
Abstract
Breast cancer is the leading cause of death among women, with approximately 1 million diagnoses annually. Triterpenoids, which have cancer preventive or anti-tumour efficacy towards various tumour cells, may play a role in breast cancer prevention. In our previous study, an acetic ether (EtOAc) fraction from the sporocarp of the edible mushroom Pleurotus eryngii (P. eryngii) exhibited significant tumour cell growth inhibition both in vitro and in vivo. In this study, three pentacyclic triterpenoid compounds (1–3) were isolated from EtOAc extracts using chromatographic separation and were identified using nuclear magnetic resonance (NMR) and mass spectrometry (MS). The compounds were 2, 3, 6, 23-tetrahydroxy-urs-12-en-28 oic acid (1), 2,3,23-trihydroxy-urs-12-en-28 oic acid (2) and lupeol (3). All three purified triterpenes showed significant inhibitory activity against breast cancer MCF-7 cell lines in vitro, with the greatest activity exhibited by compound 1, followed by compound 2 and 3. The IC50 values were 15.71, 48 and 66.89 μM, respectively. Our study may help elucidate the health benefits of P. eryngii mushroom consumption.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is one of the most frequently diagnosed malignancies and is a major cause of death in women [1]. According to statistical analyses, the morbidity has increased constantly since the 1970s, and approximately 400,000 women die from the disease each year [2]. Women in developed countries have a 1 in 8 possibility of developing breast cancer in their lifetimes [3]. Moreover, the incidence of male breast cancer is also increasing [4]. Unfortunately, there remains no effective cure for the vast majority of patients with advanced breast disease. Current treatments, such as radiotherapy, surgery, adjuvant chemotherapy, monoclonal antibody immunotherapy and hormone therapy, only partially reduce the morbidity of breast cancer. However, side effects always occur with these treatments. For example, the cytotoxic drugs used in systemic therapy are severe enough to decrease immune function and eventually damage the major organ systems [5], whereas localized radiotherapy often causes pain, oedema, fibrosis, and irreversible changes in mobility. It is therefore necessary to identify novel breast cancer chemopreventive drugs with acceptable efficacy and toxicity limits.
During the last decades, various bioactive phytochemicals and compounds based on natural products have shown preventive and therapeutic potential towards breast cancer [6–10]. One such group of phytochemicals, triterpenoids, is primarily found in a variety of plants. Increasing evidence of their wide spectrum of pharmacological effects coupled with low-toxicity profiles have sparked renewed interest regarding their effects on human health and disease. Triterpenoids are widely used in Asian traditional medicines due to their multifunctional activities, such as antibacterial, anti-inflammatory, hypolipidemic, hepatoprotective, cardiovascular, and analgesic effects [11–14]. Emerging studies indicate that triterpenoids exert anti-tumour activities in various in vitro and in vivo model systems, without manifesting toxicity in normal cells [15–17].
Mushrooms have been widely consumed as food ingredients for centuries, not only for their good flavour and texture but also for their substantial nutritional value and potential medicinal value. Recently, mushrooms were recognized as a prolific source of bioactive substances for the development of nutraceuticals and drugs. P. eryngii, known for its good taste, contains many biologically active compounds, such as peptides, polysaccharides, lipids, triterpenoids, sterols and dietary fibre, which contribute to their medicinal usage. Several recent chemical studies indicated that P. eryngii extract has several biological functions, such as antioxidant [18], hypolipidemic [19], anti-tumour and immunomodulating activities [20]. However, specific analyses of the plant bioactive compounds are lacking. Therefore, an effective analytical procedure for the separation of the major activity related compounds in P. eryngii is necessary.
The objective of this study was to isolate and characterize the triterpenoids in the EtOAc-soluble portion of P. eryngii extract (Fig. 1). Additionally, the growth inhibitory activity of these isolated compounds against breast cancer MCF-7 cells was also investigated to provide a theoretical basis for the development of new drugs.

