
Abstract
Purpose
Adverse symptom event reporting is vital as part of clinical trials and drug labeling to ensure patient safety and inform risk–benefit decision making. The purpose of this study was to assess the reliability of adverse event reporting of different clinicians for the same patient for the same visit.
Methods
A retrospective reliability analysis was completed for a sample of 393 cancer patients (42.8% men; age 26–91, M = 62.39) from lung (n = 134), prostate (n = 113), and Ob/Gyn (n = 146) clinics. These patients were each seen by two clinicians who independently rated seven Common Terminology Criteria for Adverse Events (CTCAE) symptoms. Twenty-three percent of patients were enrolled in therapeutic clinical trials.
Results
The average time between rater evaluations was 68 min. Intraclass correlation coefficients were moderate for constipation (0.50), diarrhea (0.58), dyspnea (0.69), fatigue (0.50), nausea (0.52), neuropathy (0.71), and vomiting (0.46). These values demonstrated stability over follow-up visits. Two-point differences, which would likely affect treatment decisions, were most frequently seen among symptomatic patients for constipation (18%), vomiting (15%), and nausea (8%).
Conclusion
Agreement between different clinicians when reporting adverse symptom events is moderate at best. Modification of approaches to adverse symptom reporting, such as patient self-reporting, should be considered.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Monitoring of symptoms, which may be attributable to treatment, (i.e., “adverse symptom events”) is essential during medical practice to ensure patient safety and adjust treatment planning [1–4]. It is also a key component of clinical trial conduct in order to inform investigators about the potential toxicities of interventions, to protect research participants, and to assist regulators when they balance efficacy versus risks during the approval process [5, 6]. In oncology, adverse event monitoring is particularly important, both because patients can be highly symptomatic due to underlying disease processes and because cancer drugs have traditionally been more toxic than agents used in other diseases. Moreover, patients, clinicians, and regulators are generally willing to tolerate greater toxicities due to the stakes involved in cancer treatment, particularly if survival may be improved [7].
The standard approach to adverse event monitoring in oncology is the Common Terminology Criteria for Adverse Events (CTCAE) [8]. The CTCAE is an item bank of individual questions, each of which represents a discrete adverse event (e.g., retinal tear, neutropenia, or nausea). Each item is graded using a 5-point ordinal scale, and each grade level response option is anchored to verbiage, which describes a clinical scenario felt to be representative of that level of severity [8].
During the conduct of clinical trials sponsored by the National Cancer Institute (NCI), it is mandated that adverse events be reported using CTCAE items [1]. Moreover, the CTCAE has been widely adopted in oncology beyond this context and has become standard in industry-sponsored cancer clinical trials and during routine oncology clinical practice (particularly during chemotherapy treatment) [7, 9].
The CTCAE was created and has been updated via a consensus-based process and the items have not been evaluated for validity or reliability [7]. This limitation has been acknowledged by its developers at the NCI, but nonetheless, the clinical value of this instrument has been demonstrated by its widespread adoption [10].
The CTCAE includes multiple categories of adverse event items, including lab-based toxicities such as neutropenia (which are generally sourced directly from lab reports); clinical measurement-based toxicities such as hypertension (which are typically evaluated and reported by clinicians); and symptoms such as fatigue or nausea. It is notable that this last category (i.e., symptoms) is currently reported by clinicians rather than patients. Although there is mounting evidence that patients are in a better position to report on their own symptoms than are clinicians [3], and patient-reported outcomes (PROs) have been established as the gold standard for symptom reporting in efficacy evaluation [11], the standard approach to symptom toxicity reporting in clinical trials remains clinician reporting.
We have previously reported that patient and clinician reports of adverse symptom events are discrepant [9]. Furthermore, patient reports of this information are more highly correlated with measures of underlying health status than are clinician reports [4]. Yet, a remaining argument in favor of continued reporting of this information by clinicians rather than patients is that clinicians possess expert training and perspective, which allows them to describe the patient experience with treatment within the broader context of a disease, population, or intervention of interest.
