
Abstract
Three manganese oxide catalysts (MnOx) were synthesized via a simple method, and then they were introduced into the non-thermal plasma (NTP) system for benzene removal. The XRD and EXAFS results showed the MnOx were mainly in the Mn3O4 phase, and from the analysis of N2 adsorption/desorption isotherms, we knew the MnOx calcined at 250 °C (Mn250) had the largest surface area of 274.5 m2 g−1. Besides, Mn250 also exerted higher benzene adsorption capacity (0.430 mmol g−1) according to C6H6-TPD. O2-TPD indicated that Mn250 showed better oxygen mobility than Mn300. Moreover, by analyzing XPS results, it revealed that Mn250 exhibited rich abundant of surface adsorbed oxygen species (Oads) and moderate ratio of Mn4+/Mn3+, and the reducibility temperature was also the lowest among all the MnOx catalysts drawn by H2-TPR profiles. As a result, Mn250 combined with NTP could remove 96.9% of benzene at a low input power of 3 W (benzene concentration 200 ppm, and GHSV 60,000 mL gcat.−1 h−1), performing the best catalytic activity among the three catalysts and plasma only. Furthermore, the “NTP + Mn250” system also produced the highest CO2 concentration and lowest CO concentration in downstream, and the residual O3 after catalytic reaction was also the lowest, that is to say, the synergistic effect between NTP and Mn250 was more effective than other catalysts in benzene removal.

Graphical abstract
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
As is known to all, the emission of volatile organic compounds (VOCs), such as including formaldehyde, benzene, toluene, and so on can exert severe harm to both human health and the environment (Fang et al. 2018; Kim et al. 2018; Quoc An et al. 2011; Tomatis et al. 2016). Thus, it is essential to eliminate these toxic pollutants.
In the past decades, several conventional technologies have been developed for VOCs abatement, such as non-thermal plasma (NTP), adsorption, thermal catalytic oxidation, membrane separation, photocatalytic removal, and so on (Ji et al. 2017; Liao et al. 2017; Liu et al. 2017; Mizuno 2013; Qin et al. 2016; Yao et al. 2018). Among these existed technologies, NTP is widely used for its high efficiency and low operating temperature (Veerapandian et al. 2017). But the formation of unwanted by-products (e.g., CO, CHOOH, and so on) and high energy consumption are the main bottlenecks of the NTP technology, and the combination of NTP and catalysts can overcome the disadvantages of using NTP only (Sudhakaran et al. 2017).
Many researchers have made great efforts to investigate the combination of non-thermal plasma and different kinds of catalysts. For example, Wang et al. investigated CeO2-MnOx catalysts in toluene removal using non-thermal plasma, and found that the sample CeMnOx showed the highest catalytic activity with the cooperation of plasma, as 95.94% of toluene could be removed with input power of 24 W (Wang et al. 2017). Zhu and his co-workers applied Cu-Ce catalysts into a dielectric barrier discharge reactor and found this combination can remove 94.7% of formaldehyde at a specific input energy of 486 J/L (Zhu et al. 2015b). Otherwise, Mustafa et al. investigated the synergistic effects of plasma-BaTiO3 and plasma-HZSM-5, respectively, and they observed both of the combinations can eliminate all the mixed VOCs (tetrachloroethylene, toluene, trichloroethylene) at an input power 65.8 W (Mustafa et al. 2018).
Nowadays, some researchers also introduced single manganese oxides catalysts with non-thermal plasma in VOCs elimination, as manganese oxides can not only effectively decompose ozone generated in the plasma reactor, but also exhibits high catalytic activity in the plasma-catalyst system (Li et al. 2014). For example, Vandenbroucke et al. applied a plasma-commercial MnO2 system for the abatement of trichloroethylene (TCE), and the results showed that the combination system can not only reduce the activation energy of the TCE oxidation reaction but also greatly enhance the TCE conversion as well as the COx selectivity (Vandenbroucke et al. 2014). Guo et al. reported a dielectric barrier discharge (DBD) reactor with manganese oxides in the discharge area for the purpose of toluene removing, and more than 96% of toluene could be cleared at a specific input energy of 756 J/L (Guo et al. 2006).
Considering the merits the above investigations exerted, there is also a common disadvantage: the high level of energy consumption especially in aromatic compounds removal. So in this study, MnOx catalysts with a large BET surface area were fabricated using a facile synthesis method, and then these MnOx were applied for the removal of benzene in a NTP reactor with the purpose of obtaining a NTP-catalyst system with high benzene removal efficiency as well as low energy consumption.
Experimental
Catalysts preparation
Mn (NO3)2·6H2O (≥ 98%), and (NH4)2C2O4·H2O (≥ 98%) were both purchased from Wako Pure Chemical of Japan and used without further purification.
