
Abstract
This study evaluated the effect of in vitro digestion of flaxseed products on Folin-Ciocalteu reagent reducing substances (FCRRS), its antioxidant capacity and prevention of oxidative DNA damage in human monocyte cell line U937. Flaxseed protein isolate was obtained from defatted flaxseed meal and the protein hydrolysate with high antioxidant capacity was obtained from hydrolysis of the protein isolate with Alcalase in a two factor central composite rotatable design (pH 8.5 and enzyme: substrate 1:90, w/w). The FCRRS content and antioxidant capacity measured by FRAP and ORAC in aqueous and 70 % methanol extracts were the highest in protein hydrolysate, followed by protein isolate, while the defatted meal showed the lowest values. After in vitro gastrointestinal digestion, the FCRRS content of protein isolate and hydrolysate reached similar values, however the hydrolysate had the highest antioxidant capacity, measured by FRAP while the isolate had the highest ORAC values. The defatted meal showed the lowest capacity in all assays (p < 0.05). The hydrolysate did not protect against DNA damage induced by H2O2 in U937 cells under the conditions of the present study. The results suggest that flaxseed protein isolate and hydrolysate are potential functional food ingredients with antioxidant capacity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Antioxidant compounds limit deterioration and preserve the nutritional and sensory qualities of foods. In biological systems, antioxidants such as vitamins A, C, E and phenolic compounds are involved in promoting health and prevent chronic diseases and aging [1]. In recent years, peptides with antioxidant capacity have gained significant interest. These peptides can be released from the parent protein by in vitro hydrolysis using enzymes isolated from microorganisms, plants and animals or in vivo by the gastrointestinal (GI) enzymes and/or intestinal microbiota [2].
Simulated GI digestion has been used to evaluate the effect of digestion on the antioxidant capacity of compounds and their role in the prevention of GI diseases associated to oxidative damage from reactive oxygen species (ROS) [3]. Antioxidant compounds may also protect macromolecules such as lipids and DNA against damage caused by free radicals through scavenging activity [4]. Thus, there is an increasing interest in studying natural substances that can exhibit protective activities against genotoxicity caused by oxidative stress in DNA and thus prevent pathological conditions such as inflammation, diabetes, carcinogenesis, and aging [4].
Flaxseed (Linum usitatissimum L.) is known as a rich source of α-linolenic acid, dietary fiber and lignans [5]. More recently, it has also received attention as a source of bioactive peptides [5]. In vitro studies showed that flaxseed protein hydrolysates have biological activities such as antioxidant, anti-inflammatory and cholesterol-lowering ability [5–7]. However, the stability of the antioxidant compounds from flaxseed meal, its protein isolate and hydrolysate to in vitro digestion was not studied. The aims of the present study were to investigate the effects of the protein isolation and enzymatic treatment on the antioxidant capacity of flaxseed, to evaluate the stability of its antioxidant compounds to in vitro digestion and to investigate the ability of its isolate and hydrolysate, before and after in vitro digestion, to prevent oxidative changes to the DNA of U937 human lymphocyte cells.
Materials and Methods
Materials
Partially defatted brown flaxseed meal (PDFM) was obtained from Cisbra Ltd. (Panambi, RS, Brazil). The enzymes Alcalase 2.4 L, pepsin and pancreatin, bile salts, Folin-Ciocalteu reagent, gallic acid, Trolox, 2,4.6-tri (2-pyridyl)-s-triazine (TPTZ), 2,2′-azobis (2-methylpropionamidine) dihydrochloride (AAPH), sodium dodecyl sulfate (SDS) and tricine were purchased from Sigma (St. Louis, MO, USA). The sodium fluorescein was purchased from Synth (São Paulo, Brazil). Acrylamide, bis-acrylamide and Tris were purchased from Bio-Rad (Irvine, CA, USA). Methanol, trifluoroacetic acid (TFA), acetonitrile, coomassie brilliant blue G-250 and sodium hydroxide were purchased from Merck (Hohenbrunn, Germany).
