
Abstract
Potassium is the most abundant inorganic cation that constitutes up to 10% of the total plant dry weight and plays a prominent role in plant growth and development. Plants exhibit a complex but highly organized system of channels and transporters, which are involved in absorption and distribution of K+ from soil to different parts of plants. In this study, we explored the K+ transport system in chickpea genome and identified 36 genes encoding potassium channels and transporters. The identified genes were further classified on the basis of their domain structure and conserved motifs. It includes K+ transporters (23 genes: 2 HKTs, 6 KEAs, and 15 KUP/HAK/KTs) and K+ channels (13 genes: 8 Shakers and 5 TPKs). Chromosomal localization of these genes demonstrated that various K+ transporters and channels are randomly distributed across all the eight chromosomes. Comparative phylogenetic analysis of K+ transport system genes from Arabidopsis thaliana, Glycine max, Medicago truncatula, and Oryza sativa revealed their strong conservation in different plant species. Similarly, gene structure analysis displayed conservation of family-specific intron/exon organization in the K+ transport system genes. Evolutionary analysis of these genes suggested the segmental duplication as principal route of expansion for this family in chickpea. Several abiotic stress-related cis-regulatory elements were also identified in promoter regions suggesting their role in abiotic stress tolerance. Expression analysis of selected genes under drought, heat, osmotic, and salt stress demonstrated their differential expression in response to these stresses. This signifies the importance of these genes in the modulation of stress response in chickpea. Present study provides the first insight into K+ transport system in chickpea and can serve as a basis for their functional analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Potassium ion (K+) is one of the essential inorganic macro-elements in the plant body. It can constitute up to 10% of the total plant dry weight (Clarkson and Hanson 1980) and is abundantly present cation in the cytosol (i.e., 60–150 mM) (Leigh 2001; Schroeder et al. 1994; Walker et al. 1996). The plant cells utilize K+ for different fundamental processes, i.e., osmoregulation, stomata movements, cell elongation, electrical neutralization of anionic groups, and control of membrane polarization. Potassium is also involved in the maintenance of pH of the cytosol, which is very important for normal functioning of most of the enzymes (Lebaudy et al. 2007; Véry and Sentenac 2003). In addition to cytosol, K+ is also accumulated in mitochondria and certain other organelles like vacuoles, nucleus, and chloroplasts. Storage of K+ in vacuole is very helpful in maintaining the concentration of potassium in cytosol through its exchange between these two compartments. Despite its importance and abundance in the cell, an optimum cellular concentration of K+ must be maintained for proper cellular functioning. For example, maintenance of K+ concentration in the cytoplasm is correlated with plant tolerance to salinity (Shi et al. 2000) and drought stress (Gupta et al. 1989). Moreover, it is also important for neutralization of anions involved in the maintenance and modulation of membrane potential (Leigh 2001). A complex transport system consisting of various transporters and channels is involved in homeostasis of K+ in the cytosol. This system mediates the absorption and translocation of K+ from soil to different parts of the plant (Maathuis 2009). In model plant Arabidopsis thaliana, almost 35 genes have been reported to code for K+ transport proteins (15 channels and 20 transporters) (Mäser et al. 2001; Véry and Sentenac 2003).
Potassium channels are multimeric proteins containing transmembrane (TM) segments (α-segments) that are specified based on the number of pore (P) domains. Four P domains are associated with a multimeric functional protein to make a part of the channel conduction pathway. The P domain of potassium channel proteins includes a highly conserved motif with amino acid residues “GYGD/E.” Based on topology, 15 K+ selective channels in A. thaliana are divided into three families, which include 9 voltage-gated shaker channels, 5 tandem-pore K+ channels (TPK), and 1 potassium inward rectifier (Kir-like) (Lebaudy et al. 2007; Mäser et al. 2001; Véry and Sentenac 2003). Similarly, K+ transporters have also been classified into three sub-families: Trk/HKT family of high-affinity K+ transporters (one member), KUP/HAK/KT family of K+ uptake permeases (thirteen members), and KEA family of K+ efflux antiporters (six members) (Mäser et al. 2001). A number of potassium channels and transporters have been well characterized in A. thaliana and O. sativa; however, no information is available for K+ channels and transporters in Cicer arietinum.