Firstly, the Mn (NO3)2 solution (0.2 M, 200 mL) was added into the (NH4)2C2O4 solution (0.2 M, 200 mL); then, the mixture was stirred for 40 min before being washed three times by purity water and once by anhydrous ethanol. Secondly, the washed precipitation was filtered and dried at 80 °C for 24 h by doing this manganese oxalate precursor was obtained. Finally, the precursor was calcined in static air at 200, 250, and 300 °C for 300 min (2 °C min−1), respectively. In this experiment, Mn200, Mn250, and Mn300 were used to mark the MnOx catalysts calcined at 200, 250, and 300 °C, respectively.
Catalysts characterization
The thermogravimetric analysis (TGA) of the manganese oxalate precursor and Mn250 was performed, respectively, using a TGA/DTA system (STA449 F3) in air with a heating rate of 10 °C min−1. X-ray diffraction (XRD) patterns were recorded by a Rigaku RINT 2200 system (Rigaku Co. Ltd.) with a Cu-Kα radiation in the 2θ ranging from 10 to 80 °. N2-adsorption/desorption isotherms were carried out using Micromeritics TriStar II. Field-emission scanning electron microscope images were obtained by using a scanning electron microscope (S-900H, Hitachi), while a high-resolution transmission electron microscopy (JEM-ARM200F, JEOL) was applied for detecting the microstructures and interplanar spacings. Hydrogen temperature-programmed reduction (H2-TPR) was performed using the flow-type BELCAT-30 catalyst analyzer (BEL JAPAN, Inc). The C6H6 temperature-programmed desorption studies was carried out using FTIR (Perkin-Elmer, spectrum 100). X-ray photoelectron spectroscopy (XPS, AXIS UltraDLD, Kratos group) was used to determine the surface species of the as-prepared catalysts. XAFS measurements were performed at the Photon Factory of the High Energy Accelerator Research Organization (KEK-PF) on beam line BL-7C, which has a storage ring operating energy of 2.5 GeV, and the XAFS spectra were recorded at 296 K and then analyzed using Athena and Artemis (Lian et al. 2017).
Experimental scheme and catalytic activity tests
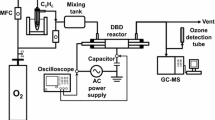
The schematic diagram used in our experiment is shown in Scheme 1. A quartz tube (shown in yellow box) with an inner diameter of 10 mm was used as a reactor (the effective reaction length was 100 mm), in which a glass rod with a copper wire coil enwound was placed in the axis of quartz tube. The diameter and length of the inner glass rod was 6 and 125 mm, respectively. The catalyst was packed in the middle of the tube (also in the discharge region) during the whole experiment. It is notable that the outer surface of the quartz tube was wrapped with aluminum foil as a counter electrode. Remarkably, the reactor was placed in an oven and the temperature inside this oven was kept at 25 °C during the whole experiment.

Schematic diagram of the experiment (several switches in both the gas piping and electrical circuits are not shown in this figure)
The non-thermal plasma system was supplied by an AC voltage power supply (Riko-Slidetrans, RSA-3) with maximum output voltage and electric current of 130 V and 3 A, respectively. A digital power meter (Yokogawa, WT310) was used to measure the input power, voltage, and electric current. All the electrical signals were monitored by a digital powermeter (Yokogawa WT310).
Gas compositions were analyzed online using a Fourier transform infrared (FTIR) spectrometer (Perkin-Elmer, resolution of 4 cm−1) equipped with a gas cell (2 m path length, PIKE Technologies).
In every test, 0.1 g of selected catalyst (pressed and sieved through 40–60 mesh) was placed in the plasma discharge region (Scheme 1). The simulated air flow (N2/O2 = 79:21) was 100 mL min−1, and initial benzene concentration was set to be 200 ppm; hence, the gas hourly space velocity (GHSV) value was 60,000 mL gcat.−1 h−1.
The removal efficiency of benzene, CO2 selectivity, and COx selectivity were defined as the following equations (Karuppiah et al. 2014; Mustafa et al. 2018):
where CC6H6, in, CC6H6, out, CCO2, and CCO are inlet and outlet concentrations of benzene, concentrations of CO2 and CO in the downstream, respectively. The CC6H6, in was 200 ppm in this experiment, and the CC6H6, out, CCO2, and CCO were the average of 20 measured values obtained after the reaction ran smoothly, respectively. The O3 in the downstream was measured by a UV photometric ozone analyzer EG-600 (Ebara Jitsugyo Co. Ltd.) after the reaction became stable.
