treatment of benzene.
The decrease in ozone generation could be explained by the decrease in discharge volume due to the presence of the catalyst which would limit ozone production; the self-decomposition of ozone on the catalyst surface; and by the utilization of ozone in the decomposition of benzene on the catalyst surface. As all PDC experiments used the same amount of catalyst, the latter would explain the differences in their catalytic performance. An alternative mechanism for ozone decomposition on metal oxides was proposed based on the following reactions [27]:
Based on these reduction–oxidation reactions, the catalytic activity towards ozone decomposition depends on the rate of Eq. 7. Higher metal oxidation states are also good for ozone adsorption and would also lead to the generation of more adsorbed oxygen (O2 −, O−, O2 2−) that has partial ionic character and is favorable for ozone decomposition [28]. Within the prepared catalysts, both DPN and DPH catalysts had multiple oxidation states which meant higher activities for ozone decomposition and were reflected on the results presented in Fig. 8. As these active oxygen species were reported to have higher activity towards VOC decomposition [16, 25, 26], this could also help explain the results from Fig. 7 where DPN and DPH catalysts were able to fully decompose benzene as compared to IP catalysts.
Energy efficiency
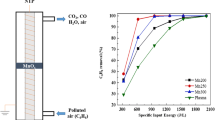
As energy consumption is related to the life span cost, which is an important parameter in assessing the capability of the process for practical applications, EE was calculated. Fig. 9 shows the effect of SIE on EE for benzene decomposition. The results show that the highest efficiency was reached at low SIE and decreased with increasing SIE. This suggests that most of the input energy at low SIE was used for benzene conversion while further increase in input energy was wasted or converted into heat or photons and was used for by-products formation [26].

Effect of specific input energy on energy efficiency during PDC decomposition of benzene. Reaction conditions standard atmospheric conditions; GHSV, 26,750 h−1; benzene initial concentration, 200 ppm; catalyst weight, 2.0 g
For the DPN and DPH catalysts, it is important to compare EE as catalysts prepared by both methods showed 100 % benzene decomposition. A maximum value of 392 g/kWh was obtained with 3 % Ag/TiO2 by DPN method compared to 220 g/kWh for 3 % Ag/TiO2 by DPH method. Therefore, maximum available value for input power in the plasma reactor should not only be determined by benzene decomposition but also by its energy efficiency.
Conclusions
Benzene decomposition was investigated at room temperature on Ag/TiO2 catalysts with different Ag loading using a low energy density PDC system. Among all the catalysts that were used, 3 % Ag/TiO2 prepared by DPN method showed the best performance reaching 100 % benzene conversion, complete ozone utilization and with the highest EE at 392 g/kWh. Catalyst properties were determined through characterization by XRD, XPS, SEM–EDS and BET method. From these results, it can be concluded that surface area together with the oxidation state and metal dispersion on the catalyst were the key factors that contributed in benzene decomposition. The low surface area of IP prepared catalysts significantly decreased its activity compared to DPN and DPH prepared catalysts. The higher oxidation state of silver found in DPN prepared catalysts was able to effectively decompose ozone into active oxygen species which further promoted benzene decomposition on the catalyst surface. Moreover, the homogeneous dispersion of the metal on the catalyst surface found in DPN and DPH prepared catalysts provided more accessible active sites for chemical reactions.