Chickpea is the second most widely cultivated, diploid (2n=2X=16) legume pulse crop, which is a rich source of proteins, fats, and carbohydrates for humans and animals (Rasool et al. 2015). This crop is grown on low input marginal lands and represents an important component of the subsistence farming. Long-term evolution and adaption to extreme conditions make chickpea a rich pool of stress tolerance genes (Singh et al. 1998). The availability of genomic and transcriptomic resources of chickpea presents valuable treasure and opportunity for comparative and evolutionary genomic analysis (Jain et al. 2013; Varshney et al. 2013). K+ is an essential plant nutrient which impacts a number of physiological and biochemical processes associated with abiotic and biotic stress tolerance in plants (Wang et al. 2013). However, our understanding about the role of K+ in whole-plant stress response mechanisms is limited, especially in chickpea. The present study was designed to explore the phylogenetic relationships of K+ transporters and channels genes in C. arietinum, A. thaliana, and O. sativa and to study their possible role in abiotic stress responses.
Materials and Methods
Protein Sequence Retrieval of Potassium Transporters and Channels
GenBank and chickpea transcriptome database (CTDB) were searched, to retrieve putative K+-transporting genes in C. arietinum. Potassium-transporting genes in chickpea were retrieved by using already known protein sequences of K+-transporting genes in A. thaliana and O. sativa (Amrutha et al. 2007; Véry and Sentenac 2003; Véry et al. 2014). For this purpose, NCBI-BLASTp program was used. To eliminate false positives, the resulting raw data were manually curated. Shorter sequences (sequences with a length < 60% of the average length between terminal motifs in respective query sequence) were discarded. Nucleotide and protein sequences of A. thaliana and O. sativa K+ transport genes were also used as query to search relevant sequences in BLASTN and TBLASTN program of CTDB, respectively. Moreover, to verify whether the screening for K+ channels in chickpea was exhaustive, its genome was screened in the six-frame translations using the TBLASTX program. Furthermore, the presence of the K+-selectivity filter motif G-Y-G was also confirmed in relevant protein sequences. Subsequently, remaining sequences were searched for redundancy. Moreover, putative K+ channels and transporters were further confirmed as described by Gomez-Porras et al. (Gomez-Porras et al. 2012). For this purpose, the identified sequences were screened for the presence of the following motifs: three motifs for K+ channels (1) [S,T]-x-xT-x-G-[Y,F,L]-G-[D,E], (2) R-[L,F]-x-R-[L,V,I,A,G]-x-[R,C,K]- [V,A,L,M], (3) [A,V,S]-Y-[L,I]-[I,L]-G-[N,I]-[M,I]-T-[N,A]-L-[V,I]); two motifs for HKTs ((4) [S,T,A]-x-[F,Y,V,L,C]-x-[D,N,S]- G, (5) [G,A]-[Y,F]-[G,A]-x-[V,A,I]-G-[L,M,Y,F]-[S,T]); and five motifs for HAK transporters (6) [A,G]-[D,S,G]-[V,L,I,M]-x-x-[S,A]-P-L-Y, (7) [A,G]- N,D,H,S]-[D,N]-x-G-[E,Q,D,N]-[A,G], (8) [A,G,S]-[D,N]-[G,S,A,C]-x-[L,I,V,F]-x-P-x-V,I,L,M]-[A,S], (9) G-[S,A,T,C]-E-[A,G]-x-[F,Y]-A-[D,N,E]-[L,I,V]-[G,C,S,A]-x-F, (10) [Y,F]-x-x-x-x-x-[H,F,Y]-G-Y-x-[E,D]). Variants of all genes were checked attentively and only largest open reading frames (ORF) were selected for further investigation, i.e., domain prediction, gene structure determination, and phylogenetic analysis. Subsequently, genes were further scrutinized via different databases, i.e., Pfam database (http://pfam.janelia.org/), NCBI conserved domain database (http: //www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi), and SMART database (http://smart.embl-heidelberg.de/). Only those genes were selected for further analysis, which contain highly conserved domains. All the genomic information, number of exons in each gene, chromosomal location, and protein length of K+ transporters and channels were taken from CTDB and NCBI.