Protein Isolate and Hydrolysate from Defatted Flaxseed Meal
PDFM was defatted with hexane (1:3, w/v) at ambient temperature (23 ± 2 °C) for 24 h with renewal of hexane every 6 h, yielding defatted flaxseed meal (DFM). To obtain the protein isolate (FPI), DFM was dispersed in deionized water (1:10), the pH was adjusted to 9 with 1 N NaOH and the mixture was stirred in a homogenizer (Fisaton, São Paulo, Brazil) at ambient temperature for 60 min, followed by centrifugation (16,274 × g for 30 min). The extraction process was repeated and supernatants of all steps were pooled. The pH was adjusted to 4.2 with 1 N HCl and the precipitated protein was separated by centrifugation, washed three times with acidified water (pH 4.2) and suspended in deionized water and pH was adjusted to 6 with 1 N NaOH. The protein isolate was freeze-dried and stored at −20 °C until analysis.
In order to obtain a flaxseed protein hydrolysate with high antioxidant capacity, a central composite rotatable design (CCRD) with 2 factors, 2 levels, 3 center points and 4 axial points with a total of 11 trials was employed. The independent variables were enzyme:substrate ratio (E/S) (1:150 to 1:30, w/w) and pH (7.5 to 9.5). The dependent variables were the antioxidant capacity and degree of hydrolysis (DH). To obtain the hydrolysates, FPI (5 % w/v protein in distilled water) was hydrolyzed with Alcalase at 60 °C for 180 min, under the conditions established for each CCRD assay. The pH was maintained by adding 0.25 N NaOH using an automatic titrator (DL50 Graphix Mettler Toledo, Schwerzenbach, Switzerland). The reaction was stopped by heating the reaction mixture at 90 °C for 10 min to inactivate the enzyme [8]. The hydrolysates were cooled (7 °C), frozen (−20 °C) and freeze-dried. A control test was performed with no Alcalase addition.
Characterization of PDFM, DFM, FPI and FPH
The ash, moisture, protein, fat, carbohydrate and dietary fiber contents of the PDFM, DFM and FPI was determined according to AOAC methods [9]. Fat and protein contents were determined by Bligh and Dyer [10] and Kjeldahl (N% × 6.25) [11] methods, respectively.
The Folin-Ciocalteu reagent reducing substances (FCRRS) content was determined in aqueous and methanolic (70 %) extracts [12]. The result was defined as the content of FCRRS, since the reagent is not specific for phenolic compounds and may react with other reducing substances, such as aromatic amino acids residues and ascorbic acid [12]. The measurements were carried out in triplicate, and the results were expressed as mg of gallic acid equivalents (GAE)/g sample.
The electrophoretic profiles of DFM and FPI were determined by SDS-PAGE in a Mini Protean II apparatus (Bio-Rad, Hercules, CA, USA) using 4–15 % polyacrylamide gradient gel under reducing conditions [13]. The samples (5 mg ml−1) were dispersed in buffer (2 % SDS and 5 % β-mercaptoethanol) and were boiled at 96 °C for 10 min. A 10 μl aliquot was applied to each well. After the run, the gels were stained with 0.1 % coomassie blue and destained in an acetic acid/methanol/distilled water solution (1:4:5). A 14.4–94.7 kDa marker kit (Bio-Rad) was used as molecular weight standard. The hydrolysate and digested samples were evaluated by Tricine-SDS PAGE [14]. The gels consisted of a resolving gel (14.6 % T, 4 % C), spacing gel (10 % T, 3 % C) and stacking gel (4 % T, 3 % C). The samples were dissolved (1 % protein w/v) in reducing buffer (0.5 M Tris–HCl, pH 6.8, 10 % SDS, 10 % glycerol, 5 % β-mercaptoethanol and 0.1 % bromophenol blue) and heated at 40 °C for 30 min. Aliquot of 10 μl was loaded in each well. After the run, the gels were fixed for 1 h in a methanol/acetic acid/water solution (5:1:4), stained with coomassie brilliant blue G-250 (0.04 % in 10 % acetic acid) and destained with 10 % acetic acid.