Structures of triterpene acids
Materials and Methods
Materials and Instruments
Fresh P. eryngii was supplied by the Tianjin Tianshou Edible Fungus Company and stored at 4 °C. Silica gel and Sephadex LH-20 (Qingdao Haiyang Chemicals, China) were used for column chromatography (CC). 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was obtained from Sigma-Aldrich. RPMI-1640 medium, foetal bovine serum (FBS) and 0.25 % trypsin-EDTA were purchased from Gibco/Life Technologies (USA). The 2, 3, 6, 23-tetrahydroxy-urs-12-en-28 oic acid, 2, 3, 23-trihydroxy-urs-12-en-28 oic acid and lupeol (purity ≥ 97, 98, and 99 %, respectively) used for cell experiments were purchased from Shanghai Yuanye Bio-Technology Company. Dimethyl sulphoxide (DMSO) and organic solvents were purchased from Tianjin JiangTian Reagent Company. High-resolution electrospray ionization mass spectrometry (HRESIMS) data were acquired on an Agilent 1200 HPLC/6520Q-TOF (American) mass spectrometer. Nuclear magnetic resonance spectra were recorded on a Bruker-III-500 spectrometer (Germany).
Preparation of the P. eryngii Extract
Sporocarps of P. eryngii were dried at 45 °C, pulverized, extracted with 100 % MeOH (15 L × 3) under reflux for 8 h and then placed in an ultrasonic apparatus for 30 min. After removal of the solvent using a rotary evaporator, the thick paste of 100 % MeOH extractives (980 g) was resuspended in H2O and then successively partitioned into petroleum ether (16 g), EtOAc (103 g) and n-BuOH (178 g) fractions.
Determination of the Triterpenoid Concentrations of the Petroleum Ether, EtOAc and n-BuOH Extracts
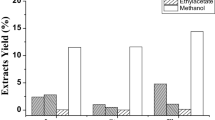
The triterpenoid contents of the petroleum ether, EtOAc and n-BuOH extracts were measured using spectrophotometry at 548 nm with oleanolic acid as the reference standard [21]. The final triterpenoid concentrations of the petroleum ether, EtOAc and n-BuOH extract were <20 mg/g, 131.47 ± 0.73 mg/g, and <20 mg/g, respectively. The EtOAc fraction, which showed the highest triterpenoid concentration, was subjected to repeated CC with a gradient elution of solvents.
Isolation of the EtOAc Extract
The EtOAc extract was subjected to silica gel (150–250 μm) CC eluted with a gradient of petroleum ether/acetone/MeOH (95:4:1→20:40:40, v/v/v). Based on the thin-layer chromatography (TLC) analysis, the collected solutions containing triterpenoid were combined into five fractions (Fr.1-Fr.5). Fraction 2, which had the highest mass per cent, was selected for the next separation.
Fraction 2 (19 g), obtained by elution with petroleum ether/acetone/MeOH (30:70:2, v/v/v), was separated on silica gel (150–250 μm) and CC eluted with CHCl3/MeOH (100:0→0:100, v/v), and seven subfractions were obtained (Fr.A–Fr.G). Fr.B and Fr.E had the relatively higher mass per cents, were selected for the next separation.
Fr.B (2.3 g), obtained by elution with CHCl3/MeOH (90:10, v/v), was separated on silica gel (150–250 μm) CC eluted with CHCl3/MeOH/H2O (10:0:0→4:4:2, v/v/v) to yield four subfractions (Fr.B1–Fr.B4). Subfraction Fr.B4 (1.2 g) was further separated by preparation lamella chromatography developed with CHCl3/MeOH/H2O (8:1:1, v/v/v) to yield compound 1 (9 mg) and compound 2 (7 mg). Fr.E, obtained by elution with CHCl3/MeOH (60:10, v/v), was separated on silica gel (150–250 μm) CC eluttion with CHCl3/MeOH/H2O (8:1:0.5→3:4:4, v/v/v) to yield three subfractions (Fr.E1–Fr.E3). Compound 3 (12 mg) was purified from subfraction Fr.E3 by Sephadex LH-20 CC eluted with CHCl3/MeOH (40:60).