Motif Recognition and Prediction of Gene Structure
Conserved motifs within the protein sequences of K+ transporters and channels were found via an online server MEME (http://meme.sdsc.edu/meme/meme.html). Default parameters were used for motif analysis. The genomic and coding sequences of all the identified genes were downloaded from NCBI. These sequences were then used for gene structure analysis. The schematic representation of their gene structures was carried out using Gene Structure Display Server (GSDS) at http://gsds.cbi.pku.edu.cn/.
Multiple Sequence Alignment, Phylogenetic Analysis, and cis-Regulatory Element Prediction
Multiple sequence alignment was performed using the ClustalW. For phylogenetic analysis, MEGA 7.0 was used. Phylogenetic trees were developed through Maximum Likelihood and Neighbor-Joining methods with 1000 bootstrap replicates. Multiple sequence alignments were graphically represented by sequence logos created with WebLogo3 (http://weblogo.threeplusone.com/) (Crooks 2004). Upstream regions (1000–1100 bp) of all genes were examined to find the conserved cis-regulatory elements. PlantCARE website (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/search_CARE.html) was used to analyze the promoter for cis-regulatory elements.
Gene Duplication, Evolutionary Analysis, and Chromosomal Mapping of K+ Transporters and Channels in C. arietinum
DNAsp was used to determine the gene duplication events. Ka (non-synonymous) and Ks (synonymous) substitution rates were determined to find out the selective pressure on duplicated genes. Divergence time was also calculated by using the Ks value to perceive the evolutionary events. The MapChart program (http://www.biometris.wur.nl/UK/Software/MapChart/download) was used to generate a chromosomal map of putative K+ transporter and channel loci.
Plant Material and Stress Imposition
Chickpea (genotype K-70005, Kabuli type) seeds were collected from National Institute of Agriculture and Biotechnology (NIAB), Faisalabad, Pakistan. Plants were grown in a growth chamber at 20 °C night/22 °C day, with a 16-h light and 8-h dark photoperiod and 65% humidity. The seedlings were raised for 15 days in pots containing peat moss, and then, they were challenged with drought, salinity, heat, and osmotic stress. Drought stress was imposed by limiting the water supply for 10 days compared to ample water supply to control plants. For salt and osmotic stress, NaCl (100 mM for 72 h) and mannitol (100 mM for 72 h) were directly applied in pots. For heat stress, plants were kept at 42 °C for 24 h. For RNA extraction, leaf samples from all the treatments were collected and were immediately frozen in liquid nitrogen and stored at − 80 °C until use.