Given the importance of clinician reporting of adverse symptoms both in clinical trials to understand toxicities and in clinical care to dictate treatment decisions, it is essential to establish whether the current approach is reliable. Therefore, we designed a study using data recorded in the medical record as part of standard care delivery at Memorial Sloan-Kettering Cancer Center (MSKCC), in which two different clinicians see the same patient on the same day consecutively and independently document symptoms using a CTCAE checklist form within a short amount of time during chemotherapy treatment. A first clinician sees the patient in a visit room and the second in the chemotherapy suite shortly thereafter. Therefore, a natural experiment is possible in which the ratings of these clinicians are compared. To conduct such an analysis, ratings were retrospectively abstracted from medical charts and compared with each other. Such a natural experiment would not be possible in other settings where this dual evaluation approach is not used.
Patients and methods
Patients
The study sample consists of a retrospective chart review of 393 English-language speaking cancer patients of mixed disease type (i.e., lung, prostate, and gynecologic) who were undergoing chemotherapy regimens at MSKCC between March 2005 and August 2009. These patients were part of an existing study protocol approved by the Institutional Review Board at MSKCC to evaluate the feasibility of a new computerized patient interface. Eligibility for this study included any patient with the stated cancer types receiving chemotherapy at MSKCC, without any other restrictions.
Study design
To investigate levels of clinician agreement, routinely documented patient electronic medical records were examined using the Health Information System of MSKCC. Medical record numbers (MRNs) and corresponding date of consent, age, gender, clinic (i.e., lung, prostate, or gynecologic), and primary oncologist were extracted for each patient. Clinician CTCAE ratings of constipation, diarrhea, dyspnea, fatigue, nausea, neuropathy, and vomiting were transcribed for each clinician rater for up to six consecutive visits for each identified patient. This information is routinely entered onto a standard paper form at visits by clinicians. For patients receiving active chemotherapy, it is standard practice for two independent clinicians to complete this information consecutively, in close time proximity, without access to each others’ reports: during a toxicity assessment in the physician’s office, and at the time of check-into the chemotherapy suite.
The form includes the name of each symptom, with a checkbox to rate the CTCAE grade, and a key providing definitions of each CTCAE grade for each symptom. The clinician raters for each patient at any given clinic visit do not have access to each others’ CTCAE reports.
For the primary analysis of agreement, data from the initial captured visit encounter was planned. A sensitivity analysis was also planned in which data from subsequent visits was analyzed to assess for the stability of agreement at different time points.
It was noted whether the patient was enrolled in a clinical trial during the recorded visits, as well as whether the person that filled out the form was a physician or nurse.
Statistical analysis
In order to quantify the level of agreement between independent clinicians, intraclass correlation coefficients (ICC) [12] were calculated for each symptom individually using the method described by Shrout and Fleiss [13]. ICCs were interpreted using the following criteria: values less than 0.40 indicate poor agreement, values between 0.40 and 0.75 indicate moderate agreement, and values greater than 0.75 indicate excellent agreement [14]. Because symptoms could be reported by either nurses or physicians, a sensitivity analysis was conducted to assess whether clinician type affected levels of agreement.
Results
Patients
Characteristics of the 393 identified patients are shown in Table 1. The median age was 63 years (range 26–91 years). Patients were diagnosed with lung (34%), prostate (29%), or gynecologic (37%) malignancies, with most having high levels of function based on the provider-reported Karnofsky Performance Status scores (i.e., 85% of patients ≥80 on a 100 point performance status scale). Twenty-three percent of the overall sample (n = 99) were enrolled in a clinical trial at the time of their visit.
The average amount of time that passed between oncology and chemotherapy visits was 68.04 min (Median = 54.00, SD = 54.91). Time between visits was not significantly associated with clinician ratings.
Table 2 shows the intraclass correlation coefficients and 95% confidence intervals for clinician ratings across patients, based on the single initial visit encounter (N = 393). The concordance of clinician rating was within the moderate range of 0.46–0.71. The concordance of clinician ratings was not significantly associated with patient gender, cancer type, race/ethnicity, or whether a patient was enrolled in a clinical trial at the time of observation.