The extraction and isolation flow schematic is shown in Fig. 2. The isolated compounds were then identified using nuclear magnetic resonance (NMR) and high-resolution electrospray ionization mass spectrometry (HRESIMS).

Extraction and isolation flow schematic
TLC Analysis of Fractions
Evaluation of the fractions was performed by thin-layer chromatography on normal phase silica gel 60 F254 plates Merck (Darmstadt, Germany) developed in ethyl acetate–methanol–water in the appropriate proportion. Visualization of the triterpenoids was performed by spraying with 5 % sulphuric acid in ethanol, and subsequent heating to 110 °C for 5 min on a hot plate.
Cell Lines
The MCF-7 human breast cancer cells were obtained from Tianjin Tumor Hospital and were maintained in RPMI-1640 medium containing 10 % v/v FBS and incubated at 37 °C in a humidified atmosphere containing 95 % air and 5 % CO2. The culture media were changed routinely every 2 days and were free of contamination during the study period.
Inhibition of MCF-7 Cell Proliferation
The proliferation inhibition activity of the three isolated compounds against the human breast cancer cell line MCF-7 was evaluated using the MTT assay [22]. As a colourimetric assay, the MTT assay uses cellular metabolic activity to convert the colourless tetrazolium to the purple-coloured formazan salt, which can be quantified by measuring the absorbance at 570 nm. The tumour cell line was cultured in RPMI 1640 medium with 10 % FBS and was then diluted to a concentration of 2.5 × 104 cells/mL prior to aliquoting (100 μL) into each well of a 96-well plate and incubation at 37 °C in a humidified atmosphere of 5 % CO2 for 10 h. After compounds 1–3 were added to the tumour cells, the plate was incubated for an additional 96 h. The concentrations of the tested compounds were 10, 25, 50, 100 and 200 μM and were dissolved in the culture medium with DMSO (the final concentration less than 0.1 %) to completely dissolve the compounds. Then, 20 μL of the MTT/medium solution (5 mg/mL in PBS) was added to each well and the tumour cells were incubated in the dark for 4 h. After removing the solution from the wells, 150 μL of DMSO was added to each well and the plate was shaken for 5 min at 60 times per minute to dissolve the precipitate. The assay plate was measured using a microplate reader at 570 nm, and the inhibitory activities were calculated. The optical density values of three independent experiments performed in triplicate were averaged. The relative cell survival rate was obtained by dividing the optical density value of the test group by the optical density value of the control group. A plot of the percentage of cell survival versus the concentrations was used to determine the half-maximal concentration of growth inhibition (IC50).
Statistical Analysis
All statistical analyses were performed using SPSS 11.5 (SPSS Inc. USA), and all data are expressed as the means ± SD. One-way ANOVA was used to determine the differences between multiple groups. Differences of P < 0.05 were considered statistically significant.
Results and Discussion
Confirmation of Chemical Structures
High-resolution ESI-MS experiments determined the molecular formulas of 1 and 2 as C30H48O6 and C30H48O5. 13C spectral assignments are provided in Table 1. Inspection of the downfield region of the proton NMR spectra indicated the presence of compounds containing an ursane-type skeleton. 13C-NMR spectral data identified urs-12-ene types for all structures 1–2, and this was also achieved by inspection of the d-values of the olefinic carbons, revealing that C-12 is deshielded by 2 ppm, otherwise C-13 is shielded by 5 ppm compared to the corresponding carbons of olean-12-enes [23]. The chemical shift differences of the double bond carbons were caused by the spatial proximity of the 19β-(equatorial)-methyl group in the urs-12-ene structure. An additional spectroscopic characteristic was position C-18 in the urs-12-enes (1–2), showing a strong downfield shift of δ11 ppm compared with the olean-12-enes due to the missing shielding effect of a 20β-(axial)-methyl-group [23].
