
Abstract
The absorbance spectrum of the Fenna-Matthews-Olson protein—a component of the antenna system of Green Sulfur Bacteria—is always one of two types, depending on the species of the source organism. The FMO from Prosthecochloris aestuarii 2K has a spectrum of type 1 while that from Chlorobaculum tepidum is of type 2. The previously reported crystal structures for these two proteins did not disclose any rationale that would explain their spectral differences. We have collected a 1.3 Å X-ray diffraction dataset of the FMO from Prosthecochloris aestuarii 2K, which has allowed us to identify an additional Bacteriochlorophyll-a molecule with chemical attachments to both sides of the central magnesium atom. A new analysis of the previously published X-ray data for the Chlorobaculum tepidum FMO shows the presence of a Bacteriochlorophyll-a molecule in an equivalent location but with a chemical attachment from only one side. This difference in binding is shown to be predictive of the spectral type of the FMO.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Fenna–Matthews–Olson protein (Olson 2004) is a bacteriochlorophyll-a containing protein that exclusively occurs in photosynthetic bacteria with a chlorosome light antenna system (Blankenship and Matsuura 2003). It is of interest, because it is a rare example of an antenna system component that is water soluble. This property has made it an attractive target for spectroscopic studies (Olson et al. 1974; Louwe et al. 1997a; Whitten et al. 1980; van Mourik et al. 1994; Francke and Amesz 1997; Vulto et al. 1998a, 1998b; Brixner et al. 2005; Engle et al. 2007), theoretical studies (Pearlstein 1992; Louwe et al. 1997b; Savikhin et al. 1997; Vulto et al. 1998a; Wendling et al. 2002; Müh et al. 2007), and X-ray diffraction structure studies (Tronrud and Matthews 1993; Camara-Artigas et al. 2003; BenShem et al. 2004). In fact, the FMO from Prosthecochloris aestuarii 2K was the first protein containing some type of chlorophyll to have its atomic structure known (Tronrud et al. 1986) and stood as the highest resolution model in this class for over 20 years. The review by Savikhin et al. (1998) is an informative recap of the spectroscopic work done up to that point.
Fenna–Matthews–Olson protein is a peptide that enfolds a core of seven Bchl-a molecules, and three copies of this complex come together to form a trimer. The excited electronic states of this particle are delocalized over multiple Bchl-a molecules in the same monomer and also, to a lesser extent, spread to other copies in the trimer. The focus of much research has been to understand this delocalization and uncover the relationship between atomic structure and the absorbance and fluorescence spectra (Brixner et al. 2005; Engle et al. 2007).
While the original X-ray structure was determined for the FMO from Ptc. aestuarii 2K, most of the spectroscopic research has been performed using the protein from Chlorobaculum tepidum (formerly known as Chlorobium tepidum (Imhoff 2003)). The structure of this variant was solved (Camara-Artigas et al. 2003) (PDB entry 1M50) (Berman et al. 2000) in an attempt to relate the differences in the spectra of the protein from these two species (Olson et al. 1974; Francke and Amesz 1997) to the differences in the conformation of their chromophores. This model of the Cbl. tepidum FMO, unfortunately, did not show any differences in the arrangement of the seven Bchl-a molecules.
Recently, another X-ray structure was described for the FMO from Cbl. tepidum (BenShem et al. 2004) (PDB entry 3BSD). This model showed the binding of an eighth Bchl-a molecule in a cleft on the surface of the trimer. This extra chromophore was located at the site where uninterpretable electron density had been reported for the Ptc. aestuarii 2K model in the remarks in PDB entry 4BCL, implying that this pocket is a binding site in both proteins.
Earlier linear dichroism studies (Melkozernov et al. 1998) and recent chemical labelling and mass spectrometry data (Wen et al. 2009) have established the orientation of the FMO protein on the membrane. The end of the protein containing Bchl-a #3 is near the membrane and the end containing Bchl-a #1 is toward the chlorosome. The newly discovered eighth Bchl-a is in the region of the protein that is toward the chlorosome and its orientation may facilitate energy transfer from the chlorosome baseplate to the reaction center. This pigment is separated from the other seven core Bchl-a’s and is located in a cleft in the protein surface. The location of this pigment bridges the distance between the baseplate pigments and the core Bchl-a’s in the FMO, and will thus increase the energy-transfer efficiency. In addition, the orientation of this pigment serves to increase the efficiency of energy transfer between the donor baseplate Bchl-a and this new pigment. This is because the Qy transition dipole moment of the eighth Bchl-a is oriented in a similar way as the Bchl-a in the baseplate, as reported on the basis of fluorescence anisotropy of single chlorosomes (Shibata et al. 2007).