RNA Isolation, Reverse Transcriptase PCR, and Quantitative Real-time PCR
The total RNA was extracted from leaf samples using TRIzol reagent (Invitrogen & Co.) following the manufacturer’s instructions and quantified using NanoDrop spectrophotometer (Colibri spectrometer, Titertek Berthold, Germany). One microgram of total RNA was reverse transcribed using Maxima H Minus First Strand cDNA synthesis kit, with dsDNase (cat#K1681) using manufacturer’s instructions. The following ingredients were added in RNase-free tube on ice: 10X dsDNase buffer, dsDNase, total RNA (1 μg), and nuclease free H2O to make 10 μL total volume. After gently mixing and spinning, the mixture was incubated at 37 °C for 2 min on hot water bath and then chilled on ice. Subsequently, first-strand cDNA synthesis reagents (oligodT primer, 10 mM dNTP mix, 5X RT buffer, Maxima H minus reverse transcriptase enzyme) were added and were mixed gently and briefly centrifuged. The mixture was then incubated for 30 min at 50 °C and for 5 min at 85 °C. In the end, the mixture was stored at − 80 °C until use. qRT-PCR was performed using a real-time PCR detection system (CFX96 Touch™ Real-Time PCR Detection System) with the iTaq Universal SYBR Green Super Mix. The gene-specific primers were designed by using the online tool “Oligo Calculator” (http://mcb.berkeley.edu/labs/krantz/tools/oligocalc.html), and primer specificity was verified by NCBI Primer-BLAST program (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). The primer sequences for the assessment of expression of different genes in response to abiotic stresses are provided in Table S3. Sample preparation and qRT-PCR analysis were conducted following manufacturer’s instructions. A 10 μL of reaction mix consisted of 5 μL of SYBR green, 2.0 μL of each primer (forward and reverse, each 10 mmol), 1 μL of template consisted of reaction product (cDNA) from reverse transcriptase PCR. The C. arietinum CarGAPDH gene (accession number: XM_004515773.2, forward primer, 5′- GAAGCTTGAGAAGGCCGCTA-3′; reverse primer, 5′-TGCCTTTCAACTTGCCCTCA-3′) was used as an internal control for normalization of the expression data. An equal amount of cDNA template was used for each sample including the internal control. PCR amplification conditions were as follows: an initial denaturation step for 10 min at 95 °C; 40 cycles of quantification consisting of denaturation for 10 s at 95 °C, annealing for 20 s at 58 °C, and extension for 30 s at 72 °C, and it was completed by melting curve analysis to confirm the specificity of the PCR product. The qPCR analysis was repeated in three independent experiments.
Results
Identification of Potassium Transporters and Channels
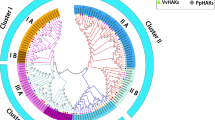
After carefully surveying the chickpea genome and duly confirming the presence of characteristic conserved domains and motifs, 36 genes were identified as potential candidates of the K+ transport system in chickpea (Table 1, Figs. 1 and 2). The analysis further categorized 23 of these genes as K+ transporters (Table 1, Fig. 3), while the remaining 13 genes were identified as K+ channels (Table 1, Fig. 4). The members of both families were renamed according to the nomenclature used for their orthologues in A. thaliana (Mäser et al. 2001). A phylogenetic tree was constructed to show five families of potassium channels and transporters in A. thaliana, O. sativa, and C. arietinum (Fig. 1).

Molecular phylogeny analysis. The evolutionary history was inferred by using the maximum likelihood method based on the JTT matrix-based model (Jones et al. 1992) tree with the highest log likelihood (− 119,897.9497) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Evolutionary analyses were conducted in MEGA7 (Kumar et al. 2016)

Conserved motif analysis. Sequence logo showing the conserved amino acids of channel and transporter protein sequences from Cicer arietinum, Oryza sativa, and Arabidopsis thaliana

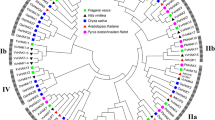