Consistent with the reference data, the 13C-NMR resonances for all triterpene acids in 1–2 were almost superimposable. The 2, 3, 6, 23-tetrahydroxy-urs-12-en-28 oic acid (1), the 6-hydroxyl derivate of 2, 3, 23-trihydroxy-urs-12-en-28 oic acid, was elucidated by the strong downfield shift of carbon C-6 with nearly 50 ppm. As expected, 13C-resonance for the methylene group C-6 observed in substance 2 (δ-value 19 ppm) was not detectable in 1. C-7, in the β-position to the hydroxyl group, was slightly deshielded by δ 6 ppm compared to a chemical shift of δ 33.7 ppm in structure 2.
The identity of compound 3 (Fig. 1) was confirmed by comparison of 1H and 13C NMR spectra in C5D5N with reference data as well as confirmation of distinct fragment peaks in the mass spectrum. The aromatic region of the 1H spectrum provided the information necessary for the elucidation of 3. Chemical shifts were observed at δ 8.01 (d, J) 7.2 Hz, (H-2, 6), 7.56 (t, H-4), and 7.45 (t, H-3, 5). A characteristic “roofing effect” was observed among peaks due to coupling between adjacent protons H-2 with H-3 and H-3 with H-4. The 13C spectrum showed two characteristic peaks at δ 151.0 (C-20) and 109.3 (C-29). The chemical shifts and peak intensity patterns displayed by three were consistent with those found in the Spectral Data Base System (SDBS) [24]. The mass spectrum of 3 (C30H50O) gave a molecular ion at m/z 425 and loss of a hydroxyl group at m/z 408 as the predominant peaks. We have found no previous reports of lupeol isolated from Pleurotus mushrooms.
Inhibition of MCF-7 Cell Proliferation
The inhibition of proliferation of human breast cancer MCF-7 cells by compounds 1–3 at different concentrations (10, 25, 50, 100 and 200 μM) are shown in Fig. 3. All of the compounds significantly inhibited MCF-7 cell proliferation, with compound 1 being the most effective anti-tumour agent (IC50 value of 15.71 μM), followed by compound 2 (IC50 value of 48 μM) and compound 3 (IC50 value of 66.89 μM). As the concentrations of compounds 1–3 increased from 10 to 200 μM, the tumour cell survival rate significantly decreased from 60.14 % to 22.99 %, 69.03 % to 29.21 % and 89.55 % to 20.12 %, respectively. Of these, lupeol has been reported to have biological activity in humans [23] and is hypothesized to have anti-tumour effect towards several cancer. Further studies of the effect of lupeol on breast cancer are underway. Lupeol slightly inhibited tumour cell growth in our assays. The varying inhibition of the three purified compounds towards tumour cells may be due to their differences in chemical structure. Several functional groups, such as hydroxyl, carboxyl, and double bonds, may contribute to the proliferation inhibition capacity of the three compounds, although their structure–activity relationships remain unclear. Several possible mechanisms indicate that triterpenoids may exert anti-tumour activities through the modulation of cellular proliferation, apoptosis, differentiation, inflammation, oxidative stress, angiogenesis and several key signalling pathways implicated in the initiation, promotion and progression of cancers [15–17].

Triterpenoids inhibit the proliferation of human breast cancer cells. Data are the means ± SD, n = 3
Compound 1 showed greater anti-tumour activity than compound 2, indicating that the presence of an additional −OH group at C-6 enhances the cytotoxic effect. This was consistent with previous reports. According to Huang et al. [24], the number of hydroxyl groups in triterpene may play a crucial role in their cytotoxicity against human tumour cells. Yu et al. [25] found that the C-16 hydroxyl group contributed to enhancing the biological activity of tubeimoside ІІ. A similar result was also observed in the study by Liu et al. [26], which indicated that the hydroxylation at the C-3 of betulinic acid is important for murine melanoma B16 cells apoptosis.
