
Abstract
The physiological significance of photosystem II (PSII) core protein phosphorylation has been suggested to facilitate the migration of oxidative damaged D1 and D2 proteins, but meanwhile the phosphorylation seems to be associated with the suppression of reactive oxygen species (ROS) production, and it also relates to the degradation of PSII reaction center proteins. To more clearly elucidate the possible protecting effect of the phosphorylation on oxidative damage of D1 protein, the degradation of oxidized D1 protein and the production of superoxide anion in the non-phosphorylated and phosphorylated PSII membranes were comparatively detected using the Western blotting and electron spin resonance spin-trapping technique, respectively. Obviously, all of three ROS components, including superoxide anion, hydrogen peroxide and hydroxyl radical are responsible for the degradation of oxidized D1 protein, and the protection of the D1 protein degradation by phosphorylation is accompanied by the inhibition of superoxide anion production. Furthermore, the inhibiting effect of 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU), a competitor to QB, on superoxide anion production and its protecting effect on D1 protein degradation are even more obvious than those of phosphorylation. Both DCMU effects are independent of whether PSII membranes are phosphorylated or not, which reasonably implies that the herbicide DCMU and D1 protein phosphorylation probably share the same target site in D1 protein of PSII. So, altogether it can be concluded that the phosphorylation of D1 protein reduces the oxidative damage of D1 protein by decreasing the production of superoxide anion in PSII membranes under high light.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Photosystem II (PSII) is a large multi-subunit protein–pigment complex consisting of more than 25 protein species. Inside the PSII complex, 32 kDa D1 protein is an intrinsic component which contains five membrane-spanning helixes (Michel and Deisenhofer 1988; Ferreira et al. 2004). The D1 protein can be damaged or degraded under high light, and meanwhile the electron transport in thylakoid membranes is partially inhibited, which is referred to as photoinhibition of PSII (Kyle et al. 1984; Ohad et al. 1984; Aro et al. 1993). Strong-light photoinhibition in PSII is caused by several different mechanisms, such as the acceptor-side mechanism, the donor-side mechanism, the singlet oxygen mechanism, the manganese-dependant (or two-step) mechanism (Aro et al. 1993; Yamamoto 2001; Jung and Kim 1990; Hakala et al. 2005; Ohnishi et al. 2005). Besides the abovementioned mechanisms, our group has further developed a new hypothesis to explain the inhibition of oxygen-evolution activity induced by successive reactive oxygen species (ROS) under high light (Song et al. 2006). Meanwhile, it has been reported that ROS including superoxide anion (O ·−2 ), hydrogen peroxide (H2O2), and hydroxyl radical (HO·) are involved in the D1 protein degradation of PSII under strong illumination (Miyao 1994; Miyao et al. 1995). They concluded that ROS is one of the main factors that initiate the D1 protein degradation.
More recently, it has been suggested that phosphorylation of PSII reaction center protein D1 is a prerequisite for efficient migration of damaged PSII complexes from grana to stroma lamellae for repair (Tikkanen et al. 2008). After the migration, the damaged D1 protein is dephosphorylated and successively degraded by a D1 specific protease. Without the phosphorylation of D1 protein, accumulation of photodamaged PSII complexes will result in oxidative damage of photosynthetic proteins in the thylakoid membranes. On the other hand, however, the phosphorylation may alter conformation of D1 protein, and, in turn, results in reducing the ability of quinones and herbicides to bind the QB site (Giardi et al. 1992). Because the QB site of D1 protein is considered to play an important role in the production of O ·−2 (Kyle 1987; Zhang et al. 2003), the conformation change in the QB-binding pocket caused by the phosphorylation of D1 protein may modulate ROS production. We therefore assume that, besides benefit to migration of damaged PSII complexes, the protecting effect of D1 protein phosphorylation on oxidative damage of D1 protein is probably also related to the down-regulation of O ·−2 production in PSII via influencing the binding affinity of the QB site.
To confirm the above assumption, herein, the effects of phosphorylation of D1 protein and QB site occupation by herbicide 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU) on the production of O ·−2 and the degradation of D1 protein in PSII membranes have been investigated using the electron spin resonance (ESR) spin-trapping technique and the Western blotting assay, respectively.
Materials and methods
Preparation of thylakoid membranes from spinach
The spinach plants were purchased from a local market and kept in darkness at 4 °C for more than 12 h. Thylakoid membranes were prepared from the spinach leaves as described previously (Robinson and Yocum 1980). The isolated thylakoid membranes were suspended in medium A containing 0.4 M sucrose, 10 mM NaCl, 50 mM 4-Morpholineethanesulfonic acid (Mes)–NaOH (pH 6.5) and kept frozen in liquid nitrogen before use. Chlorophyll concentration was determined as described by Arnon (1949).