Table 2 also shows ICCs with 95% confidence intervals capturing ratings of symptoms between physicians and nurses, as well as between nurses and nurses. All 95% confidence intervals overlapped, which is a crude indication of no discernable differences in the ICC estimates for different types of raters (post hoc analysis) [15]. ICCs are also shown for the subsample of 99 patients enrolled in treatment trials and are not statistically different from the overall sample.
In order to investigate the stability of the concordance in clinician versus clinician ratings across multiple clinic visits, concordance estimates were examined in a subsample of patients who had data available from visits on six separate dates (n = 132). ICCs did not statistically differ across these six observation points for any of the symptoms (i.e., constipation, diarrhea, dyspnea, fatigue, nausea, neuropathy, and vomiting), indicating these estimates are stable over time.
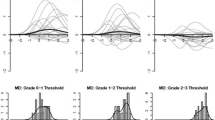
Levels of agreement between raters may appear to be more favorable when patients are perceived as being “asymptomatic” and therefore rated as zero by both clinicians. It has been suggested that it may be more challenging for clinicians to reach agreement on a severity grade when a patient is “symptomatic” and grades greater than zero must be selected [4]. Therefore, we wished to separately analyze clinician–clinician agreement for those patients who were considered as being symptomatic, defined as patients for whom at least one clinician graded severity above zero. For such an analysis, ICC’s could not be meaningfully interpreted because of limited variability due to the restricted range of ratings (i.e., most patients were rated as a 1 or 2) [13]. Consequently, agreement in the “symptomatic” sample is shown descriptively in Fig. 1. Levels of absolute agreement are low, ranging between 15 and 43%, depending on the symptom. In particular, the frequency of disagreement of 2 or more points between raters ranged between 1 and 18%, variable by symptom (e.g., vomiting: 15%; constipation 18%).

Agreement between Independent clinicians by symptom—symptomatic patients only
Discussion
This natural experiment demonstrates lower than desired levels of agreement between clinician reporting of adverse symptom events via the CTCAE. It is of particular concern that a two-point difference between raters’ scores for “symptomatic” patients was observed in 18% of cases for constipation, 15% for vomiting, and more than 5% of the time for nausea, dyspnea, and fatigue. A two-point difference on the narrow CTCAE scale is sufficient to dictate meaningful treatment changes such as chemotherapy dose reductions or eligibility for continued treatment. Since the CTCAE is the standard mechanism for documenting adverse events in cancer clinical trials and is frequently used in routine care, this brings into question the reliability and meaningfulness of this information. Notably, 23% of our patients were enrolled in therapeutic clinical trials in which CTCAE data inform investigators, drug safety monitors, and regulators about the safety profile of drugs. If this information is inconsistently reported between raters, this may compromise the risk–benefit evaluations made by these stakeholders. Moreover, a downstream implication is that unreliable information will be represented in publications and drug labels and may be used by clinicians and patients to inform treatment decisions.
Is the lack of reliability observed in this study attributable to the CTCAE itself, or is it an inherent limitation of clinician symptom reporting? Notably, the CTCAE was developed via a consensus process without any formal assessment of the validity or reliability of its items [7, 10]. Conceivably, a symptom checklist for clinicians could be developed with an up-front evaluation of measurement properties [11].
However, we question whether any clinician symptom assessment will be capable of reliably representing patients’ subjective experiences. Prior research demonstrates the challenge of any individual accurately representing another individual’s symptoms [3, 4, 9, 16]. Although clinicians have expert training and experience, they are still inherently limited in this sense. Clinicians appear to interpret or filter patients’ reported symptom information based on their own experiences, which can lead to different ratings between clinicians [2, 4, 5, 9, 17].
What are the possible reasons that clinicians may filter patients’ reports of symptoms? Various mechanisms have been postulated, including limited time at visits to fully explore symptoms; clinician downgrading because they understand a continuum of severity along which a given patient is contextualized; clinician downgrading in medical documentation to justify continuing a treatment; and patient understatement of symptoms during clinical encounters to “please” clinicians or to remain in a study [3, 17–19]. Regardless of the mechanism, clinicians appear to systematically under-endorse patients’ symptoms compared to patients themselves [2, 4, 9, 20]—and as shown in this study, they do so to a varying degree between themselves.