Compounds 1 and 2, with the carboxylic acid at C-28, showed slightly more potent inhibitory activities in MCF-7 cells than compound 3, which lacks a carboxy group at C-28. These results suggest that a free carboxylic group at C-28 may be important to exert antiproliferative activity. Yang et al. [27] compared the anti-proliferative activity of triterpenes from the leaves and twigs of Juglans sinensis and found that the inhibitory activity on HSC-T6 cells decreased when the −COOH at C-28 was replaced by a glucopyranoside moiety. In addition, different carbon ring skeletons of the three purified compounds may be responsible for their dissimilar inhibitory activities. The ursane-type compounds 1 and 2 showed more potent antiproliferative activities than the lupane-type compound 3. This is similar to the report that ursane-type triterpenoids have greater inhibitory activities in general than those of the oleanane type for the proliferation of HSC-T6 cells [27]. A study also showed that most of the oleanane-type triterpene saponins exhibit slight greater cytotoxicity than dammarane-type triterpene saponins towards cervical cancer HeLa cells [24]. Moreover, the weaker anti-tumour activity of compound 3 may be related to the absence of a double bond at C-12,13, which is similar to the report that a 5,6-double bond plays a significant role in biological effects (apoptosis and cell cycle arrest) against the human 1547 cell line [28]. Taken together, the carbon ring skeleton, the absence of an hydroxyl group at C-6 and a free carboxylic group at C-28, results in the weakest inhibitory activity of compound 3 for MCF-7 cell proliferation, compared with compound 1 and compound 2.
In conclusion, our results provide new insight into the anticarcinogenic action of triterpene acids in human breast cancer. Moreover, the triterpene compound 1 and compound 2 were isolated for the first time from EtOH extracts of the edible mushroom P. eryngii. Therefore, our study may help elucidate the health benefits of P. eryngii mushroom consumption.
References
Ogunleye AA, Xue F, Michels KB (2010) Green tea consumption and breast cancer risk or recurrence: a meta-analysis. Breast Cancer Res Treat 119:477–484
Veroensi U, Boyle P (2008) Breast cancer. International encyclopedia of public health. v. 5. Academic, San Diego, pp 348–357
Lillie SE, Brewer NT, O’Neill SC, Morrill EF, Dees EC, Carey LA, Rimer BK (2007) Retention and use of breast cancer recurrence risk information from genomic tests: the role of health literacy. Cancer Epidemiol Biomark 16:249–255
Stang A, Thomssen C (2008) Decline in breast cancer incidence in the United States: what about male breast cancer. Breast Cancer Res Treat 112:595–596
Liu B, Yang M, Li R, Ding YT, Qian XP, Yu LX, Jiang XQ (2008) The antitumor effect of novel docetaxel-loaded thermosensitive micelles. Eur J Pharm Biopharm 69:527–534
Harlev E, Nevo E, Solowey E, Bishayee A (2013) Cancer preventive and curative attributes of plants of the Cactaceae family: a review. Planta Med 79:713–722
Pan MH, Ho CT (2008) Chemopreventive effects of natural dietary compounds on cancer development. Chem Soc Rev 37:2558–2574
Zhao X, Li L, Wang Z (2005) Chemoprevention of breast cancer: current status and future prospects. Front Biosci 11:2249–2256
Naithani R, Huma LC, Moriarty RM, McCormick DL, Rajendra GM (2008) Comprehensive review of cancer chemopreventive agents evaluated in experimental carcinogenesis models and clinical trials. Curr Med Chem 15:1044–1071
Bishayee A, Waghray A, Patel MA, Chatterjeed M (2010) Vanadium in the detection, prevention and treatment of cancer: the in vivo evidence. Cancer Lett 294:1–12
Yadav VR, Prasad S, Sung B, Bharat BA (2010) Targeting inflammatory pathways by triterpenoids for prevention and treatment of cancer. Toxins 2:2428–2466