Phosphorylation treatment of thylakoid membranes
The thylakoid membranes were thawed in a water bath at 25 °C. The thylakoids were illuminated with weak light at 100 μE m−2 s−1 in the presence of ATP to phosphorylate thylakoid proteins according to the method as described previously (Mizusawa et al. 1999) to obtain phosphorylated thylakoid membranes. This treatment causes phosphorylation of more than 90 % of the D1 protein (Mizusawa et al. 1999). Non-phosphorylated thylakoid membranes (control treatment) were performed as phosphorylation treatment (Mizusawa et al. 1999), but thylakoid membranes were illuminated in the absence of ATP. Chlorophyll concentration was determined as described by Arnon (1949).
Preparation of non-phosphorylated and phosphorylated PSII membranes
Non-phosphorylated and phosphorylated PSII membranes were isolated from non-phosphorylated and phosphorylated thylakoid membranes by the method of Berthold et al. (1981) with the modification of Yruela et al. (1991), respectively. Samples were suspended in medium A and kept frozen in liquid nitrogen until use. Chlorophyll concentration was determined as described by Arnon (1949).
High light treatment for PSII membranes
High light treatment was performed according to Henmi et al. (2004) with slight modifications. PSII membranes were suspended to a concentration of 0.4 mg Chl ml−1 in medium A. The suspension was placed in a glass container and illuminated with white light from a halogen lamp at 4,000 μE m−2 s−1 for 3 h. The suspension was stirred gently and the temperature was maintained at 25 °C with circulating water under thermostatic control during the high light treatment. After treatment, the suspension was used to analyze the degradation of D1 protein. In some experiments, the high light treatment was performed in the presence of some scavengers such as 20 U ml−1 superoxide dismutase (SOD), 20 U ml−1 catalase and 2 % (v/v) dimethyl sulfoxide (DMSO), or some inhibitors, such as 5 μM tetracyanoethylene (TCNE) and 5 μM DCMU.
Protein separation and Western blotting analysis
Proteins from isolated PSII membranes before and after high light treatment were separated on 15 % sodium dodecyl sulfate (SDS)-polyacrylamide gels and transferred to a nitrocellulose membrane (Pall). The membranes were then blocked with non-fat dry milk and incubated with rabbit primary antibodies against D1 protein (Agrisera) according to the method as described previously (Aro et al. 2004). The blots were incubated with the horseradish peroxidase-conjugated secondary antibody and developed using the enhanced chemiluminescence system (Millipore). Quantification of protein levels was done with ImageJ software (Abramoff et al. 2004).
Spin trapping-ESR measurement for superoxide anion
The spin-trapping was accomplished by 5-tert-butoxycarbonyl-5-methyl-1-pyrroline N-oxide (BMPO). For ESR measurements, the PSII membranes were suspended in medium A at 0.5 mg Chl ml−1 with 2 mM diethylene-triaminepentaacetic acid (DTPA), 10 mM BMPO, and 5 μM TCNE in the presence or absence of 50 μM DCMU. ESR spectra were recorded on a Bruker ESP 300 operating at X-band. High light treatment was performed by continuous He–Ne laser (25 mW, 663 nm) that produced strong irradiation without heating for 5 min. The ESR spectra were recorded simultaneously with the illumination at ambient temperature (20 °C). The instrument settings used were as follows: modulation amplitude, 1.02 G; receiver gain, 2 × 105; modulation frequency, 100 kHz; microwave power, 12.8 mW; sweep width, 100 G; time constant, 0.164 s; sweep time, 41.94 s.
BMPO synthesis
BMPO was synthesized according to a literature procedure (Tsai et al. 2003).