Results
We have used the X-ray data of Camara-Artigas et al. 2003 (PDB entry 1M50) to create an improved model of the Cbl. tepidum FMO (deposited in the PDB as entry 3ENI). The new model shows that there was an extra Bchl-a molecule in this crystal form, although probably at lower occupancy than that seen in entry 3BSD. Our new model also corrects some errors in the seven core Bchl-a molecules, and these corrections increase the similarity between this FMO and that from Ptc. aestuarii 2K.
The diffraction dataset for model 1M50 (and the new 3ENI) extends to 2.2 Å resolution and that of 3BSD only to 2.3 Å resolution. While with this quality of data, one can trace the chain of the polypeptide, determine side chain rotomers, and observe the location, orientation, and conformation of Bchl-a molecules, it is very difficult to estimate occupancy. Both models 3BSD and 3ENI were created with the constraint that the occupancy of the eighth Bchl-a molecule be unity. Such a constraint is typically applied in models with this resolution even when the molecule is not believed to occur with full occupancy. We believe the occupancy of the eighth Bchl-a molecule is less than unity in 3ENI because the density for this molecule is not as strong as that of the protein and the refined B factors are much higher (~80 Å2) compared to the surrounding protein (between 30 and 40 Å2). How much lower than unity is difficult to estimate.
We have grown new crystals of the FMO protein from Ptc. aestuarii 2K in an attempt to create a higher resolution model and investigate this newly recognized binding site. These crystals typically diffract at a synchrotron to resolutions between 1.4 and 1.3 Å. We have created a model based on a 1.3 Å dataset and deposited in the Protein Data Bank (deposited as entry 3EOJ).
Refinement statistics for both 3ENI and 3EOJ are listed in Table 1.
Overall there are few differences in conformation between the new Ptc. aestuarii 2K model and the new Cbl. tepidum model. If one superimposes the two, using just the central atoms of the seven Bchl-a rings as a guide, the r.m.s. agreement of the Cα atoms of the protein is 0.8 Å, and of the atoms in the seven core rings is 0.6 Å. The largest difference in the main chain of the protein is the region about residue 58. This is a loop between two β strands where some atoms are shifted by as much as 9 Å between the two species. Because this loop has both sequence changes and differing crystal environments it is difficult to determine if this structural change is an artifact of the differing crystal packing. Most of the differences in the seven core Bchl-a molecules are located in their Phytol tails, with the largest difference at atom C4 of ring number 6. This atom is flipped by 180° because the nearby residue 66 changes from an Isoleucine in Cbl. tepidum to a Phenylalanine in Ptc. aestuarii 2K. Neither of these structural changes is near the conjugated ring systems of the optically active parts of the molecule. The atoms of the seven core Bchl-a molecules, other than the Phytol tails, agree to an r.m.s. of 0.3 Å.
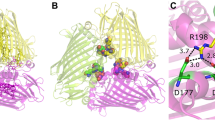
While our models confirm that a Bchl-a molecule binds at the “eighth site” in both the Ptc. aestuarii 2K and Cbl. tepidum variants of FMO, they show that the details of the binding interaction are significantly different. In both variants, the carbonyl oxygen of residue 123 binds to the central magnesium atom on one side of the Bchl-a ring while an α-helix (residues 155–172) covers the other side. Our new model for Ptc. aestuarii 2K shows a unique, bidentate interaction between the protein and this Bchl-a molecule (Fig. 1a). The additional link is between the central magnesium atom and the Oγ atom of Serine 168. The two links to this Bchl-a molecule originate from two different monomers within the biological trimer.