Antioxidant Capacity
The antioxidant capacity was measured in aqueous and methanolic (70 %) extracts. The ferric reducing antioxidant power (FRAP) was performed according to Thaipong et al. [15] with adaptations. FRAP values of the extracts were determined from a standard curve of ferrous sulfate solution and the results were expressed in mg FeSO4 (FS)/g sample. The oxygen radical absorbance capacity (ORAC) method was performed according to the methodology described by Dávalos et al. [16]. Fluorescein reacted with free radicals generated by AAPH, yielding a non-fluorescent product. The fluorescence measurements were made using a Synergy™ HT Multi-Mode Microplate Reader with an excitation wavelength of 485 nm and an emission wavelength of 520 nm. The antioxidant activity was expressed as μmol of Trolox equivalent (TE)/g sample. The assays were carried out in triplicate.
Degree of Hydrolysis (DH)
The DH of the hydrolysates was determined using the pH-stat procedure [8]. The OPA method [17] was used to determine the DH of the samples before and after in vitro digestion. All measurements were performed in duplicate.
Simulated GI Digestion
The simulated GI digestion of the products was carried out as described by Martos et al. [18] with adaptations. To simulate gastric digestion, the samples were dispersed in a simulated gastric fluid (35 mM NaCl), the pH was adjusted to 2 by adding 1 N HCl and the volume was adjusted to reach a final concentration of 5.9 mg protein/ml. The mixture was incubated in a shaking water bath at 37 °C, 60 rpm. After 15 min, pepsin was added (E/S 1:20, w/w), the pH was adjusted to 2 and the mixture was incubated at 37 °C for 60 min under continuous stirring. The intestinal digestion was simulated by using an aliquot of 2.9 ml of gastric digest adjusted to pH 6.5 by adding 1 M NaHCO3, 1 M CaCl2, and 240 μl of bile salts at a concentration 9 mg/ml. The mixture was incubated at 37 °C for 15 min under stirring. Pancreatin (E/S 1:25, w/w) was added and after stirring at 60 rpm for 60 min at 37 °C the reaction was stopped by heating to 90 °C for 10 min. At the end of the simulated GI digestion, the digest presented the following final concentrations: 3.9 mg/ml protein, 7.6 mM CaCl2, 0.07 mg/ml bile salts, 0.16 mg/ml pancreatin and 0.2 mg/ml pepsin. The digest was centrifuged (11,000 × g for 15 min) and the supernatant containing the digested product was stored at −20 °C. The procedure was performed in triplicate.
Cell Culture and DNA Damage (Comet Assay)
Human monocytic U937 cells were obtained from the European Collection of Animal Cell Cultures (ECACC) and maintained in RPMI-1640 medium enriched with 10 % FBS (v/v). Cells were incubated at 37 °C in a humidified atmosphere of CO2 in air in the absence of antibiotics. U937 cells (1 × 105 cells/ml) were supplemented with the test compounds for 24 h in 6-well plates to a final volume of 2 ml. After incubation, the cells were exposed to 40 μM H2O2 for 30 min at 37 °C. Cells were harvested, embedded in a low melting point agarose (1 %) and placed on microscope slides coated with a layer of normal gelling agarose. Slides were placed in lysis solution [2.5 M NaCl, 100 mM EDTA, 10 mM Tris, pH 10, 1 % (w/v) sodium sarcosinate, 1 % (v/v) Triton X-100 and 10 % (v/v) DMSO; 4 °C] for 2 h. Slides were then placed in a horizontal gel electrophoresis tank (Horizon 20 25, GIBCO BRL, Life Technologies, Scotland) containing fresh electrophoresis solution (300 mM NaOH, 1 mM EDTA) for 30 min. Electrophoresis was performed at 25 V and 300 mA for 25 min at 4 °C. After electrophoresis, the slides were washed with neutralizing buffer (0.4 M Tris, pH 7.5). Slides were stained with ethidium bromide (20 μg/ml) and then covered with coverslips. Cells were visualized at a magnification of 200X (Nikon Optiphot-2) and the Komet 5.5 image analysis software program (Andor Technology, Northern Ireland) was used to determine the level of DNA damage, which was expressed as percentage of tail DNA. The results represent mean ± standard error (SE) of three independent experiments.