Results
Effect of phosphorylation on the degradation of D1 protein in the presence of ROS enhancer or scavengers
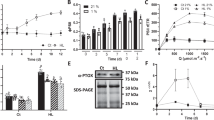
To obtain phosphorylated PSII membranes, we phosphorylated thylakoid membranes with weak light at 100 μE m−2 s−1 in the presence of ATP, and the phosphorylated PSII membranes were isolated from the phosphorylated thylakoid membranes. In the phosphorylated PSII membranes, roughly more than 90 % of D1 protein was phosphorylated (Supplementary Fig. S1) and the D1 protein was assigned as the phosphorylated D1 protein. In the non-phosphorylated PSII membranes, more than 80 % of D1 protein remained unphosphorylated form (Supplementary Fig. S1) and the D1 protein was assigned as the non-phosphorylated D1 protein. As mentioned in the introduction, O ·−2 , H2O2, and HO· are generated in PSII membranes under high light, which are involvement in the oxidative damage of the D1 protein. On the other hand, the phosphorylation of D1 protein may reduce the degradation of D1 protein (Koivuniemi et al. 1995; Chen et al. 2011). To simultaneously compare the effects of ROS production and phosphorylation on the degradation of 32 kDa-D1 protein, non-phosphorylated and phosphorylated PSII membranes were illuminated with white light at 4,000 μE m−2 s−1 for 3 h in the presence of SOD (O ·−2 scavenger), TCNE (O ·−2 enhancer), catalase (H2O2 scavenger), and DMSO (HO· scavenger) (Scaduto 1995; Shimmura et al. 1999; Sahni and Locke 2006), respectively. The photo-induced degradation of the D1 protein was monitored by Western blotting with specific antibodies against the C-terminal part of the D1 protein (Fig. 1a, b). High light caused the degradation of both non-phosphorylated and phosphorylated D1 proteins, but the extent of degradation of the non-phosphorylated form of D1 protein was obviously more severe than that of the phosphorylated form (see the second column group in Fig. 1c, d). All three ROS scavengers, including SOD, catalase and DMSO, significantly reduced the degradation of D1 protein from high light (over 85 %) and had the same effect on each form of D1 protein (i.e., the non-phosphorylated and phosphorylated D1 proteins), as shown in Fig. 1. However, TCNE, an enhancer of superoxide anion via its inhibiting effect on endogenous SOD-like activity of PSII (Ananyev et al. 1994; Song et al. 2006; Tiwari and Pospísil 2009), to some extent, accelerated the degradation of the non-phosphorylated D1 protein. All of the above results appeared to indicate that besides the damaging effect of ROS on the non-phosphorylated D1 protein as proposed previously (Miyao 1994; Miyao et al. 1995), they were somewhat involved in the degradation of the phosphorylated D1 protein. In addition, the phosphorylation of D1 protein can partially protect the D1 protein of PSII against photo-damage caused by endogenous ROS.

Effect of phosphorylation on the degradation of D1 protein in the presence of SOD, TCNE, catalase, DMSO, respectively. a Effect of phosphorylation on the degradation of D1 protein in the presence of SOD, TCNE, respectively. b Effect of phosphorylation on the degradation of D1 protein in the presence of catalase, DMSO, respectively. c Protein levels detected by Western blotting (a) were quantified using ImageJ software. d Protein levels detected by Western blotting (b) were quantified using ImageJ software. Values are the means of three measurements ± SD normalized to the control value in the non-phosphorylated and phosphorylated D1 proteins. After non-phosphorylated and phosphorylated PSII membranes were illuminated with light at 4,000 μE m−2 s−1 for 3 h in the absence (No add.) and in the presence of 20 U ml−1 SOD, 5 μM TCNE, 20 U ml−1 catalase and 2 % (v/v) DMSO, respectively, samples corresponding to 0.2 μg of chlorophyll were loaded in the gel and the D1 protein was identified with specific antibodies against the C-terminal part of the D1 protein. D1 and D1* denote the non-phosphorylated and phosphorylated D1 proteins, respectively. Control, non-phosphorylated and phosphorylated PSII membranes were kept in darkness for 3 h
Modulation of superoxide anion production by D1 protein phosphorylation and DCMU
To reveal whether the production of superoxide anion can be modulated by D1 protein phosphorylation and how it works, the light-induced ESR signal of O ·−2 from PSII membranes was examined by the spin trapping method using BMPO as a trapping reagent (Tsai et al. 2003; Chiang et al. 2010). As shown in trace I from Fig. 2a, there was no observable ESR signal under the dark condition. However, ESR signals can be immediately recorded under light illumination (Fig. 2a, traces II, III, IV, V). These ESR spectra are in accordance with the typical spectrum obtained from a mixture of xanthine oxidase (XOD) and hypoxanthine (HX) (picture not shown), thereby indicating that the trapped radicals can be recognized as O ·−2 . Both traces (II and III) were obtained from the non-phosphorylated PSII membranes, and the other two (traces IV and V) from the phosphorylated PSII membranes. Moreover, to examine the inhibiting effect of QB site occupation on the O ·−2 production, ESR spectra were obtained on the addition of DCMU, a competitor to QB, in trace III and trace V, respectively.