More broadly, adverse symptom event reporting in the cancer clinical trials enterprise is troubled beyond our observation of unreliable reporting at the point-of-care. Adverse symptom events in most industry trials outside of oncology are reported using items from the Medical Dictionary of Regulatory Activities (MedDRA). Mechanistically, clinicians document adverse symptoms in charts, then this information is abstracted by non-clinical data managers and via mapping verbatim symptom terms to MedDRA. But data are often lost or transformed during this process [21, 22]. Therefore, the value of reported adverse symptom event information in clinical trials must be generally questioned, and the downstream ability of regulators to fully appreciate the symptom toxicity profiles of drugs must similarly be questioned.
An alternate approach to adverse symptom event monitoring and documentation is direct, unfiltered, and patient self-reporting. Collecting such information directly from patients bypasses the multiple pitfalls of clinician reporting. This approach would be consistent with the US Food and Drug Administration’s recent Guidance for Industry on the Use of Patient-Reported Outcome Measures in Medical Development to Support Labeling Claims, which has generally been applied to evaluation of symptoms for evaluation of treatment benefit (i.e., efficacy), but could be further expanded in its application to encompass evaluation of tolerability and safety [11]. Well-developed patient-reported instruments generally have high levels of reliability [23]. Efforts are underway at the National Cancer Institute to develop a patient version of the CTCAE called the Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE),Footnote 1 and several of the authors are included in this effort. It remains unclear if patient self-reports in this context would supplement of replace the current standard approach of clinician reporting, and this is an area of active investigation.
Limitations of our study include its conduct in a single, tertiary cancer center with limited diversity of the patient population in terms of race and ethnicity, and inclusion of only three cancer type populations (lung, prostate, gynecologic). Clinicians were unaware that these comparisons would be made, and no special instructions were provided to clinicians how to complete the forms. Therefore, it is not known if training would improve levels of agreement. Nonetheless, more than a quarter of the patients were enrolled in clinical trials, in which meticulous attention is generally paid to adverse event reporting, and no differences from the larger patterns were seen in these patients.
In conclusion, the current approach to adverse symptom event reporting appears unreliable, and alternative approaches such as direct patient reporting should be considered for use in both clinical research and routine care. Improving the understanding of the patient experience with treatment will allow multiple stakeholders, including patients, clinicians, industry sponsors, regulators, and payers, to feel more confident in available information when balancing risk versus benefit for treatment and policy decisions.
Notes
Additional information on the PRO-CTCAE initiative can be found at https://wiki.nci.nih.gov/x/cKul
Abbreviations
- CTCAE:
-
Common Terminology Criteria for Adverse Events
- ICC(s):
-
Intraclass correlation coefficient(s)
- MedDRA:
-
Medical dictionary of regulatory activities
- MRN(s):
-
Medical record number(s)
- NCI:
-
National Cancer Institute
- PRO(s):
-
Patient-reported outcome(s)
- PRO-CTCAE:
-
Patient-reported outcomes version of the Common Terminology Criteria for Adverse Events
References
NCI: National Cancer Institute. (2001). Cancer therapy evaluation program. NCI guidelines—Expedited adverse event reporting requirements for NCI investigational agents. Bethesda: National Cancer Institute.
Basch, E., Iasonos, A., Barz, A., et al. (2007). Long-term toxicity monitoring via electronic patient-reported outcomes in patients receiving chemotherapy. Journal of Clinical Oncology, 25, 5374–5380.
Basch, E. (2010). The missing voice of patients in drug-safety reporting. New England Journal of Medicine, 362, 865–869.
Basch, E., Jia, X., Heller, G., et al. (2009). Adverse symptom event reporting by patients vs clinicians: Relationships with clinical outcomes. Journal of the National Cancer Institute, 101, 1624–1632.
Belknap, S. M., Georgopoulos, C. H., West, D. P., et al. (2010). Quality of methods for assessing and reporting serious adverse events in clinical trials of cancer drugs. Clinical Pharmacology and Therapeutics, 88, 231–236.