Statistical Analysis
A paired t-test was performed to determine the difference between the contents of the FCRRS and the antioxidant capacity before and after in vitro digestion. The FCRRS content and antioxidant capacity were analyzed by ANOVA followed by the Tukey’s test. The software STATISTICA version 7.0 (Stat Soft, Tulsa, USA) was used to analyze the CCRD.
Results and Discussion
The composition of PDFM, DFM and FPI is shown in Table 1. The protein content of FPI (71.8 %) was lower than that reported in literature [11, 19]. The difference in the contents may be either due to the removal of the seed coat prior to obtaining the flaxseed meal or the extraction method [11]. The dietary fiber content of DFM and FPI samples were 38.8 % and 13 %, respectively. Due to its high dietary fiber content, particularly mucilage, flaxseed products can play an important role in preventing diseases such as diabetes, obesity and cancer [11].
Flaxseed Hydrolysates
The antioxidant capacity and the DH of the hydrolysates produced under the CCRD conditions are shown in Table 2. The highest FRAP values in aqueous and methanolic extracts were 41.7 mg FS/g FPH (E/S: 1:133, pH 9.2) and 38.6 mg FS/g FPH (E/S: 1:150, pH 8.5), respectively. The highest ORAC values shown by the FPHs were 338.0 μmol TE/g FPH (E/S: 1:150, pH 8.5) in the aqueous extract and 353.5 μmol TE/g FPH (E/S: 1:90, pH 8.5) in the methanol extract. Based on mathematical models derived from the CCRD (Online Resource 1), the hydrolysis condition for obtaining hydrolysate with the highest antioxidant capacity in aqueous and methanolic extracts, measured by ORAC and FRAP, was E/S 1:90 and pH 8.5 (Online Resource 2). The hydrolysate obtained under this condition was designated as FPH 0.
The DH of the hydrolysates ranged from 12.7 to 19.3 %. The hydrolysates with the highest antioxidant capacity measured by FRAP and ORAC methods showed DH values of 13.6 and 14.7 %, respectively, which are in the middle of this range. Marambe et al. [6] reported that increasing the DH value led to decreased hydroxyl radical scavenging activity of flaxseed protein hydrolysate with Flavourzyme. These results are in accordance with other studies, which suggested that there is an optimal range of DH values to produce peptides with antioxidant capacity [20]. As the peptide size increases, the access of reactive species to nucleophilic centers becomes more difficult while low molecular weight peptides and free amino acids exhibit low antioxidant capacity, since they have reduced solubility in lipophilic medium [2]. The antioxidant activity is a synergistic effect of the combination of amino acid with antioxidant potential and their position in the peptide sequence [2].
Effect of In Vitro Digestion on the Content of Reducing Substances and Antioxidant Capacity
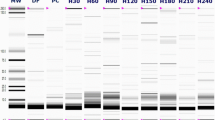
Electrophoretic profile of DFM and FPI showed bands between 6–67 kDa (Fig. 1a) and of FPH 0 consisted of a diffused band at approximately 6 kDa (Fig. 1b), which indicates that none of the original fractions found in FPI (Fig. 1a, b) was resistant to the Alcalase action. Following the digestion, the bands >14 kDa were no longer visible, and two bands between 14 and 6 kDa remained visible. The existence of these bands in the FPH 0 after digestion suggests that these fractions are at least partially resistant to hydrolysis.