Result and discussion
TGA/DSC profiles
Figure 1 displays the thermogravimetric analysis of the manganese oxalate precursor and Mn250, respectively. It can be concluded from Fig. 1a that for the manganese oxalate precursor there are two main weight loss stages in the temperature range from 40 to 450 °C. The first weight loss stage at about 150 °C should be assigned to the dehydration of the precursor, while the second weight loss stage starting from about 250 °C can be contributed to the decomposition of the manganese oxalate (Tang et al. 2014). The thermogravimetric curve of Mn250 in Fig. 1b reveals that the total weight loss in the range from 40 to 450 °C is less than 4%, indicating the complete decomposition of the manganese oxalate precursor after being calcined at 250 °C.

TGA/DSC curves of the manganese oxalate precursor (a) and sample Mn250 (b)
XRD patterns, N2 adsorption/desorption isotherms, and SEM and TEM images
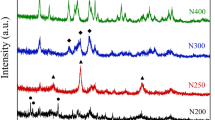
XRD patterns of the prepared catalysts are shown in Fig. 2. It can be inferred that there exist Mn3O4 (JCPDS PDF 018-0803) in all the three manganese oxide catalysts, but for Mn200 there is also some peaks which can be assigned to MnC2O4. The XRD patterns of these three catalysts are in agreement with the TGA/DSC analysis results that the manganese oxalate precursor could not be completely decomposed at 200 °C.

XRD patterns of obtained MnOx catalysts
Figure 3 displays the N2 adsorption/desorption isotherms and pore size distributions of obtained catalysts. It can be clearly seen that every sample exhibited a classical type IV isotherm with a typical hysteresis loop which is often associated with narrow slit-like micro- and meso-pores in the catalysts (Tang et al. 2014). From the data shown in Table 1, Mn250 exhibits a surface area (418.2 m2 g−1), far larger than Mn200 (172.4 m2 g−1) and Mn300 (138.2 m2 g−1), and the pore volume shows the same trend as the surface area. It can also conclude from Table 1 that as the calcination temperature increases, the pore volume increased from 0.29 cm3 g−1 (Mn200) to 0.37 cm3 g−1 (Mn250) and then declined to 0.27 cm3 g−1 (Mn300). All the above facts indicate that the calcination temperature can strongly affect the structure of a catalyst.

N2 adsorption/desorption isotherms of MnOx catalysts (a) and pore size distribution (b)
The field emission scanning electron microscopy (FESEM) images of the three catalysts are shown in Fig. 4. It can be observed in Fig. 4a that Mn200 exhibited rough surface with some aggregation, but the aggregation disappeared in Mn250 (Fig. 4b). Moreover, the Mn250 also developed more porous surface than the other two samples seen from Fig. 4. Increasing the calcination temperature to 300 °C (Mn300) resulted in a great number of aggregations on the surface as shown in Fig. 4c. It further confirmed that the calcination temperature can affect the morphology of catalysts obviously.

FE-SEM images of Mn200 (a), Mn250 (b), and Mn300 (c)
Figure 5 shows the TEM bright-field image and high-resolution TEM (HRTEM) images of Mn250. The interplanar spacings in Fig. 5c were measured to be 0.312, 0.283, and 0.272 nm which are in agreement with the [1 1 2], [2 0 0], and [1 0 3] crystal planes of Mn3O4 (JCPDS PDF 018-0803), respectively, and these data are also in fairly accordance with the XRD patterns of Mn250.

TEM bright-field image (a), HRTEM images (b), and high-angle annular-dark field STEM image (c) of Mn250
C6H6-TPD
It is reported that the adsorption of VOCs on surface of catalysts plays a vital role in the catalytic process (Ikhlaq and Kasprzyk-Hordern 2017). Figure 6 shows the C6H6-TPD curves of Mn250 and Mn300 (sample Mn200 is mainly consisted with MnC2O4 which will be decomposed when the temperature is higher than 200 °C (Gao and Dollimore 1993; Ma et al. 2017)), and Table 2 presents the summary of desorbed C6H6 and CO2 from Mn250 and Mn300, respectively.

C6H6-TPD curves of Mn250 (a) and Mn300 (b)
From Fig. 6, it can be seen that both Mn250 and Mn300 could release C6H6 and CO2 as the temperature increased. The desorbed C6H6 can be attributed to physisorption as a result of the van der Waals interactions between the molecule of C6H6 and surface manganese metal, while the CO2 can be assigned to the chemical reaction of adsorbed C6H6 and MnOx at higher temperature (Bauer et al. 2018; Liu et al. 2012; Tao et al. 2018). Table 2 revealed that the desorbed C6H6 of Mn250 and Mn300 are 0.205 and 0.175 mmol g−1, respectively. As to the desorbed CO2, Mn250 and Mn300 are 1.347 and 0.845 mmol g−1, respectively. That is to say, Mn250 can adsorb more C6H6 (0.430 mmol g−1 in total) than Mn300 (0.316 mmol g−1 in total) which are in agreement with their BET surface area.