Patlolla JMR, Rao CV (2012) Triterpenoids for cancer prevention and treatment: current status and future prospects. Curr Pharm Biotechnol 13:147–155
Bishayee A, Thoppil RJ, Waghray A, Kruse JA, Novotny NA, Darvesh AS (2012) Dietary phytochemicals in the chemoprevention and treatment of hepatocellular carcinoma: in vivo evidence, molecular targets, and clinical relevance. Curr Cancer Drug Targets 12:1191–1232
Ilyina A, Arredondo-Valdés R, Farkhutdinov S, Segura-Ceniceros EP, Martínez-Hernández JL, Zaynullin R, Kunakova R (2014) Effect of betulin-containing extract from birch tree bark on α-Amylase activity in vitro and on weight gain of broiler chickens in vivo. Plant Foods Hum Nutr 69:65–70
Petronelli A, Pannitteri G, Testa U (2009) Triterpenoids as new promising anticancer drugs. Anti-Cancer Drug 20:880–892
Laszczyk MN (2009) Pentacyclic triterpenes of the lupane, oleanane and ursane group as tools in cancer therapy. Planta Med 75:1549–1560
Parikh NR, Mandal A, Bhatia D, Siveen KS, Sethi G, Bishayee A (2014) Oleanane triterpenoids in the prevention and therapy of breast cancer: current evidence and future perspectives. Phytochem Rev 13:793–810
Jayakumar T, Ramesh E, Geraldine P (2006) Antioxidant activity of the oyster mushroom, Pleurotus ostreatus, on CCl4-induced liver injury in rats. Food Chem Toxicol 44:1989–1996
Chen J, Mao D, Yong Y, Li JL, Wei H, Lu L (2012) Hepatoprotective and hypolipidemic effects of water-soluble polysaccharidic extract of Pleurotus eryngii. Food Chem 130:687–694
Jeong YT, Jeong SC, Gu YA, Islam R, Song CH (2010) Antitumor and immunomodulating activities of endo-biopolymers obtained from a submerged culture of Pleurotus eryngii. Food Sci Biotechnol 19:399–404
Xin X, Yu Z, Fan QS, Wei Q (2009) Separation and purification of triterpenoid in artemisia selengensis turcz and its anti-oxidation and bacteriostatic effect in vitro. Nat Prod Res Dev 21:312–318
Fernández-Pérez F, Belchí-Navarro S, Almagro L, Bru R, Pedreño MA, Gómez-Ros LV (2012) Cytotoxic effect of natural trans-resveratrol obtained from elicited vitis vinifera cell cultures on three cancer cell lines. Plant Foods Hum Nutr 67:422–429
Mohato SB, Kundu AP (1994) 13C NMR spectra of pentacyclic triterpenoids-A compilation and some salient feature. Phytochemistry 37:1517–1575
Huang HC, Wu MD, Tsai WJ, Liao SC, Liaw CC, Hsu LC, Wu YC, Kuo YH (2008) Triterpenoid saponins from the fruits and galls of Sapindus mukorossi. Phytochemistry 69:1609–1616
Yu TX, Ma RD, Yu LJ (2001) Structure–activity relationship of tubeimosides in anti-inflammatory, antitumor and antitumor-promoting effects. Acta Phamacol Sin 22:463
Liu WK, Ho JC, Cheung FW, Liu BP, Ye WC, Che CT (2004) Apoptotic activity of betulinic acid derivatives on murine melanoma B16 cell line. Eur J Pharmacol 498:71–78
Yang H, Jeong EJ, Kim J, Sung SH, Kim YC (2011) Antiproliferative triterpenes from the leaves and twigs of Juglans sinensis on HSC-T6 cells. J Nat Prod 74:751–756
Trouillas P, Corbière C, Liagre B, Duroux JL, Beneytout JL (2005) Structure–function relationship for saponin effects on cell cycle arrest and apoptosis in the human 1547 osteosarcoma cells: a molecular modelling approach of natural molecules structurally close to diosgenin. Bioorg Med Chem 13:1141–1149
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No.31271979) and the Natural Science Foundation of Tianjin (No.15JCYBJC30100).
This article does not contain any studies with human or animal subjects.
Conflict of Interest
We have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Xue, Z., Li, J., Cheng, A. et al. Structure Identification of Triterpene from the Mushroom Pleurotus eryngii with Inhibitory Effects Against Breast Cancer. Plant Foods Hum Nutr 70, 291–296 (2015). https://doi.org/10.1007/s11130-015-0492-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11130-015-0492-7