Comparison of the binding of the “eighth” Bchl-a in Ptc. aestuarii 2K and Cbl. tepidum. The “eighth” Bchl-a molecule is shown wedged between the α-helix of one monomer (in green) of the trimer and an irregular peptide strand from another (in orange). a In Ptc. aestuarii 2K, the carbonyl oxygen of residue 123 forms the first link to the magnesium atom while the Oγ atom of Serine 168 forms the second link. This model was refined with riding hydrogen atoms and these hydrogen atoms are also shown. b In Cbl. tepidum, the second link to the magnesium atom (from below) cannot be made. In both cases, atoms from the apo conformation are not shown
While the bidentate ligation of chlorin compounds by proteins has been suggested by spectroscopic studies (Fiedor 2006), this is the first instance reported in an X-ray model, despite over 100 models in the Protein Databank and many hundreds of individual chlorin-protein examples.
The variant of FMO from Ptc. aestuarii 2K has three, critical, differences in sequence in this helix when compared to Cbl. tepidum. First, residue 165 changes from a Threonine to a Phenylalanine. Secondly, residue 168 changes from an Alanine to a Serine. Finally, there is an insertion of an Alanine in the loop that links the end of the helix back to the body of the protein. Since the occupancy of the extra Bchl-a molecule in the Ptc. aestuarii 2K crystal is only 0.341 molecules/site (with an estimated standard uncertainty of 0.004 molecules/site), both the apo and holo conformations can be observed in the same crystal structure. In the apo form, the Phenylalanine side chain extends into the base of the binding pocket. When a Bchl-a molecule binds, this side chain must rotate out of its way, and this rotation requires that the side chain of residue 167 (an Isoleucine) move, and its motion results in the entire end of the α-helix moving toward the binding cleft, bringing the Oγ atom of Serine 168 close enough to the magnesium atom to allow it to make an additional link (Figs. 2 and 3).

Conformational change upon binding in Ptc. aestuarii 2K. The apo configuration of the helix is shown in light blue while the holo configuration is shown in green. Collisions between the two conformations indicate that they are mutually exclusive. These collisions indicate a progression of falling dominos that begin with the binding of the Bchl-a molecule and end with the creation link between the serine and the magnesium atom. (a) The Bchl-a molecule slides into the pocket. (b) This forces the side chain of Phenylalanine 165 to move to the right, to get out of the way. (c) This requires that Isoleucine 169 has to move, which pushes its main chain to the left. (d) This pushes Serine 168 to the left causing the Oγ atom to bind to the magnesium atom

Omit difference map showing the second ligation. The electron density map that allowed the identification of the second link to the Bchl-a molecule is shown. The apo model of the α-helix (here in blue) along with the Bchl-a molecule itself were included in the refinement but the holo conformation of the α-helix was not. The difference electron density map was contoured at ±3 sigma. The locations of the main chain atoms for the holo conformation are clearly defined by the continuous density and the striking bumps for the carbonyl oxygen atoms. The location of the holo conformation of the side chain of Serine 168 is also unambiguous
We believe that the occupancy of the eighth Bchl-a in the crystal of the FMO from Cbl. tepidum is also less than unity, however, there is no evidence of a significant change in the protein structure upon binding. The smaller Threonine side chain of residue 166 does not have to move when a Bchl-a molecule binds, and the shorter, more ordered, loop at the end of the α-helix restricts any motion of the helix. The FMO of Cbl. tepidum cannot make the second link to the Bchl-a molecule (Fig. 1b).
There is no obvious reason why the eighth Bchl-a molecule is not present at 100% occupancy. This molecule does bind in a very different way than the other seven. It is located in a cleft at the surface of the complex, while the others are completely protected from solvent. A consequence of the exposure is that the Phytol tail is not visible in the electron density map and we presume it is disordered. While it is possible that a fraction of the molecules binding at this site are lost during purification, we cannot eliminate the possibility that this binding site is not fully occupied when the FMO complex is in its functional state.