Electrophoresis of samples before and after in vitro digestion. a SDS PAGE: 1 - molecular weight standards, 2 - defatted flaxseed meal (DFM), 3 - flaxseed protein isolate (FPI). b tricine SDS-PAGE: 1 - molecular weight standards, 2 - defatted flaxseed meal (DFM), 3 - flaxseed protein isolate (FPI), 4 - flaxseed protein hydrolysate (FPH 0), 5 - control digestion, 6 - DFM, 7 - FPI, 8 - FPH 0, after in vitro digestion
Prior to the in vitro digestion, the FCRRS content of the DFM in the methanol extract (0.7 mg GAE/g) was lower than that reported for whole flaxseed meal (2.7 to 3.25 mg GAE/g) [12]. These differences may be due to the loss of phenolic compounds and lignans during the defatting process [21]. The FCRRS content of FPI was two to three times higher than that of DFM in the aqueous and methanol extracts, and up to 9 times lower than the hydrolysates (Table 3). The increase in the FCRRS content in FPI compared to the meal, which has also been observed for amaranth [22] and buckwheat [20], may be due to the solubilization of phenolic compounds during the alkaline extraction step [11]. The highest FCRRS content was shown by FPH (24.2 mg GAE/g FPH). The hydrolysis of FPI catalyzed by Alcalase was expected to release small peptides and phenolic compounds associated with flaxseed protein by breaking down the protein-polyphenol complexes [3], leading to increased FCRRS content. In addition, the alkaline pH and temperature used for Alcalase activity may have caused a partial hydrolysis of the lignans, releasing phenolic compounds [23, 24], as was shown by the control assay, which exhibit higher antioxidant capacity than the FPI (p < 0.05).
The releasing of FCRRS during the protein concentration and hydrolysis processes affected the ability of neutralizing free radicals. FPI showed FRAP value two times higher in the aqueous extract and three times higher in the methanol extract and up to two times higher ORAC values than the DFM. The antioxidant capacity of FPI measured by FRAP and ORAC was up to four to six times lower than of FPH (p < 0.05).
After in vitro digestion, the FCRRS content of DFM increased 9 times in the aqueous extract and 17 times in the methanol extract (p < 0.05), while that of FPI increased 20 times in both extracts, reaching values similar to the digested hydrolysates (p < 0.05). DFM also showed an increase in antioxidant capacity, being four times higher in the aqueous extract and 10 times in the methanol extract. In addition, FPI exhibited an increase in both extracts, being three times higher as determined by FRAP and eight times by ORAC. During in vitro hydrolysis, DFM and FPI generated peptides with molecular weight lower than 6 kDa (Fig. 1), which have higher antioxidant capacity than the parent proteins [25]. These results suggest that flaxseed antioxidant compounds remained stable under the simulated GI conditions, possibly due to the protection exerted by the food matrix. Siracusa et al. [26] observed that the behavior of phenolic compounds toward simulated digestion strongly depends on the matrix which exerts a sort of “self-protection” effect on its components when submitted to the digestion process. The actions of the digestive enzymes and bile salts can also promote the release of phenolic compounds stored in plant cells or bound to other compounds [27].
Overall, the FPH exhibited less dramatic changes after in vitro digestion, showing small but significant, increases in FCRRS content in both extracts and a 1.4-fold increase (p < 0.05) in antioxidant capacity (FRAP and ORAC). These results suggest that compounds with antioxidant capacity present in the hydrolysates were already accessible and stable under the digestion conditions. The low effect of digestive enzymes on antioxidant capacity of FPH should be due to the broad specificity of Alcalase, which may have already cleaved the specific sites required for pepsin and pancreatin action [28] and released phenolic compounds during hydrolysis. Furthermore, after in vitro digestion, the antioxidant capacity of FPI as determined by ORAC was 30 % higher than of the FPH but 10 % lower when determined by FRAP (p < 0.05). The ORAC method mimics both oxidative deterioration and the mechanism of action of antioxidants that occur in vivo [29]. Thus, the results suggest that FPI may have the potential to maintaining the redox equilibrium in the GI tract. These results are relevant since FPI may contribute to prevent diseases caused by ROS production by immune cells during the digestive process [3, 30] or by a pro-oxidation environment induced by a diet rich in saturated fat and meat [31].