XPS spectra
Figure 7 and Table 3 report the XPS spectra and analysis results of XPS spectra, respectively.

XPS spectra of as-calcined catalysts: a Mn 2p3/2 and b O 1 s
As illustrated in Fig. 7a, for each catalyst, the peak of Mn 2p3/2 can be divided into two components which are centered at binding energy (BE) of 641.6–641.8 eV and 643.3–643.9 eV, corresponding to Mn3+ and Mn4+, respectively (Fang et al. 2018; Piumetti et al. 2015; Zuo et al. 2018). It can be concluded from Table 3 that the ratio of surface Mn4+ on those catalysts decreased with the increasing of calcination temperature, for example, the proportion of surface Mn4+ on Mn200 is 28.91%, while for Mn250 and Mn300 the corresponding values are 20.19 and 12.88%, respectively. Thus, the ratio of Mn4+/Mn3+ decreases from 0.407 for Mn200 to 0.253 for Mn250 and 0.148 for Mn300, respectively. As it has been reported that higher Mn4+/Mn3+ ratio implied MnOx can sustain manganese content at higher oxidation states in catalytic reactions, and furthermore this would exert an important influence on the catalytic activities of these catalysts (Li et al. 2017).
Figure 7b presents the O 1 s XPS spectra of the calcined catalysts. The asymmetrical O 1 s signal can be fitted with two components: the peaks at BE of 529.6–529.8 eV can be ascribed to the lattice oxygen (O2−) which is usually named Olatt, and the peaks at BE of 531.0–531.2 eV can be identified as surface adsorbed oxygen (O2−, O−, O22−) that is frequently denoted as Oads (Chen et al. 2018; Fang et al. 2018). Also, from Table 3, we can see that the ratio of surface Oads showed the same decreasing trend as surface Mn4+, and Mn200 exhibited the highest content of Oads (39.60%), while Mn250 (33.89%) and Mn300 (28.75%) both contained lower Oads than Mn200. Some researcher had proved that a higher manganese ion (Mn4+) would lead to more surface adsorbed oxygen species (Oads), and our research is in good agreement with this conclusion (Tang et al. 2014). Oads exhibits advantages in mobility compared with Olatt; thus, the former plays a more important role than the latter in catalytic removal of VOCs (Piumetti et al. 2015).
O2-TPD
O2-TPD was usually used to detect oxygen species and oxygen mobility, and the related O2-TPD profiles of Mn250 and Mn300 were presented in Fig. 8.

O2-TPD profiles of Mn250 and Mn300
Seen from Fig. 8, Mn250 showed a desorption peak at 274 °C with a little shoulder at 183 °C, which could be assigned to chemically adsorbed and physically adsorbed oxygen species, respectively, and the peak at 498 °C could attribute to the desorption of lattice oxygen. As to Mn300, no desorption of physically adsorbed oxygen could be seen and the desorption of the chemically adsorbed oxygen was seen at 375 °C; finally, the desorption peaks at 479 °C and 505 °C corresponded to lattice oxygen (Cai et al. 2014; He et al. 2015).
Both physically and chemically adsorbed oxygen species were believed to play an important role in a catalytic reaction (Zhang et al. 2018). Mn250 contained some physically adsorbed oxygen species, and the chemically adsorbed oxygen species also exerted better mobility than Mn300; these results would be beneficial to the catalytic activity of Mn250.
H2-TPR
The redox properties of the catalysts were investigated, and the results are presented in Fig. 9. There exist two main separated peaks in each curve. From analyzing XRD spectra, we know that Mn200 is consisted of manganese oxalate and Mn3O4, and manganese oxalate could decompose when the temperature is beyond 200 °C. So we can conclude that the first peak of Mn200 might refer to the decomposition of manganese oxalate and the reduction of Mn3+ located in the lattice sites of Mn3O4, and the second peak should be contributed to the final reduction of Mn3O4 to MnO (Gao and Dollimore 1993; Piumetti et al. 2015; Yang et al. 1993). The reduction orders of Mn250 and Mn300 are somewhat the same as Mn200, except the decomposition of manganese oxalate. Besides, the first sharp peak of Mn200 indicates there is a phase transition during the decomposition of manganese oxalate and formation of manganese oxide (Strohmeier and Hercules 1985).

H2-TPR curves of the gained catalysts
Notably, the first peak of Mn250 is at 274 °C and second peak is at 421 °C; both are lower than the corresponding peaks of Mn200 (first 277 °C and second 430 °C) and Mn300 (first 278 °C and second 426 °C), indicating Mn250 has better low-temperature reducibility that is helpful for its catalytic activity.