Discussion
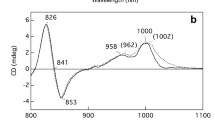
In 1974, it was observed that the absorbance and CD spectra for the FMOs from different species of Green Sulfur Bacteria can be two types when measured at less than 77 K (Olson et al. 1974) (Fig. 4). As can be seen in the figure, the FMO from Ptc. aestuarii 2K is type 1, while that of Cbl. tepidum is type 2. Although intense theoretical calculations ((Pearlstein 1992; Louwe et al. 1997b; Savikhin et al. 1997; Vulto et al. 1998a, 1998b; Müh et al. 2007) have been attempted in the past years to describe the tuning of the electronic structure by the pigment–protein interaction, the structural differences that lead to these spectral differences are not yet understood. Since the other seven Bchl-a molecules in this complex are nearly identical in these two variants, the presence or absence of the second link to the eighth is possibly the key to their spectral differences. If this is the case, the FMO variants from other species should have a spectral class consistent with the presence or absence of the amino acid sequence signature of the bidentate interaction.

Absorption spectra of FMO from three species of Green Sulfur Bacteria. The peaks on the right side of the spectra are from the Qy transition of the Bchl-a molecules in the complex. For Ptc. aestuarii 2K and Pel. phaeum, the 815 nm peak is stronger than the 806 nm peak, while for Cbl. tepidum the reverse is true. The former are members of spectral class 1 while the latter is class 2. These spectra were measured at 77 K with 80% glycerol as a cryoprotectant
A search of the SwissProt gene databank (Bairoch et al. 2004) for the sequences of FMO proteins, followed by alignment using the 3DCoffee web server (O’Sullivan et al. 2004), shows that these three mutations tend to appear together (Fig. 5). All members of the genus Prosthecochloris contain a sequence consistent with the bidentate Bchl-a binding mode. (The genus assignments and species names of the Green Sulfur bacteria were recently reassessed (Imhoff 2003). The species Pelodictyon phaeum was not considered at that time because the necessary sequences were unknown. We have recently sequenced the FMO gene and the 16S rRNA of this species, and found them similar to that found in members of the genus Prosthecochloris.) None of the species of the other two genera contain a sequence compatible with the bidentate interaction.

Sequence alignment of several species’ FMO proteins. This sequence alignment (O’Sullivan et al. 2004) shows that the variants from differing species of Green Sulfur Bacteria can be segregated into two categories. The first category contains the amino acid types that are consistent with a bidentate interaction between the protein and the extra Bchl-a molecule, as is the case with Ptc. aestuarii 2K. This pattern is defined by a Phenylalanine at position 165, a Serine at 168, and an insertion at 174. The second category contains sequences that are inconsistent with a bidentate interaction, as is the case with Cbl. tepidum. Although the FMO proteins from Cbl. parvum and Chlorobium phaeobacteroides contain a Serine at position 168, the presence of a Threonine at position 165, as well as deletions relative to first class which further constrict the motion of the helix, makes bidentate ligation unlikely. The final column of the table indicates the spectral classification for the six species that have been determined. Those sequences that produce spectra in spectral class 1 are all consistent with the presence of a bidentate ligation, while those of spectral class 2 are inconsistent with bidentate ligation. The spectral classification of Pel. phaeum, as well as Ptc. aestuarii 2K, and Cbl. tepidum, is demonstrated in Fig. 4. The classification of Ptc vibrioformis and Chlorobium phaeovibrioides is taken from Francke and Amesz (1997), and that of Cbl. thiosulfatiphilum is from Whitten et al. (1980)
Figure 5 shows the correspondence between the amino acid sequences and the spectral classes for six species of Green Sulfur Bacteria. All of the FMO variants that have been classified with type 1 spectra also have the sequence signature of a bidentate Bchl-a ligation. All of the FMO proteins with type 2 spectra are not consistent with this configuration. No other aspect of the structure of the FMO protein has been found to correlate with the spectral classification.
These observations, together, imply that it is very likely that the type of ligation of the eighth Bchl-a molecule in FMO determines the spectral classification of the protein’s absorption spectrum at low temperature.