Spin trapping of superoxide anion radical in the photo-induced PSII membranes. a ESR spectra of BMPO-OOH spin adducts. b Relative ESR signal intensities of BMPO-OOH adducts in the non-phosphorylated and phosphorylated PSII membranes in the presence and absence of DCMU, respectively. The ESR signal intensity was determined by measuring the relative height of the central doublet peak of the first derivative of absorption spectrum. I PSII membranes were kept in darkness; II, IV denote the non-phosphorylated and phosphorylated PSII membranes under light illumination, respectively. III, V denote the non-phosphorylated and phosphorylated PSII membranes in the presence of 50 μM DCMU under light illumination, respectively. NP, P denote the non-phosphorylated and phosphorylated PSII membranes, respectively. The average value of three independent measurements is given. The error bars show the standard deviation
The quantitative ESR analysis of O ·−2 production was further demonstrated in Fig. 2b. As expected, the ESR signal level in phosphorylated PSII membranes was approximately 28 % lower than that in non-phosphorylated PSII membranes (control). However, the signal strengths of both non-phosphorylated and phosphorylated forms of PSII membranes were further decreased to approximately the same value, i.e., roughly half of that in the control group, on the addition of DCMU.
Effect of DCMU on the degradation of D1 protein
DCMU can bind to the QB site of D1 protein, and this binding could retard the degradation of D1 protein under high light (Mattoo et al. 1984). However, to our knowledge, there is no report related to the effect of DCMU on the degradation of the phosphorylated D1 protein. To test if the phosphorylation affects DCMU effect on the degradation of D1 protein, after non-phosphorylated and phosphorylated PSII membranes were illuminated with white light at 4,000 μE m−2 s−1 for 3 h in the presence and absence of DCMU, respectively, the degradation of the D1 protein in both non-phosphorylated and phosphorylated forms was monitored by Western blotting analysis with specific antibodies against the C-terminal part of the D1 protein. As shown in Fig. 3, when DCMU was absent, the light-induced degradation of D1 protein in the non-phosphorylated form was evidently observed, but the phosphorylation of D1 protein significantly reduced the degradation. In the presence of DCMU, the level of D1 protein in either non-phosphorylated or phosphorylated PSII membranes was apparently increased, and, moreover, both levels of D1 protein were almost identical (Fig. 3).

The effect of DCMU on the degradation of the non-phosphorylated and phosphorylated D1 proteins. a The profiles of the degradation of the non-phosphorylated and phosphorylated D1 proteins. After non-phosphorylated and phosphorylated PSII membranes were illuminated with light at 4,000 μE m−2 s−1 for 3 h in the absence of DCMU and in the presence of 5 μM DCMU, respectively, samples corresponding to 0.2 μg of chlorophyll were loaded in the gel, and the D1 protein was identified with specific antibodies against the C-terminal part of the D1 protein. b Protein levels detected by Western blotting (a) were quantified using ImageJ software. Values are means of three measurements ± SD normalized to the control value in the non-phosphorylated and phosphorylated D1 proteins. D1, D1* denote the non-phosphorylated and phosphorylated D1 proteins, respectively. Control non-phosphorylated and phosphorylated PSII membranes were kept in darkness for 3 h. No add. non-phosphorylated and phosphorylated PSII membranes were illuminated in the absence of DCMU. DCMU non-phosphorylated and phosphorylated PSII membranes were illuminated in the presence of 5 μM DCMU
Discussion
When plants encounter light intensities that exceed their photosynthetic capacity, a part of the excess light energy is used to produce ROS and/or other highly oxidizing species in the PSII. The presence of these active species results in photoinhibition of PSII electron transport and degradation of intrinsic proteins in PSII membranes. On the other hand, the reversible phosphorylation of chloroplast thylakoid membrane proteins, such as D1, D2 and light-harvesting complex II (LHCII), is pre-required for regulating PSII electron transport, facilitating D1 protein turnover and the repair of photo-damaged PSII, as well as adjusting the energy distribution on thylakoid membranes under high light (Michael and John 1991; Tikkanen et al. 2008; Tikkanen et al. 2011). Consequently, it is vital to understand more clearly how the protein phosphorylation plays an important role in protection against the oxidative damage.