Fine structure analysis
Extended X-ray absorption fine structure (EXAFS) studies can further reveal the manganese oxidation state. Figure 10a shows the Fourier transformed EXAFS signals of Mn3O4 (reference), Mn200, Mn250, and Mn300. For the spectra of Mn3O4, the peak corresponding to the Mn-O bond in the first shell appears at about 1.5 Å (phase uncorrected; denoted by pu) and those peaks at about less than 2.4 to 3.1 Å (pu) corresponding to Mn-Mn bonds and smaller Mn-O contributions appear in the higher shells (Einaga et al. 2013; Wang et al. 2018). In contrast, for Mn200, the Mn-O peak was observed at ~ 1.5 Å (pu), while the Mn-Mn peaks were at lower intensities than the reference. Increasing calcination temperature, the Mn-O peak would be observed a smaller radial distance and the intensities of those Mn-Mn peaks also showed an increasing trend.

Mn K-edge EXAFS spectra of calcined manganese oxides and reference Mn3O4 (a), XANES spectra of calcined manganese oxides and reference Mn3O4: normalized spectra (b), and the first derivatives of the spectra (c)
The quantitative analysis performed on the EXAFS and fitting data are summarized in Table 4. For Mn200, the Mn-O and Mn-Mn coordination numbers (CN) were 2.42 and 3.62, respectively, which might be the reason that the main component of the sample is MnC2O4. As the calcination temperature increased, the Mn-O and Mn-Mn coordination numbers also increased from 4.48 (Mn250) to 4.74 (Mn300). Additionally, the Mn-O bond distance (R = 1.87–1.90 Å) was essentially unchanged as the calcination temperature increased, while the Mn-Mn bond distance showed an increasing trend.
The manganese oxidation state is strongly correlated with the absorption edge of the Mn XANES spectra (Einaga et al. 2013). Figure 10b, c shows the XANES spectra of obtained catalysts and reference Mn3O4. The absorption edges for Mn200, Mn250, and Mn300 were 6546.2, 6546.5, and 6545.8 eV, respectively, and these values were close to the absorption edge of Mn3O4 (6544.3 eV), confirming the formation of Mn3O4 in every sample that is in great agreement with XRD results.
Catalytic performance
Figure 11 presents the benzene removal effects of obtained catalysts combined with NTP and plasma only, respectively. It can be inferred from Fig. 11a that although rising the power (plasma only) can enhance the benzene removal efficiency, the participation of MnOx catalysts promoted the elimination of benzene more effectively. As we all know, the non-thermal plasma can cause plasma gas phase reactions in benzene oxidation, and the synergistic effects between catalyst and plasma include not only plasma gas phase reactions but also plasma-assisted surface reactions; furthermore, the adsorption of benzene on MnOx catalysts also increased the residence time of benzene in the plasma reactor (Hamada et al. 2018; Zhu et al. 2015b). The catalytic performance of the three catalysts and plasma only follow the orders Mn250 > Mn300 > Mn200 > plasma only, that is to say, Mn250 combined with plasma exerted the highest catalytic activity than the others. For example, at 3 W, the benzene removal efficiency of Mn200, Mn250, Mn300, and plasma only are 70.7, 96.9, 80.6, and 54%, respectively.

Benzene removal efficiency (a), CO2 concentration (b), CO concentration (c), COx (CO2 and CO) selectivity (d), CO2 selectivity (e), and O3 concentration (f) as a function of input power, respectively (initial benzene concentration 200 ppm, GHSV 60,000 ml gcat.−1 h−1)
From Fig. 11b, c, we can see that if plasma was used without any catalyst, it can produce much lower CO2 and higher CO than combining catalysts and plasma together. Thus, we can conclude that the appearance of catalysts can promote CO2 formation and suppress CO production simultaneously compared with the application of plasma only. Among all the samples, Mn250 showed the highest CO2 and the lowest CO production in the whole experiment. As the input power increased, the production of CO2 is also increased at first but then decreased for all the catalysts, while the CO shows a monotonical increasing trend all the time, which may be ascribed to the absence of ozone and only some other active species left at high power (Zhang et al. 2016). At an input power of 6 W, the CO2 concentration reached the maximum for all the catalysts, and Mn250 produced the highest CO2 of 592.6 ppm, comparing with 524.9 ppm (Mn200), 487.1 ppm (Mn300), and 375.1 ppm (plasma only), respectively. Of course, at 6 W, the sample Mn250 also showed the highest COx (CO2 and CO) selectivity (62.5%) and CO2 selectivity (49.4%) in this experiment (Fig. 10d, e).
For all the catalysts and plasma only, their selectivity of COx (including CO2 and CO) during the catalytic reactions are all less than 100%, the possible reason may due to the formation of non-volatile organic compounds on the surface of the catalysts (Sivachandiran et al. 2015; Trinh and Mok 2015).