Materials and methods
FMO purification from Ptc. aestuarii 2K
Cells of the Green Sulfur Bacterium Ptc. aestuarii 2K were grown anaerobically at room temperature with a light intensity of 150 μE for 2 days in two 15 l carboys. The cells were harvested by centrifugation at 7,500 rpm for 15 min. The cells were resuspended and washed with 20 mM Tris/HCl (pH = 8.0) buffer, and then run through the centrifuge again. This pellet was resuspended and then broken by sonication. A 4.0 M Na2CO3 solution was slowly added until a 0.2 M final concentration was reached, and the solution was stirred gently for 20 h in the dark at 4°C. Cell debris and unbroken cells were then removed by centrifuging at 10,000g for 15 min. More Na2CO3 solution was added until a final concentration of 0.4 M was reached. The solution was gently stirred in the dark for 20 h. The solution was ultracentrifuged for 2 h. The supernatant liquid containing the FMO protein was carefully decanted and dialyzed against 20 mM Tris/HCl (pH = 8.0) for a day. The solution was loaded in a SuperQ-650S ion-exchange column and washed with NaCl step gradients. The FMO protein was eluted with around 80–100 mM NaCl elution solution. The FMO protein was loaded on an S-300 gel filtration column and the fractions with OD267/OD371 <0.6 were selected and pooled. The final product was concentrated using the Amicon YM30.
Crystallization of FMO from Ptc. aestuarii 2K
Crystals were grown using the hanging drop method. The well solution was 10 mM Tris/HCl pH 8.0, 1.0 M NaCl, and 10% w/v (NH4)2SO4. The protein was transferred to a buffer of 10 mM Tris/HCl pH 7.8, and 1.0 M NaCl and concentrated to 14 mg/ml using a Biospin 30. Six wells were created. On each cover slip, we placed a 5 μl drop of the protein solution. To the first drop, we add 0.5 μl of well solution, and then 1, 2, 3, 4, and 5 μl of well solution to each of the other drops and closed the wells. The crystallization tray was stored at room temperature. Crystals grew in every drop.
X-ray diffraction data collection for the FMO from Ptc. aestuarii 2K
A crystal was swished through a cryroprotectant solution and frozen by placing the loop in the cryostream. The cryroprotectant solution consisted of mother liquor with ethylene glycol added to a concentration of 22% v/v of ethylene glycol. This crystal was taken to the Advance Light Source and mounted on the 8.2.2 beamline. Two sweeps of images were collected, with the second sweep having one half the exposure time per frame. The images were processed using HKL2000 (Otwinowski and Minor 1997). The resolution cut-off of 1.3 Å resolution was determined because that was the resolution where the I/σ(I) dropped below 2 and the completeness of the dataset began to drop off quickly at higher resolution.
Structure solution of FMO from Ptc. aestuarii 2K
Our goal was to create a model with minimal bias from the previous refinement efforts. In addition, to avoid model bias, the region where it was believed that some interesting structure would appear was left uninterpreted for as long as possible, along with the region of the map containing the residues from 123 to 126.
The starting model consisted only of the Bchl-a core as published in 1977 (Fenna et al. 1977). This model was given to Arp/Warp (Perrakis et al. 1999), which managed to trace nearly the entire peptide, despite having discarded the Bchl-a core it was originally given. The Bchl-a core had been replaced by dummy atoms, so we removed these atoms and restored the starting model for the Bchl-a core. Refinement then commenced with autoBuster (Bricogne 1997) and Coot (Emsley and Cowtan 2004), and was completed with Shelxl (Sheldrick 2008) and Coot. Eventually, the apo and holo conformations of 123–126 were built and refined with partial occupancy. Ethylene glycol molecules were then added to the model. At this point, the unknown density could finally be identified as a Bchl-a molecule. The “eighth” Bchl-a was then built into the density. In the final stages of refinement the alpha-helix on the other side of the Bchl-a was refined with its occupancy tied to the apo occupancy. The resulting map clearly indicated the conformation of the holo conformation (see Fig. 3) and the nature of the bidentate binding of the Bchl-a became clear. Details of the final model are given in Table 1.
Refinement of FMO from Cbl. tepidum
The model of FMO from Cbl. tepidum previously in the Protein Databank (Camara-Artigas et al. 2003), 1M50, does not contain any atoms in the region where BenShem et al. (2004) reported an eighth Bchl-a molecule. In addition, a map calculated with the deposited structure factors and phases calculated from 1M50 did not show the presence of unexplained density in that region.
In order to see if a modern refinement program, using Maximum Likelihood, would produce a different result, we created a new model based on the data deposited with 1M50. Care was taken to ensure that no water molecules were built in the region where binding of the extra Bchl-a molecule suspected.