Our ESR observation indicated that after phosphorylation of D1 protein, the average signal strength of O ·−2 decreased about 28 % (Fig. 2b), but Western blotting analysis showed that the D1 protein level increased roughly 100 % under a comparable condition (the second column group in Fig. 1c). The big difference in percentage change may be due to the degradation of D1 protein that is caused by multiple ROS components attack (i.e., besides O ·−2 , H2O2, and HO· can attack the protein). It was found that under strong illumination the endogenous O ·−2 was a primary ROS component, then O ·−2 can be converted to H2O2, and sequentially, HO· was produced via a Fenton reaction catalyzed by the integrated transition metal ions (Pospísil et al. 2004; Liu et al. 2004; Song et al. 2006). Although all of three ROS components are responsible for the degradation of D1 protein in both the non-phosphorylated and phosphorylated forms (Fig. 1), each component of ROS may have a specific damaging capacity. Thus, if the primary ROS component, i.e., O ·−2 , is partially inhibited, the other two components will correspondingly decrease and the protective effect on the degradation of D1 protein is probably more evident. D1 protein phosphorylation may play a similar role under high light, owing to down-regulation of O ·−2 .
The superoxide O ·−2 was stated to be the primary product of molecular oxygen reduction in the photosynthetic electron transport chain. Although the main site of O ·−2 formation in thylakoid membranes was supposed to be PSI (Asada 1999), the photo-induced production of O ·−2 in PSII was also experimentally demonstrated (Ananyev et al. 1994; Cleland and Grace 1999). The primary electron acceptor (Pheo−) and the quinone acceptors (Q −A and Q −B ) were proposed to reduce molecular oxygen. Because the well-known binding site of DCMU on the reducing side of PSII membranes is just located on the QB-binding site of D1 protein (Takahashi et al. 2010), the electron transporting from Q −B to O2 can be totally blocked in the presence of herbicide DCMU. As a result, DCMU could roughly inhibit by 50 % the production of superoxide anion in either non-phosphorylated or phosphorylated PSII membranes (Fig. 2). Meanwhile, Pospísil et al. (2006) also found that DCMU could roughly inhibit by 50 % the production of superoxide anion in PSII membranes. Considering the anionic plastosemiquinone radical located in the QB site of PSII would react with molecular oxygen to generate O ·−2 (Kyle 1987), the other 50 % superoxide anion production may come from Q −A and/or other electron acceptors, such as Pheo− (Fufezan et al. 2002; Arató et al. 2004; Pospísil 2009).
The previous studies indicated that the phosphorylation of D1 protein reduced the ability of quinines and herbicides to bind the QB site (Giardi et al. 1992), in line with the conformation changes in the QB site of the phosphorylated D1 protein. Hodges et al. (1987) also found that although the phosphorylation of thylakoid membrane protein did not affect the electron transfer from Q −A to QB, the phosphorylation-induced destabilization of Q −B , leading to a lower concentration of Q −B . It implies that the phosphorylation results in lower O ·−2 concentration originated from Q −B . As a result, the inhibiting effect of the D1 protein phosphorylation on O ·−2 production was not as strong as that by DCMU-treatment (i.e., concentration of O ·−2 decreases <50 %, Fig. 2b), resulting in the protective effect of the phosphorylation on D1 protein degradation in PSII membranes that was not as efficient as the DCMU treatment did (Fig. 3b).
In comparison to the phosphorylation, the inhibiting effect of DCMU on O ·−2 production and its protecting effect on D1 protein degradation were even more obvious, and both DCMU effects were independent of whether PSII membranes were phosphorylated or not (Figs. 2, 3). Therefore, we can deduce that both DCMU incubation and D1 protein phosphorylation probably have the same target location in PSII membranes, or more exactly, they share the QB-binding site in D1 protein.
When plants were exposed to high light in vivo, the major physiological significance of PSII core protein phosphorylation (D1, D2, CP43, and PsbH) is supposed to facilitate the migration of oxidative damaged proteins (Tikkanen et al. 2008). Although a direct protective effect of the phosphorylation on the degradation of D1 protein was probably not found in vivo, the phosphorylation-induced diminution in ROS generation may prevent or reduce the accumulation of photodamaged PSII complexes and thus enhance the protection of plants against light-induced oxidative damage. Moreover, suppression of partial ROS can enhance the repair of PSII via acceleration of the synthesis of the D1 protein (Allakhverdiev et al. 2005; Nishiyama et al. 2006, 2011; Murata et al. 2007, 2012). Tikkanen et al. (2008) also found that the proteins of the photosynthetic machinery in wild type Arabidopsis were less carbonylated than that in stn7 stn8 (i.e., an Arabidopsis mutant with defects in D1 and other PSII core protein phosphorylation), possibly implying that production of ROS is down-regulated during the light-dependent phosphorylation of D1 protein in vivo. On the other hand, ROS initiated the D1 protein degradation may occur under extreme conditions when the repair processes are unable to cope with the rate of damage. Without the interference from endogenous antioxidants in chloroplasts and the influence of the protease on the degradation of D1 protein, as shown in Scheme 1, all the observations on the production of O ·−2 and the degradation of D1 protein in the PSII membranes can be useful to get an overall understanding of the protecting effect of phosphorylation on oxidative damage and the phosphorylation-related suppression of ROS production. It can further be used to simulate the effect of ROS on the D1 protein degradation under extreme conditions.