The NTP can produce O3 and therefore can help to promote the abatement of benzene, but the residual O3 in the downstream is another kind of pollutant, so it is essential to detect O3 in the downstream (Zhu et al. 2017). As is shown in Fig. 11f, for all the “NTP + catalyst” systems and plasma only, the remained O3 in the downstream was first increased at low power and then declined even to 0 ppm at high power. The enhancement of O3 could be assigned to the increase of input power, but at high power the excess high-energy electrons can suppress O3 formation and even decompose O3 meanwhile (Hamada et al. 2018; Qin et al. 2016; Zhu et al. 2015a). The highest O3 concentration (1493 ppm) is produced by using plasma only at 3 W, and introducing catalysts can reduce the O3 in the downstream greatly as the MnOx can decompose O3 effectively (Einaga et al. 2013; Liu and Zhang 2017). It is noteworthy that the introduction of Mn250 led to the lowest O3 concentration, the main reason may be that Mn250 can decompose a larger proportion of O3 into active species which can react with benzene than other catalysts considering its high catalytic activity.
Based on N2 adsorption/desorption curves, Mn250 has the largest BET surface area among all the catalysts, so the amounts of surface active sites are also more than others’ (Einaga et al. 2013). Besides, the large BET surface area could also prolong the retention time of benzene molecular in the discharge region (Wang et al. 2017). Furthermore, it exerts a larger benzene adsorption capacity and relatively low-temperature reducibility which are considered to be helpful to the catalytic activity (Einaga and Ogata 2009). Besides, Mn250 also shows a moderate ratio of Mn4+/Mn3+ and surface adsorbed oxygen species that are good for its catalytic activity. All these above advantages should contribute to the best performance of the “NTP + Mn250” system.
Conclusions
In summary, the removal of benzene in a “NTP + catalyst” system with MnOx calcined at different temperatures have been investigated concerning removal efficiency, formation of CO2, CO, and O3 as well as energy consumption. Comparing with the plasma only, the introduction of MnOx into the NTP reactor can not only improve the benzene removal efficiency significantly, but also promote the CO2 formation and suppress the CO simultaneously. Among all the obtained MnOx catalysts, the sample Mn250 showed the highest catalytic activity when placed in the NTP reactor, as it could eliminate 96.9% of benzene at an input power of 3 W (benzene concentration 200 ppm, GHSV 60,000 mL gcat.−1 h−1). Moreover, the “NTP + Mn250” system also produced the most amounts of CO2 as well as the lowest CO and O3 after reaction. At an input power of 6 W, the “NTP + Mn250” system exhibited the highest CO2 selectivity (49.4%) and COx selectivity (62.5%) in this experiment. The O3 generated in the discharge area increased at low input power but decreased at high power in the whole research.
References
Bauer U, Gleichweit C, Höfert O, Späth F, Gotterbarm K, Steinrück H-P, Papp C (2018) Reactivity studies of ethylene, benzene and cyclohexane on carbide-modified Mo (110) using high resolution X-ray photoelectron spectroscopy. Surf Sci 678:11–19
Cai W, Zhong Q, Zhao W, Bu Y (2014) Focus on the modified CexZr1-xO2 with the rigid benzene-muti-carboxylate ligands and its catalysis in oxidation of NO. Appl Catal B Environ 158-159:258–268
Chen J, Chen X, Chen X, Xu W, Xu Z, Jia H, Chen J (2018) Homogeneous introduction of CeOy into MnOx-based catalyst for oxidation of aromatic VOCs. Appl Catal B Environ 224:825–835
Einaga H, Ogata A (2009) Benzene oxidation with ozone over supported manganese oxide catalysts: effect of catalyst support and reaction conditions. J Hazard Mater 164:1236–1241
Einaga H, Teraoka Y, Ogata A (2013) Catalytic oxidation of benzene by ozone over manganese oxides supported on USY zeolite. J Catal 305:227–237
Fang R, Huang H, Ji J, He M, Feng Q, Zhan Y, Leung DYC (2018) Efficient MnOx supported on coconut shell activated carbon for catalytic oxidation of indoor formaldehyde at room temperature. Chem Eng J 334:2050–2057
Gao X, Dollimore D (1993) The thermal decomposition of oxalates: part 26. A kinetic study of the thermal decomposition of manganese (II) oxalate dihydrate. Thermochim Acta 215:47–63