A model was constructed as a probe for Molecular Replacement. This model was based on the model with the lab name of fmo-pa_fm28_v010 (This model is the result of an incomplete refinement of the FMO from Ptc. aestuarii 2K where the X-ray diffraction data were measured from crystal fm28. The final model derived from this data set will be reported elsewhere). The model was modified by removing all hydrogen atoms, defining all ADP’s as isotropic, and removing the atoms in residues 123 through 126, as well as removing all solvent molecules except those completely buried within the protein. This model was given to the program Phaser (McCoy et al. 2005) in the Phenix (Adams et al. 2002) package of computer programs. The program confirmed the Allen Laboratory’s choice of space group P4332. The molecular replacement solution was rebuilt by the AutoBuild (Terwilliger et al. 2008) program, again from the Phenix package. This program performed several iterations of building anew the peptide portion of the model along with the water molecules, and passed this along with the seven core Bchl-a molecules to phenix.refine (Adams et al. 2002). The resulting model was examined using the model building program Coot and the validation tool MolProbity (Lovell et al. 2003). Refinement was continued using the autoBuster refinement program (Bricogne 1997). Eight macrocycles of refinement followed by manual model rebuilding were performed.
Once all problems unrelated to the site of possible Bchl-a binding were corrected, it was possible to identify the density in that site as a Bchl-a molecule. This molecule was added to both copies in the asymmetric unit and a final round of refinement ran. The final statistics of the refinement are listed in Table 1.
Measuring the absorbance spectra of the FMO Protein
Each FMO variant was dissolved in 20 mM Tris/HCl buffer (pH = 8.0) and diluted into 80% glycerol and was slowly cooled to 77 K in a temperature-controlled cryostat (OptistatDN, Oxford Instruments, UK). The spectra were taken by Lamda 950UV/Vis spectrophotometer (Perkin Elmer, USA).
Abbreviations
- Bchl-a :
-
Bacteriochlorophyll-a
- Cbl :
-
Chlorobaculum
- FMO:
-
Fenna–Matthews–Olson Protein
- PDB:
-
Protein Data Bank
- Pel :
-
Pelodictyon
- Ptc :
-
Prosthecochloris
- r.m.s.:
-
Root mean square
References
Adams PD, Grosse-Kunstleve RW, Hung L-W, Ioerger TR, McCoy AJ, Mariarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D58:1948–1954
Bairoch A, Boeckmann B, Ferro S, Gasteiger E (2004) Swiss-Prot: juggling between evolution and stability. Brief Bioinform 5:39–55. doi:10.1093/bib/5.1.39
BenShem A, Frolow F, Nelson N (2004) Evolution of photosystem I—from symmetry through pseudosymmetry to asymmetry. FEBS Lett 564:274–280. doi:10.1016/S0014-5793(04)00360-6
Berman HM, Westbrook J, Feng Z, Gilliand G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242. doi:10.1093/nar/28.1.235
Blankenship RE, Matsuura K (2003) Antenna complexes from green photosynthetic bacteria. In: Green BR, Parson WW (eds) Light-harvesting antennas. Kluwer Academic Publishers, Dordrecht, The Netherlands, pp 195–217
Bricogne G (1997) The Bayesian statistical viewpoint on structure determination: basic concepts and examples. In: Carter CW, Sweet RM (eds) Methods in enzymology 276A. New York, Academic Press, pp 361–423
Brixner T, Stenger J, Vaswani HM, Minhaeng C, Blankenship RE, Fleming GR (2005) Two-dimensional spectroscopy of electronic couplings in photosynthesis. Nature 434:625–628. doi:10.1038/nature03429
Camara-Artigas A, Blankenship RE, Allen JP (2003) The structure of the FMO protein from Chlorobium tepidum at 2.2 Å resolution. Photosynth Res 75:49–55. doi:10.1023/A:1022406703110
Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi:10.1107/S0907444904019158
Engh RA, Huber R (1991) Accurate bond and angle parameter for X-ray protein structure refinement. Acta Crystallogr A47:392–400