A proposed mechanism for the protecting effect of phosphorylation on oxidative damage of D1 protein
In the light of the above discussion, we provide an assumption for light-induced oxidation of the D1 protein and suggest that the phosphorylation of D1 protein reduces oxidative damage of D1 protein via down-regulating the production of O ·−2 in PSII membranes.
Abbreviations
- PSII:
-
Photosystem II
- ROS:
-
Reactive oxygen species
- QB :
-
Secondary quinone electron acceptor
- DCMU:
-
3-(3,4-Dichlorophenyl)-1,1-dimethylurea
- ESR:
-
Electron spin resonance
- Mes:
-
4-Morpholineethanesulfonic acid
- Chl:
-
Chlorophyll
- SOD:
-
Superoxide dismutase
- DMSO:
-
Dimethyl sulfoxide
- TCNE:
-
Tetracyanoethylene
- SDS-PAGE:
-
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- BMPO:
-
5-Tert-butoxycarbonyl-5-methyl-1-pyrroline N-oxide
- DTPA:
-
Diethylene-triaminepentaacetic acid
- XOD:
-
Xanthine oxidase
- HX:
-
Hypoxanthine
- LHCII:
-
Light-harvesting complex II
- Pheo:
-
Pheophytin-primary electron acceptor
- QA :
-
Primary quinone electron acceptor
References
Abramoff MD, Magelhaes PJ, Ram SJ (2004) Image processing with ImageJ. Biophotonics Int 11:36–42
Allakhverdiev SI, Nishiyama Y, Takahashi S, Miyairi S, Suzuki I, Murata N (2005) Systematic analysis of the relation of electron transport and ATP synthesis to the photodamage and repair of photosystem II in synechocystis. Plant Physiol 137:263–273
Ananyev GM, Renger G, Wacker U, Klimov VV (1994) The photoproduction of superoxide radicals and the superoxide dismutase activity of photosystem II. The possible involvement of cytochrome b559. Photosynth Res 41:327–338
Arató A, Bondarava N, Krieger-Liszkay A (2004) Production of reactive oxygen species in chloride- and calcium-depleted photosystem II and their involvement in photoinhibition. Biochim Biophys Acta 1608:171–180
Arnon DI (1949) Copper enzymes in isolated chloroplasts. Polyphenoloxidase in Beta vulgaris. Plant Physiol 24:1–15
Aro EM, Virgin I, Andersson B (1993) Photoinhibition of photosystem II. Inactivation, protein damage and turnover. Biochim Biophys Acta 1143:113–134
Aro EM, Rokka A, Vener AV (2004) Determination of phosphoproteins in higher plant thylakoids. In: Carpentier R (ed) Photosynthesis research protocols. Methods in molecular biology. Humana Press, Totowa, pp 177–193
Asada K (1999) The water–water cycle in chloroplasts: scavenging of active oxygens and dissipation of excess photons. Annu Rev Plant Physiol Plant Mol Biol 50:601–639
Berthold DA, Babcock GT, Yocum CF (1981) A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes. EPR and electron-transport properties. FEBS Lett 134:231–234
Chen LB, Jia HY, Du LB, Tian Q, Gao YL, Liu Y (2011) Release of the oxygen-evolving complex subunits from photosystem II membranes in phosphorylation condition under light stress. Chin J Chem 29:2631–2636
Chiang HM, Yin JJ, Xia QS, Zhao YW, Fu PP, Wen KC, Yu HT (2010) Photoirradiation of azulene and guaiazulene-formation of reactive oxygen species and induction of lipid peroxidation. J Photochem Photobiol A 211:123–128
Cleland RE, Grace SC (1999) Voltammetric detection of superoxide production by photosystem II. FEBS Lett 457:348–352
Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S (2004) Architecture of the photosynthetic oxygen-evolving center. Science 303:1831–1838
Fufezan C, Rutherford AW, Krieger-Liszkay A (2002) Singlet oxygen production in herbicide-treated photosystem II. FEBS Lett 532:407–410
Giardi MT, Rigoni F, Barbato R (1992) Photosystem II core phosphorylation heterogeneity, differential herbicide binding, and regulation of electron transfer in photosystem II preparations from spinach. Plant Physiol 100:1948–1954