Phylogeny of potassium transporters. Phylogenetic relationships of K+ transporter proteins in C. arietinum, A. thaliana, O. sativa, G. max, and M. truncatula. a Phylogenetic tree of HKT transporters, b phylogenetic tree of KEA transporter family, and c phylogenetic tree of HAK/KU/KT transporter family. The tree is derived by neighbor-joining method with bootstrap analysis (1000 replicates) from alignment of protein sequences. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Evolutionary analyses were conducted in MEGA7 (Kumar et al. 2016)

Phylogeny of potassium channels. Phylogenetic relationships of K+ channel proteins in C. arietinum, A. thaliana, O. sativa, G. max, and M. truncatula. The tree is derived by neighbor-joining method with bootstrap analysis (1000 replicates) from alignment of protein sequences. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Evolutionary analyses were conducted in MEGA7 (Kumar et al. 2016)
Potassium Transporters
KUP/HAK/KT Family
Different acronyms are used for the members of this family like high-affinity K+ transporters (HAKs) in fungi, K+ uptake permeases (KUP) from bacteria, and potassium transporters (KT), but a collective name KUP/HAK/KT was used in the present study to express all of them in a single family. We identified 15 members of this family in C. arietinum genome; the number is relatively greater than A. thaliana (13 members) and smaller as compared to rice (25 members). At protein level, the length of KUP/HAK/KTs ranged from 416 aa (amino acids) to 881 aa (Table 1). At genomic level, the introns varied from 6 to 9. All members of KUP/HAK/KT family contain 10 to 14 TM domains in A. thaliana and rice except OsKUP/HAK/KT3. Domain analysis showed that members of KUP/HAK/KT family in C. arietinum contain 12 TM domains and an additional potassium transporter domain (K_trans) (Table 1). An unrooted phylogenetic tree has been created to show the intra-family relations (Fig. S1A). This analysis shows relative distance in different members of the same family.
Multiple sequence alignment of KUP/HAK/KT protein sequences in C. arietinum indicates the presence of many conserved regions (Fig. S1B), which may play a crucial role in controlling their function. A stretch of 12 residues (GGTFALYSLLCR) was observed at position 192 in addition to a stretch of three conserved residues (LGG) at position 375. A characteristic motif of K+ transporters (GVVYGDLGTSPLY) was found at position 129. The motifs SPLY and LGG are also conserved in C. arietinum. Conservation of these motifs showed a close relation between the KUP/HAK/KT family of A. thaliana and C. arietinum.
Trk/HKT Family
In current genome-wide analysis, two members (CarHKT1.1 and CarHKT1.2) of HKT/Trk family were found in C. arietinum. There are 524 to 571 aa in protein sequences CarHKTs. The genomic sequences contain two introns each. An unrooted phylogenetic tree of CarHKTs was created to show their relation with HKT members of different species (Fig. S2A). At protein level, CarHKT1.1 and CarHKT1.2 showed 46 and 41% identity to AtHKT1;1, respectively. With the differences in conserved residues in the first pore domain (PD) of the protein, the HKT transporters are divided into subfamilies I and II (Platten et al. 2006; Tholema et al. 2005). There is a serine (S) at this position in subfamily I. A glycine (G) residue is present in other three PDs forming S-G-G-G motif, whereas members of subfamily II have “G” residue in all PDs forming a G-G-G-G motif. The residue S is linked with a preference for Na+ conductance. The residue G is related to either Na+ and/or K+ conductance (for review, see (Almeida et al. 2013)). Multiple sequence alignment of first MPM-loop indicates that the “G” present at the end of first loop is replaced by S in C. arietinum. Therefore, these were placed in subfamily I but other three G residues are conserved at the end of remaining P-loops (CarHKT1.1: G-243, G-367, G-469 and CarHKT1.2: G-272, G-396, G-500).
Glycosylation site analysis was done by searching the tripeptide (NXS/T, where X represents any amino acid) (Koizumi et al. 1999; Lerouge et al. 1998). Only one putative N-glycosylation site (NIT) was found at position 429–431 in case of AtHKT1;1, two N-glycosylation sites were found in CarHKT1.1 at different positions (165–167 and 452–454), and five N-glycosylation sites were found in CarHKT1.2 at five different positions (9–11, 167–169, 181–183, 195–197, and 563–565) (Fig. S2B).