Engle GS, Calhoun TR, Read EL, Ahn T-K, Mančal T, Cheng Y-C, Blankenship RE, Fleming GR (2007) Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature 446:782–786. doi:10.1038/nature05678
Fenna RE, Ten Eyck LF, Matthews BW (1977) Atomic coordinates for the chlorophyll core of a bacteriochlorophyll a-protein from green photosynthetic bacteria. Biochem Biophys Res Commun 75:751–755. doi:10.1016/0006-291X(77)91536-4
Fiedor L (2006) Hexacoordination of bacteriochlorophyll in photosynthetic antenna LH1. Biochemistry 45:1910–1918. doi:10.1021/bi0514055
Francke C, Amesz J (1997) Isolation and pigment composition of the antenna system of four species of green sulphur bacteria. Photosynth Res 52:137–146. doi:10.1023/A:1005845828676
Imhoff JF (2003) Phylogenetic taxonomy of the family Chlorobiaceae on the basis of 16S rRNA and fmo (Fenna-Matthews-Olson protein) gene sequences. Int J Syst Evol Microbiol 53:941–951. doi:10.1099/ijs.0.02403-0
Louwe RJW, Vrieze J, Aartsma TJ, Hoff AJ (1997a) Toward an integral interpretation of the optical steady-state spectra of the FMO-complex of Prosthecochloris aestuarii. 1. An investigation with linear-dichroic absorbance-detected magnetic resonance. J Phys Chem B 101(51):11273–11279. doi:10.1021/jp972215+
Louwe RJW, Vrieze J, Hoff AJ, Aartsma TJ (1997b) Toward an integral interpretation of the optical steady-state spectra of the FMO-complex of Prosthecochloris aestuarii. 2. Exciton simulations. J Phys Chem B 101(51):11280–11287. doi:10.1021/jp9722162
Lovell SC, Davis IW, Arendall WBIII, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC (2003) Structure validation by C-alpha geometry: phi, psi, and C-beta deviation. Proteins 50:437–450. doi:10.1002/prot.10286
McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr 61:458–464. doi:10.1107/S0907444905001617
Melkozernov AN, Olson JM, Li YF, Allen JP, Blankenship RE (1998) Orientation and excitonic interactions of the Fenna–Matthews–Olson Protein in membranes of the green sulfur bacterium Chlorobium tepidum. Photosynth Res 56:315–328. doi:10.1023/A:1006082513522
Müh F, Madjet ME-A, Adolphs J, Abdurahman A, Rabenstein B, Ishikita H, Knapp E-W, Renger T (2007) α-Helices direct excitation energy flow in the Fenna–Matthews–Olson protein. Proc Natl Acad Sci USA 140:16862–16867. doi:10.1073/pnas.0708222104
O’Sullivan O, Suhre K, Abergel C, Higgins DG, Notredame C (2004) 3DCoffee: combining protein sequences and structures within multiple sequence alignments. J Mol Biol 340:385–395. doi:10.1016/j.jmb.2004.04.058
Olson JM (2004) The FMO protein. Photosynth Res 80:181–187. doi:10.1023/B:PRES.0000030428.36950.43
Olson JM, Ke B, Thompson KH (1974) Exciton interaction among chlorophyll molecules in bacteriochlorophyll a protein and bacteriochlorophyll a reaction center complexes from green bacteria. Biochim Biophys Acta 430:524–537 Errata (1976). 440, 763
Otwinowski ZW, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM (eds) Methods in Enzymology, 276A. Academic Press, New York, pp 307–326
Pearlstein RM (1992) Theory of the optical spectra of the bacteriochlorophyll a antenna protein trimer from Prosthecochloris aestuarii. Photosynth Res 31:213–226. doi:10.1007/BF00035538
Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6:458–463. doi:10.1038/8263
Savikhin S, Buck DR, Struve WS (1997) Pump-probe anisotropies of Fenna–Matthews–Olson protein trimers from Chlorobium tepidum: a diagnostic for exciton localization? Biophys J 73:2090–2096. doi:10.1016/S0006-3495(97)78239-0
Savikhin S, Buck DR, Struve WS (1998) Toward level-to-level energy transfers in photosynthesis: the Fenna–Matthews–Olson protein. J Phys Chem B 102:5556–5565. doi:10.1021/jp981186f
Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr A 64:112–122. doi:10.1107/S0108767307043930