Hakala M, Tuominen I, Keränen M, Tyystjärvi T, Tyystjärvi E (2005) Evidence for the role of the oxygen-evolving manganese complex in photoinhibition of photosystem II. Biochim Biophys Acta 1706:68–80
Henmi T, Miyao M, Yamamoto Y (2004) Release and reactive-oxygen-mediated damage of the oxygen-evolving complex subunits of PSII during photoinhibition. Plant Cell Physiol 45:243–250
Hodges M, Boussac A, Briantais JM (1987) Thylakoid membrane protein phosphorylation modifies the equilibrium between photosystem II quinone electron acceptors. Biochim Biophys Acta 894:138–145
Jung J, Kim HS (1990) The chromophores as endogenous sensitizers involved in the photogeneration of singlet oxygen in spinach thylakoids. Photochem Photobiol 52:1003–1009
Koivuniemi A, Aro EM, Andersson B (1995) Degradation of the D1- and D2-proteins of photosystem II in higher plants is regulated by reversible phosphorylation. Biochemistry 34:16022–16029
Kyle DJ (1987) The biochemical basis for photoinhibition of photosystem II. In: Kyle DJ, Osmond CB, Arntzen CJ (eds) Topics in photosynthesis: photoinhibition. Elsevier, Amsterdam, pp 197–226
Kyle DJ, Ohad I, Arntzen CJ (1984) Membrane protein damage and repair: selective loss of a quinone-protein function in chloroplast membranes. Proc Natl Acad Sci USA 81:4070–4074
Liu K, Sun J, Song YG, Liu B, Xu YK, Zhang SX, Tian Q, Liu Y (2004) Superoxide, hydrogen peroxide and hydroxyl radical in D1/D2/cytochrome b-559 photosystem II reaction center complex. Photosynth Res 81:41–47
Mattoo AK, Hoffman-Falk H, Marder JB, Edelman M (1984) Regulation of protein metabolism: coupling of photosynthetic electron transport to in vivo degradation of the rapidly metabolized 32-kilodalton protein of the chloroplast membranes. Proc Natl Acad Sci USA 81:1380–1384
Michael AH, John FA (1991) Light-dependent phosphorylation of photosystem II polypeptides maintains electron transport at high light intensity: separation from effects of phosphorylation of LHC-II. Biochim Biophys Acta 1058:289–296
Michel H, Deisenhofer J (1988) Relevance of the photosynthetic reaction center from purple bacteria to the structure of photosystem II. Biochemistry 27:1–7
Miyao M (1994) Involvement of active oxygen species in degradation of the D1 protein under strong illumination in isolated subcomplexes of photosystem II. Biochemistry 33:9722–9730
Miyao M, Ikeuchi M, Yamamoto N, Ono T (1995) Specific degradation of the D1 protein of photosystem II by treatment with hydrogen peroxide in darkness: implications for the mechanism of degradation of the D1 protein under illumination. Biochemistry 34:10019–10026
Mizusawa N, Yamamoto N, Miyao M (1999) Characterization of damage to the D1 protein of photosystem II under photoinhibitory illumination in non-phosphorylated and phosphorylated thylakoid membranes. J Photochem Photobiol B Biol 48:97–103
Murata N, Takahashi S, Nishiyama Y, Allakhverdiev SI (2007) Photoinhibition of photosystem II under environmental stress. Biochim Biophys Acta 1767:414–421
Murata N, Allakhverdiev SI, Nishiyama Y (2012) The mechanism of photoinhibition in vivo: re-evaluation of the roles of catalase, α-tocopherol, non-photochemical quenching, and electron transport. Biochim Biophys Acta. doi:10.1016/j.bbabio.2012.02.020
Nishiyama Y, Allakhverdiev SI, Murata N (2006) A new paradigm for the action of reactive oxygen species in the photoinhibition of photosystem II. Biochim Biophys Acta 1757:742–749
Nishiyama Y, Allakhverdiev SI, Murata N (2011) Protein synthesis is the primary target of reactive oxygen species in the photoinhibition of photosystem II. Physiol Plant 142:35–46
Ohad I, Kyle DJ, Arntzen CJ (1984) Membrane protein damage and repair: removal and replacement of inactivated 32-kilodalton polypeptides in chloroplast membranes. J Cell Biol 99:481–485