Guo Y-F, Ye D-Q, Chen K-F, He J-C, Chen W-L (2006) Toluene decomposition using a wire-plate dielectric barrier discharge reactor with manganese oxide catalyst in situ. J Mol Catal A Chem 245:93–100
Hamada S, Hojo H, Einaga H (2018) Effect of catalyst composition and reactor configuration on benzene oxidation with a nonthermal plasma-catalyst combined reactor. Catal Today. https://doi.org/10.1016/j.cattod.2018.07.055
He C, Liu X, Shi J, Ma C, Pan H, Li G (2015) Anionic starch-induced Cu-based composite with flake-like mesostructure for gas-phase propanal efficient removal. J Colloid Interface Sci 454:216–225
Ikhlaq A, Kasprzyk-Hordern B (2017) Catalytic ozonation of chlorinated VOCs on ZSM-5 zeolites and alumina: formation of chlorides. Appl Catal B Environ 200:274–282
Ji J, Xu Y, Huang H, He M, Liu S, Liu G, Xie R, Feng Q, Shu Y, Zhan Y, Fang R, Ye X, Leung DYC (2017) Mesoporous TiO2 under VUV irradiation: enhanced photocatalytic oxidation for VOCs degradation at room temperature. Chem Eng J 327:490–499
Karuppiah J, Reddy EL, Reddy PMK, Ramaraju B, Subrahmanyam C (2014) Catalytic nonthermal plasma reactor for the abatement of low concentrations of benzene. Int J Environ Sci Technol 11:311–318
Kim M, Park E, Jurng J (2018) Oxidation of gaseous formaldehyde with ozone over MnOx/TiO2 catalysts at room temperature (25 °C). Powder Technol 325:368–372
Li H, Lee S, Wang Z, Huang Y, Ho W, Cui L (2017) Peroxymonosulfate activated by amorphous particulate MnO2 for mineralization of benzene gas: redox reaction, weighting analysis, and numerical modelling. Chem Eng J 316:61–69
Li Y, Fan Z, Shi J, Liu Z, Zhou J, Shangguan W (2014) Removal of volatile organic compounds (VOCs) at room temperature using dielectric barrier discharge and plasma-catalysis. Plasma Chem Plasma Process 34:801–810
Lian S, Browne MP, Domínguez C, Stamatin SN, Nolan H, Duesberg GS, Lyons MEG, Fonda E, Colavita PE (2017) Template-free synthesis of mesoporous manganese oxides with catalytic activity in the oxygen evolution reaction. Sustain Energy Fuels 1:780–788
Liao Y, Zhang X, Peng R, Zhao M, Ye D (2017) Catalytic properties of manganese oxide polyhedra with hollow and solid morphologies in toluene removal. Appl Surf Sci 405:20–28
Liu G, Ji J, Huang H, Xie R, Feng Q, Shu Y, Zhan Y, Fang R, He M, Liu S, Ye X, Leung DYC (2017) UV/H2O2: an efficient aqueous advanced oxidation process for VOCs removal. Chem Eng J 324:44–50
Liu W, Carrasco J, Santra B, Michaelides A, Scheffler M, Tkatchenko A (2012) Benzene adsorbed on metals: concerted effect of covalency and van der Waals bonding. Phys Rev B 86(24):245405
Liu Y, Zhang P (2017) Removing surface hydroxyl groups of Ce-modified MnO2 to significantly improve its stability for gaseous ozone decomposition. J Phys Chem C 121:23488–23497
Ma X, Li J, Rankin MA, Croll LM, Dahn JR (2017) Highly porous MnOx prepared from MnC2O4·3H2O as an adsorbent for the removal of SO2 and NH3. Microporous Mesoporous Mater 244:192–198
Mizuno A (2013) Generation of non-thermal plasma combined with catalysts and their application in environmental technology. Catal Today 211:2–8
Mustafa MF, Fu X, Liu Y, Abbas Y, Wang H, Lu W (2018) Volatile organic compounds (VOCs) removal in non-thermal plasma double dielectric barrier discharge reactor. J Hazard Mater 347:317–324
Piumetti M, Fino D, Russo N (2015) Mesoporous manganese oxides prepared by solution combustion synthesis as catalysts for the total oxidation of VOCs. Appl Catal B Environ 163:277–287
Qin C, Huang X, Dang X, Huang J, Teng J, Kang Z (2016) Toluene removal by sequential adsorption-plasma catalytic process: effects of Ag and Mn impregnation sequence on Ag-Mn/γ-Al2O3. Chemosphere 162:125–130
Quoc An HT, Pham Huu T, Le Van T, Cormier JM, Khacef A (2011) Application of atmospheric non thermal plasma-catalysis hybrid system for air pollution control: toluene removal. Catal Today 176:474–477
Sivachandiran L, Thevenet F, Rousseau A (2015) Isopropanol removal using MnxOy packed bed non-thermal plasma reactor: comparison between continuous treatment and sequential sorption/regeneration. Chem Eng J 270:327–335
Strohmeier BR, Hercules DM (1985) Surface spectroscopic characterization of manganese/aluminum oxide catalysts. Chemischer Informationsdienst 16:4922–4929