In order to investigate the antioxidant potential of the extracts at cellular level, the human macrophage cell line U937 was employed. The conditions of oxidative stress were simulated in the cells by the addition of H2O2, which produces hydroxyl radicals in the presence of iron, by means of the Fenton reaction [32]. The addition of 40 μM H2O2 to U937 cells increased the level of DNA damage in the cell from a baseline level of approximately 5 to 40 % tail DNA (Fig. 2) as measured by the comet assay. The preincubation of the cells with aqueous and methanolic FPH extracts, before and after digestion, did not significantly protect (p > 0.05) the cells against H2O2-induced DNA single strand breaks (Fig. 2). A similar effect was obtained at concentrations of 0.125, 0.25 or 1.0 μg/ml (data not shown). Despite good radical scavenging activity shown by the extracts and digests (Table 3), they failed to demonstrate any radical scavenging in U937 cells. Plant extracts have demonstrated an ability to scavenge ROS and prevent hydroxyl radical induced damage to macromolecules including DNA in human cells [4]. Thus, neither the main polyphenols found in flaxseed, the lignans, which require hydrolysis by colonic microflora to exert biological effects in vivo [33] nor the peptides released by Alcalase and digestive enzymes showed protective activity against DNA damage.

DNA damage in U937 cells treated with flaxseed protein hydrolysate (FPH) extracts before and after simulated GI digestion. DNA damage was expressed as percentage of tail DNA. Data represent the mean ± standard deviation of three independent experiments
These results suggest that the use of flaxseed protein isolate and hydrolysate as ingredients to formulate functional foods and nutraceuticals may be a promising alternative. However, studies that characterize the antioxidant compounds in the aqueous and methanol extracts are necessary to better understand the contribution of peptides and phenolic compounds to the antioxidant activity of flaxseed protein products.
Abbreviations
- CCRD:
-
Central composite rotatable design
- DFM:
-
Defatted flaxseed flour
- FCRRS:
-
Folin-Ciocalteu reagent reducing substances
- FPI:
-
Flaxseed protein isolate
- FPH:
-
Flaxseed protein hydrolysate
- FRAP:
-
Ferric reducing antioxidant power
- GI:
-
Gastrointestinal
- ORAC:
-
Oxygen radical absorbance capacity
- PDFM:
-
Partially defatted brown flaxseed meal
- ROS:
-
Reactive oxygen species
References
Shahidi F (2000) Antioxidants in food and food antioxidants. Nahrung 44:158–163
Samaranayaka AGP, Li-Chan ECY (2011) Food-derived peptidic antioxidants: A review of their production, assessment, and potential applications. J Funct Foods 3:229–254
Bouayed J, Hoffmann L, Bohn T (2011) Total phenolics, flavonoids, anthocyanins and antioxidant activity following simulated gastro-intestinal digestion and dialysis of apple varieties: Bioaccessibility and potential uptake. Food Chem 128:14–21
Jang I-C, Park J-H, Park E et al (2008) Antioxidative and antigenotoxic activity of extracts from cosmos (Cosmos bipinnatus) flowers. Plant Foods Hum Nutr 63:205–210
Rabetafika HN, Van Remoortel V, Danthine S et al (2011) Flaxseed proteins: Food uses and health benefits. Int J Food Sci Technol 46:221–228
Marambe P, Shand P, Wanasundara J (2008) An in-vitro investigation of selected biological activities of hydrolysed flaxseed (Linum usitatissimum L.) proteins. J Am Oil Chem Soc 85:1155–1164
Udenigwe CC, Lu Y-L, Han C-H et al (2009) Flaxseed protein-derived peptide fractions: Antioxidant properties and inhibition of lipopolysaccharide-induced nitric oxide production in murine macrophages. Food Chem 116:277–284
Adler-Nissen J (1986) Enzymic hydrolysis of food proteins, 1st edn. Elsevier Applied Science Publishers, New York, pp 110–131
AOAC (1995) Official methods of analysis, 18th edn. Association of Official Analytical Chemists, Arlington
Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917
Wanasundara P, Shahidi F (2003) Flaxseed proteins: Potential food applications and process-induced changes. In: Thompson LU, Cunnane SC (eds), Flaxseed in human nutrition, 2nd edn. AOCS Press, Champaign, pp 387–403