References
Van Durme J, Dewulf J, Leys C, Van Langenhove H (2008) Combining non-thermal plasma with heterogeneous catalysis in waste gas treatment: a review. Appl Catal B Environ 78:324–333
Chen HL, Lee HM, Chen SH, Chang MB, Yu SJ, Li SN (2009) Removal of volatile organic compounds by single-stage and two-stage plasma catalysis systems: a review of the performance enhancement mechanisms, current status, and suitable applications. Environ Sci Technol 43:2216–2227
Whitehead JC (2010) Plasma catalysis: a solution for environmental problems. Pure Appl Chem 82:1329–1336
TOXNET Hazardous Substances Data Bank (2014) United States National Library of Medicine. http://toxnet.nlm.nih.gov. Accessed 23 Oct 2014
Atkinson R, Baulch DL, Cox RA, Hampson RF Jr, Kerr JA, Rossi MJ, Troe J (1997) Evaluated kinetic and photochemical data for atmospheric chemistry, organic species: Supplement VII. J Phys Chem Ref Data 26:1329
Fridman A, Kennedy LA (2004) Plasma physics and engineering. Taylor & Francis, New York
Urashima K, Chang JS (2000) Removal of volatile organic compounds from air streams and industrial flue gases by non-thermal plasma technology. IEEE Trans Dielectr Electr Insul 7:602–614
Chikorov A, Gutsol A, Fridman A (2005) Atmospheric pressure plasma of dielectric barrier discharges. Pure Appl Chem 77:487–495
Petipas G, Rollier JD, Darmon A, Gonzales-Aguilar J, Metkemeijer R, Fulcheri L (2007) A comparative study of non-thermal plasma assisted reforming technologies. Int J Hydrog Energy 32:2848–2867
Vandernbroucke AM, Morent R, De Geyter N, Leys C (2011) Non-thermal plasmas for non-catalytic and catalytic VOC abatement. J Hazard Mat 195:30–54
Oda T (2003) Non-thermal plasma processing for environmental protection: decomposition of dilute VOCs in air. J Electrostat 57:293–311
Ogata A, Einaga H, Kabashima H, Futamura S, Kushiyama S, Kim HH (2003) Effective combination of nonthermal plasma and catalysts for decomposition of benzene in air. Appl Catal B Environ 46:87–95
Han SB, Oda T, Ono R (2005) Improvement of the energy efficiency in the decomposition of dilute trichloroethylene by the barrier discharge plasma process. IEEE Trans Ind Appl 41:1343–1349
Subrahmanyam C, Magureanu M, Renken A, Kiwi-Minsker L (2006) Catalytic abatement of volatile organic compounds assisted by non-thermal plasma: Part 1. A novel dielectric barrier discharge reactor containing catalytic electrode. Appl Catal B Environ 65:150–156
Mista W, Kacprzyk R (2008) Decomposition of toluene using non-thermal plasma reactor at room temperature. Catal Today 137:345–349
Madhu Kumar P, Badrinarayanan S, Sastry M (2000) Nanocrystalline TiO2 studied by optical, FTIR and X-ray photoelectron spectroscopy: correlation to presence of surface states. Thin Solid Films 358:122–130
Kuo YL, Su TL, Chuang KJ, Chen HW, Kung FC (2011) Preparation of platinum- and silver-incorporated TiO2 coatings in thin-film photoreactor for the photocatalytic decomposition of o-cresol. Environ Technol 32:1799–1806
Hoflund GB, Hazos ZF, Salaita GN (2000) Surface characterization study of Ag, AgO, and Ag2O using X-ray photoelectron spectroscopy and electron energy-loss spectroscopy. Phys Rev B 62:11126–11133
Weaver JF, Hoflund GB (1994) Surface characterization study of the thermal decomposition of AgO. J Phys Chem 98:8519–8524
Feng N, Wang Q, Zheng A, Zhang Z, Fan J, Liu SB, Amoereux JP, Deng F (2013) Understanding the high photocatalytic activity of (B, Ag)-codoped TiO2 under solar-light irradiation with XPS, solid-state NMR, and DFT calculations. J Am Chem Soc 135:1607–1616
Einaga H, Futamura S (2004) Catalytic oxidation of benzene with ozone over alumina-supported manganese oxides. J Catal 227:304–312
Huang H, Ye D, Guan X (2008) The simultaneous catalytic removal of VOCs and O3 in a post-plasm. Catal Today 139:43–48
Kim HH, Ogata A, Futamura S (2008) Oxygen partial pressure-dependent behavior of various catalysts for the total oxidation of VOCs using cycled system of adsorption and oxygen plasma. Appl Catal B Environ 79:356–367
Dhandapani B, Oyama ST (1996) Gas phase ozone decomposition catalysts. Appl Catal B Environ 11:129–166
Dimitrova S, Ivanov G, Mehandjiev D (2004) Metallurgical slag as support of catalysts for complete oxidation in the presence of ozone. Appl Catal A Gen 266:81–87
Fan X, Zhu TL, Wang MY, Li XM (2009) Removal of low-concentration BTX in air using combined plasma catalysis system. Chemosphere 75:1301–1306
Zhihua L, Ma J, He H (2015) Decomposition of high-level ozone under high humidity over Mn-Fe catalyst: the influence of iron precursors. Catal Commun 59:156–160
Tang WX, Liu HD, Wu XF, Chen YF (2014) Higher oxidation state responsible for ozone decomposition at room temperature over manganese and cobalt oxides: effect of calcination temperature. Ozone Sci Eng 36:502–512
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pangilinan, C.D.C., Kurniawan, W., Salim, C. et al. Effect of Ag/TiO2 catalyst preparation on gas-phase benzene decomposition using non-thermal plasma driven catalysis under oxygen plasma. Reac Kinet Mech Cat 117, 103–118 (2016). https://doi.org/10.1007/s11144-015-0920-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-015-0920-6