Sudhakaran MSP, Trinh HQ, Karuppiah J, Hossian MM, Mok YS (2017) Plasma catalytic removal of p-xylene from air stream using γ-Al2O3 supported manganese catalyst. Top Catal 60:1–11
Tang W, Wu X, Li D, Wang Z, Liu G, Liu H, Chen Y (2014) Oxalate route for promoting activity of manganese oxide catalysts in total VOCs’ oxidation: effect of calcination temperature and preparation method. J Mater Chem A 2:2544–2554
Tao J, Tang H, Patra A, Bhattarai P, Perdew JP (2018) Modeling the physisorption of graphene on metals. Phys Rev B 97(16):165403
Tomatis M, Xu H-H, He J, Zhang X-D (2016) Recent development of catalysts for removal of volatile organic compounds in flue gas by combustion: a review. J Chem 2016:1–15
Trinh QH, Mok YS (2015) Effect of the adsorbent/catalyst preparation method and plasma reactor configuration on the removal of dilute ethylene from air stream. Catal Today 256:170–177
Vandenbroucke AM, Mora M, Jiménez-Sanchidrián C, Romero-Salguero FJ, De Geyter N, Leys C, Morent R (2014) TCE abatement with a plasma-catalytic combined system using MnO2 as catalyst. Appl Catal B Environ 156-157:94–100
Veerapandian S, Leys C, De Geyter N, Morent R (2017) Abatement of VOCs using packed bed non-thermal plasma reactors: a review. Catalysts 7(4):113
Wang B, Chi C, Xu M, Wang C, Meng D (2017) Plasma-catalytic removal of toluene over CeO2-MnOx catalysts in an atmosphere dielectric barrier discharge. Chem Eng J 322:679–692
Wang H, Huang Z, Jiang Z, Jiang Z, Zhang Y, Zhang Z, Shangguan W (2018) Trifunctional C@MnO catalyst for enhanced stable simultaneously catalytic removal of formaldehyde and ozone. ACS Catal 8:3164–3180
Yang Y, Huang R, Chen L, Zhang J (1993) Redox behavior of trimanganese tetroxide catalysts. Appl Catal A Gen 101:233–252
Yao P, Liu H, Wang D, Chen J, Li G, An T (2018) Enhanced visible-light photocatalytic activity to volatile organic compounds degradation and deactivation resistance mechanism of titania confined inside a metal-organic framework. J Colloid Interface Sci 522:174–182
Zhang X, Junhui Y, Jing Y, Ting C, Bei X, Zhe L, Kunfeng Z, Ling Y, Dannong H (2018) Excellent low-temperature catalytic performance of nanosheet Co-Mn oxides for total benzene oxidation. Appl Catal A Gen 566:104–112
Zhang Z, Jiang Z, Shangguan W (2016) Low-temperature catalysis for VOCs removal in technology and application: a state-of-the-art review. Catal Today 264:270–278
Zhu R, Mao Y, Jiang L, Chen J (2015a) Performance of chlorobenzene removal in a nonthermal plasma catalysis reactor and evaluation of its byproducts. Chem Eng J 279:463–471
Zhu X, Gao X, Qin R, Zeng Y, Qu R, Zheng C, Tu X (2015b) Plasma-catalytic removal of formaldehyde over Cu-Ce catalysts in a dielectric barrier discharge reactor. Appl Catal B Environ 170-171:293–300
Zhu X, Tu X, Chen M, Yang Y, Zheng C, Zhou J, Gao X (2017) La0.8M0.2MnO3 (M = Ba, Ca, Ce, Mg and Sr) perovskite catalysts for plasma-catalytic oxidation of ethyl acetate. Catal Commun 92:35–39
Zuo H, Xu D, Liu W, Dan H, Liu X, Lin S, Hou P (2018) Heat-treated dolomite-palygorskite clay supported MnOx catalysts prepared by various methods for low temperature selective catalytic reduction (SCR) with NH3. Appl Clay Sci 152:276–283
Acknowledgments
The authors would like to thank Hamada Shoma-san in Einaga-Hojo-lab for adjusting the non-thermal plasma system in this experiment. This work was financially supported by the National Key Research & Development Plan (2017YFC0211804).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Responsible editor: Philippe Garrigues
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
• MnOx with Mn3O4 phase was obtained after being calcined.
• MnOx calcined at 250 °C had far larger BET surface area than others’.
• The synergistic effect between MnOx calcined at 250 °C and NTP was most effective in benzene removal.
Rights and permissions
About this article

Cite this article
Guo, H., Liu, X., Hojo, H. et al. Removal of benzene by non-thermal plasma catalysis over manganese oxides through a facile synthesis method. Environ Sci Pollut Res 26, 8237–8247 (2019). https://doi.org/10.1007/s11356-019-04264-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-019-04264-5