Medina MB (2011) Simple and rapid method for the analysis of phenolic compounds in beverages and grains. J Agric Food Chem 59:1565–1571
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head bacteriophage T4. Nature 27:680–685
Schäger H, Jagow G (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem 166:368–379
Thaipong K, Boonprakob U, Crosby K et al (2006) Comparison of ABTS, DPPH, FRAP, and ORAC assays for estimating antioxidant activity from guava fruit extracts. J Food Compos Anal 19:669–675
Dávalos A, Gómez-Cordovés C, Bartolomé B (2004) Extending applicability of the oxygen radical absorbance capacity (ORAC-fluorescein) assay. J Agric Food Chem 52:48–54
Nielsen PM, Petersen D, Dambmann C (2001) Improved method for determining food protein degree of hydrolysis. Food Chem Toxicol 66:642–646
Martos G, Contreras P, Molina E et al (2010) Egg white ovalbumin digestion mimicking physiological conditions. J Agric Food Chem 58:5640–5648
Krause J-P, Schultz M, Dudek S (2002) Effect of extraction conditions on composition, surface activity and rheological properties of protein isolates from flaxseed (Linum usitativissimum L). J Sci Food Agric 82:970–976
Tang C-H, Peng J, Zhen D-W et al (2009) Physicochemical and antioxidant properties of buckwheat (Fagopyrum esculentum Moench) protein hydrolysates. Food Chem 115:672–678
Siger A, Nogala-Kalucka M, Lampart-Szczapa E (2008) The content and antioxidant activity of phenolic compounds in cold-pressed plant oils. J Food Lipids 15:137–149
Gamel TH, Linssen JP, Mesallam AS et al (2006) Seed treatments affect functional and antinutritional properties of amaranth flours. J Sci Food Agric 86:1095–1102
Ho CHL, Cacace JE, Mazza G (2007) Extraction of lignans, proteins and carbohydrates from flaxseed meal with pressurized low polarity water. LWT-Food Sci Technol 40:1637–1647
Kosińska A, Penkacik K, Wiczkowski W et al (2011) Presence of caffeic acid in flaxseed lignan macromolecule. Plant Foods Hum Nutr 66:270–274
Arcan I, Yemenicioglu A (2010) Effects of controlled pepsin hydrolysis on antioxidant potential and fractional changes of chickpea proteins. Food Res Int 43:140–147
Siracusa L, Kulisic-Bilusic T, Politeo O et al (2011) Phenolic composition and antioxidant activity of aqueous infusions from Capparis spinosa L. and Crithmum maritimum L. before and after submission to a two-step in vitro digestion model. J Agric Food Chem 59:12453–12459
Saura-Calixto F, Serrano J, Goñi I (2007) Intake and bioaccessibility of total polyphenols in a whole diet. Food Chem 101:492–501
Orsini Delgado MC, Tironi VA, Añón MC (2011) Antioxidant activity of amaranth protein or their hydrolysates under simulated gastrointestinal digestion. LWT-Food Sci Technol 44:1752–1760
Ou B, Huang D, Hampsch-Woodill M et al (2002) Analysis of antioxidant activities of common vegetables employing oxygen radical absorbance capacity (ORAC) and ferric reducing antioxidant power (FRAP) assays: a comparative study. J Agric Food Chem 50:3122–3128
Tagliazucchi D, Verzelloni E, Bertolini D et al (2010) In vitro bio-accessibility and antioxidant activity of grape polyphenols. Food Chem 120:599–606
Babbs CF (1990) Free radicals and the etiology of colon cancer. Free Radical Bio Med 8:191–200
Barbouti A, Doulias P-T, Zhu B-Z et al (2001) Intracellular iron, but not copper, plays a critical role in hydrogen peroxide-induced DNA damage. Free Radical Bio Med 31:490–498
Eeckhaut E, Struijs K, Possemiers S et al (2008) Metabolism of the lignan macromolecule into enterolignans in the gastrointestinal lumen as determined in the simulator of the human intestinal microbial ecosystem. J Agric Food Chem 56:4806–4812
Acknowledgments
The authors thank FAPESP (2009/51580-1 and 2010/52680-7) for the financial support and CAPES for the scholarship granted to F.G.D. Silva.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Silva, F.G.D., O’Callagahan, Y., O’Brien, N.M. et al. Antioxidant Capacity of Flaxseed Products: The Effect of In vitro Digestion. Plant Foods Hum Nutr 68, 24–30 (2013). https://doi.org/10.1007/s11130-012-0329-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11130-012-0329-6