Ohnishi N, Allakhverdiev SI, Takahashi S, Higashi S, Watanabe M, Nishiyama Y, Murata N (2005) Two-step mechanism of photodamage to photosystem II: step 1 occurs at the oxygen-evolving complex and step 2 occurs at the photochemical reaction center. Biochemistry 44:8494–8499
Pospísil P (2009) Production of reactive oxygen species by photosystem II. Biochim Biophys Acta 1787:1151–1160
Pospísil P, Arató A, Krieger-Liszkay A, Rutherford AW (2004) Hydroxyl radical generation by photosystem II. Biochemistry 43:6783–6792
Pospísil P, Snyrychová I, Kruk J, Strzałka K, Naus J (2006) Evidence that cytochrome b559 is involved in superoxide production in photosystem II: effect of synthetic short-chain plastoquinones in a cytochrome b559 tobacco mutant. Biochem J 397:321–327
Robinson HH, Yocum CF (1980) Cyclic photophosphorylation reactions catalyzed by ferredoxin, methyl viologen and anthraquinone sulfonate. Use of photochemical reactions to optimize redox poising. Biochim Biophys Acta 590:97–106
Sahni M, Locke BR (2006) Quantification of hydroxyl radicals produced in aqueous phase pulsed electrical discharge reactors. Ind Eng Chem Res 45:5819–5825
Scaduto RC (1995) Oxidation of DMSO and methanesulfinic acid by the hydroxyl radical. Free Radic Biol Med 18:271–277
Shimmura S, Masumizu T, Nakai Y, Urayama K, Shimazaki J, Bissen-Miyajima H, Kohno M, Tsubota K (1999) Excimer laser-induced hydroxyl radical formation and keratocyte death in vitro. Invest Ophthalmol Vis Sci 40:1245–1249
Song YG, Liu B, Wang LF, Li MH, Liu Y (2006) Damage to the oxygen-evolving complex by superoxide anion, hydrogen peroxide, and hydroxyl radical in photoinhibition of photosystem II. Photosynth Res 90:67–78
Takahashi R, Hasegawa K, Takano A, Noguchi T (2010) Structures and binding sites of phenolic herbicides in the QB pocket of photosystem II. Biochemistry 49:5445–5454
Tikkanen M, Nurmi M, Kangasjärvi S, Aro EM (2008) Core protein phosphorylation facilitates the repair of photodamaged photosystem II at high light. Biochim Biophys Acta 1777:1432–1437
Tikkanen M, Grieco M, Aro EM (2011) Novel insights into plant light-harvesting complex II phosphorylation and ‘state transitions’. Trends Plant Sci 16:126–131
Tiwari A, Pospísil P (2009) Superoxide oxidase and reductase activity of cytochrome b559 in photosystem II. Biochim Biophys Acta 1787:985–994
Tsai P, Ichikawa K, Mailer C, Pou S, Halpern HJ, Robinson BH, Nielsen R, Rosen GM (2003) Esters of 5-carboxyl-5-methyl-1-pyrroline N-oxide: a family of spin traps for superoxide. J Org Chem 68:7811–7817
Yamamoto Y (2001) Quality control of photosystem II. Plant Cell Physiol 42:121–128
Yruela I, Montoya G, Alonso PJ, Picorel R (1991) Identification of the pheophytin-QA-Fe domain of the reducing side of the photosystem II as the Cu(II)-inhibitory binding site. J Biol Chem 266:22847–22850
Zhang S, Weng J, Pan J, Tu T, Yao S, Xu C (2003) Study on the photo-generation of superoxide radicals in photosystem II with EPR spin trapping techniques. Photosynth Res 75:41–48
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Nos. 20875093 and 90813021) and the Pilot Project of Knowledge Innovation Program of the Chinese Academy of Sciences (KGCX2-EW-308-1’). The authors are very grateful to Dr. Ronald Charles Kurtenbach for the careful reading and the correction of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Chen, L., Jia, H., Tian, Q. et al. Protecting effect of phosphorylation on oxidative damage of D1 protein by down-regulating the production of superoxide anion in photosystem II membranes under high light. Photosynth Res 112, 141–148 (2012). https://doi.org/10.1007/s11120-012-9750-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-012-9750-9