Ahmad, S. R. (2003). Adverse drug event monitoring at the Food and Drug Administration. Journal of General Internal Medicine, 18, 57–60.
Trotti, A., Colevas, A. D., Setser, A., et al. (2007). Patient-reported outcomes and the evolution of adverse event reporting in oncology. Journal of Clinical Oncology, 25, 5121–5127.
Trotti, A., Colevas, A. D., Setser, A., et al. (2003). CTCAE v3.0: Development of a comprehensive grading system for the adverse events of cancer treatment. Seminars in Radiation Oncology, 13, 176–181.
Basch, E., Iasonos, A., McDonough, T., et al. (2006). Patient versus clinician symptom reporting using the National Cancer Institute Common Terminology Criteria for Adverse Events: Results of a questionnaire-based study. The Lancet Oncology, 7, 903–909.
Bruner, D. W., Bryan, C. J., Aaronson, N., et al. (2007). Issues and challenges with integrating patient-reported outcomes in clinical trials supported by the National Cancer Institute-sponsored clinical trials networks. Journal of Clinical Oncology, 25, 5051–5057.
US Food and Drug Administration. (2009). Guidance for industry. Patient-reported outcome measures: Use in medical development to support labeling claims. Available from http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM193282.pdf. Accessed 9 Dec 2010
Uebersax, J. S. (1987). Diversity of decision-making models and the measurement of interrater agreement. Psychological Bulletin, 101, 140–146.
Shrout, P. E., & Fleiss, J. L. (1979). Intraclass correlations: Uses in assessing rater reliability. Psychological Bulletin, 86, 420–428.
Rosner, B. (2005). Fundamentals of biostatistics. Belmont, CA: Duxbury Press.
McGill, R., Tukey, J. W., & Larsen, W. A. (1978). Variation of box plots. The American Statistician, 32, 12–16.
Zegers, M., de Bruijne, M. C., Wagner, C., et al. (2010). The inter-rater agreement of retrospective assessments of adverse events does not improve with two reviewers per patient record. Journal of Clinical Epidemiology, 63, 94–102.
Weingart, S. N., Gandhi, T. K., Seger, A. C., et al. (2005). Patient-reported medication symptoms in primary care. Archives of Internal Medicine, 165, 234–240.
Berry, D. L., Moinpour, C. M., Jiang, C. S., et al. (2006). Quality of life and pain in advanced stage prostate cancer: Results of a Southwest Oncology Group randomized trial comparing docetaxel and estramustine to mitoxantrone and prednisone. Journal of Clinical Oncology, 24, 2828–2835.
Pakhomov, S., Jacobsen, S. J., Chute, C. G., et al. (2008). Agreement between patient-reported symptoms and their documentation in the medical record. The American Journal of Managed Care, 14, 530–539.
Hahn, E. A., Cella, D., Chassany, O., et al. (2007). Precision of health-related quality-of-life data compared with other clinical measures. Mayo Clinic Proceedings, 82, 1244–1254.
Ioannidis, J. P., Evans, S. J., Gøtzsche, P. C., et al. (2004). Better reporting of harms in randomized trials: An extension of the CONSORT statement. Annals of Internal Medicine, 141, 781–788.
Institute of Medicine, National Academy of Sciences. (2006). The future of drug safety: Promoting and protecting the health of the public. Washington, DC: National Academies Press.
Kirkova, J., Davis, M. P., Walsh, D., et al. (2006). Cancer symptom assessment instruments: A systematic review. Journal of Clinical Oncology, 24, 1459–1473.
Acknowledgments
This project was supported by a National Institutes of Health Research Training Grant (T32 CA009461-25); a National Institutes of Health Support Grant (P30-CA-008748). The findings in this manuscript were partially reported at the 31st Annual Meeting and Scientific Sessions of the Society of Behavioral Medicine, Seattle, WA, April 7–10, 2010.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Atkinson, T.M., Li, Y., Coffey, C.W. et al. Reliability of adverse symptom event reporting by clinicians. Qual Life Res 21, 1159–1164 (2012). https://doi.org/10.1007/s11136-011-0031-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11136-011-0031-4