Shibata Y, Saga Y, Tamiaki H, Itoh S (2007) Polarized fluorescence of aggregated bacteriochlorophyll c and baseplate bacteriochlorophyll a in single chlorosomes isolated from Chloroflexus aurantiacus. Biochemistry 46:7062–7068. doi:10.1021/bi0623072
Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung L-W, Read RJ, Adams PD (2008) Iterative model building, structure refinement, and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr 64:61–69. doi:10.1107/S090744490705024X
Tronrud DE, Matthews BW (1993) Refinement of the structure of a water-soluble antenna complex from green photosynthetic bacteria by incorporation of the chemically determined amino acid sequence. In: Deisenhofer J, Norris JR (eds) The photosynthetic reaction center, vol 1. Academic Press, Inc., San Diego
Tronrud DE, Schmid MF, Matthews BW (1986) Structure and X-ray amino acid sequence of a bacteriochlorophyll a protein from Prosthecochloris aestuarii refined at 1.9 Å resolution. J Mol Biol 188:443–454. doi:10.1016/0022-2836(86)90167-1
van Mourik F, Verwijst RR, Mulder JM, van Grondelle R (1994) Singlet-triplet spectroscopy of the light-harvesting BChl a complex of Prosthecochloris aestuarii. The nature of the low-energy 825 nm transition. J Phys Chem 98(40):10307–10312. doi:10.1021/j100091a054
Vulto SIE, de Baat MA, Louwe RJW, Permentier HP, Neef T, Miller M, van Amerongen H, Aartsma TJ (1998a) Exciton simulations of optical spectra of the FMO complex from the green sulphur bacterium Chlorobium tepidum at 6 K. J Phys Chem B 102(47):9577–9582. doi:10.1021/jp982095l
Vulto SIE, Neerken S, Louwe RJW, de Baat MA, Amesz J, Aartsma TJ (1998b) Excited-state structure and dynamics in FMO antenna complexes from photosynthetic green sulphur bacteria. J Phys Chem B 102(51):10630–10635. doi:10.1021/jp983003v
Wen J, Zhang H, Gross ML, Blankenship RE (2009) Membrane orientation of the FMO antenna protein from Chlorobaculum tepidum as determined by mass spectrometry-based footprinting. Proc Natl Acad Sci USA 106(15):6134–31639. doi:10.1073/pnas.0901691106
Wendling M, Przyjalgowski MA, Gülen D, Vulto SIE, Aartsma TJ, van Grondelle R, van Amerongen H (2002) The quantitative relationship between structure and polarized spectroscopy in the FMO comples of Prosthecochloris aestuarii: refining experiments and simulations. Photosynth Res 71:99–123. doi:10.1023/A:1014947732165
Whitten WB, Olson JM, Pearlstein RM (1980) Seven-fold exciton splitting of the 810-nm band in bacteriochlorophyll a-proteins from green photosynthetic bacteria. Biochim Biophys Acta 591:203–207. doi:10.1016/0005-2728(80)90234-0
Acknowledgments
The authors would like to thank a number of individuals for their assistance and advice in performing this work. Roger Fenna provided advice on the purification and crystallization of this protein that was helpful in recreating the crystal form he had used in his earlier work. George Sheldrick answered a great many questions about the operation of his program ShelxL. Tom Terwilliger helped out with the operation of his Phenix.AutoBuild program. Tom Womack, Clemens Vonrhein, and others at Global Phasing, Ltd helped with the operation of AutoBuster. Anthony Addlagatta provided advice and assistance in crystal handling and data collection. X-ray diffraction data were collected at ALS beamline 8.2.2. Figures 1, 2 and 3 were created using the program PyMOL (http://www.pymol.org/). R. E. Blankenship gratefully acknowledges support from grant #DE-FG02-07ER15846 from the Energy Biosciences program of the Basic Energy Sciences division of the US Department of Energy. D. E. Tronrud wishes to thank B. W. Matthews for agreeing to host this project in his lab.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tronrud, D.E., Wen, J., Gay, L. et al. The structural basis for the difference in absorbance spectra for the FMO antenna protein from various green sulfur bacteria. Photosynth Res 100, 79–87 (2009). https://doi.org/10.1007/s11120-009-9430-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-009-9430-6