KEA Family
KEA family consists of six K+/H+ antiporter members in A. thaliana (Munro et al. 1991). KEAs are named from KEA1 to KEA6. Genome-wide analysis indicates the presence of six KEA members in C. arietinum. The length of KEA family members ranged from 572 aa to 1197 aa. The genomic sequence contains an average of 19 introns. An unrooted phylogenetic tree of the individual family was created to show the relationship of different members within the family (Fig. S3A). Multiple sequence alignment indicates that mostly G residues are conserved at different positions among the KEA members of A. thaliana and C. arietinum (Fig. S3B).
Potassium Channels
Shaker Family
Members of shaker family were first K+ channels, which were studied at the molecular level. Nine members of this family have been found in A. thaliana (Pilot et al. 2003b). Our results revealed that C. arietinum has eight genes, which encode the members of shaker family. At protein level, the length of shaker family members ranged from 617 to 891 aa. At genomic level, 10 to 13 introns were present in these genes. Shaker family is further divided into five main groups: GORK type (one member), AKT1 type (three members), KAT1 type (two members), KAT3 type (one member), and AKT2 type (one member) (Table 1; Fig. 4). The domain analysis shows the same domain pattern (cNMP, KHA, and Ank) as their counterparts in A. thaliana (Table 1). GORK and AKTs contain Ank repeat which is absent in KATs. Conserved motifs (1) T-X-X-T-X-G-Y-G-D, (2) R-[LF]-[WY]-R-[LA]-X-R-V, and (3) [AS]-Y-[LI]-I-G-N-M-T-[NA]-L-[VI]-V were identified among all members (Fig. 1, Fig. S4B).
TPK and Kir-Like Family
Members of TPK family contain a hydrophobic core, four TM domains and two P-loops (also known as KCO-2P), while members of Kir-like family contain a hydrophobic core consisting of two TM domains and one P-loop (KCO-1P). In both families, there is no voltage sensor TMS. Mostly, the members of both families contain one or two EF (Ca2+-binding sites) motifs at their C-terminal (Hedrich 2012; Moshelion et al. 2002). A conserved motif (RSXpSXP) is present at the N-terminal, and it is used to interact with 14-3-3 proteins (Latz et al. 2007). In C. arietinum, five members of TPK family are found (Table 1, Fig. 4). The length of TPKs proteins ranged from 364 to 428 aa. The number of introns ranges from 1 to 3 per sequence. Motif analysis indicates that all members contain EF motif, except CarTPK1.2 and CarTPK3.2. Multiple sequence alignment showed that all five members contain conserved motif (GYGD), necessary for highly selective potassium channels. Analysis of N-terminal motif showed that serine/threonine residue is highly conserved in all members except CarTPK1.1 (Fig. S5).
Gene Structure Analysis and Chromosomal Distribution of Potassium Transport-Related Genes
Intron and exon position is generally very well conserved in orthologous genes over long evolutionary time intervals, whereas intron/exon structure is slightly less but sufficiently conserved in paralogous genes (Li et al. 2009, 2014). To investigate the gene structural diversity of K+ transporters and channels in C. arietinum, we analyzed the exon/intron organization. Overall, there was considerable diversity in the number of exons (2–21) and the length of exons in the K+ transporters and channels (Fig. 5). However, it was noticed that members within a subfamily shared similar gene structure in terms of intron number, exon length, and/or intron phases. In shaker family, CarAKT1.1, CarAKT1.2, and CarAKT1.3 have almost the same intron/exon pattern, except CarAKT1.1, with lengthy introns.

Structural analysis of potassium channel and transporter genes. Exons and introns of 36 potassium transport genes are represented by black boxes and black lines, respectively. Gene models are based on CDC Frontier genome Cav1.0 gene annotations
In C. arietinum, 34 genes out of 36 were mapped on eight chromosomes (Fig. 6). Positions of two genes (CarKUP/HAK/KT4 and CarKUP/HAK/KT5.1) were not found. They are present on scaffolds, which have not yet assigned to any linkage group. Thus, they are not shown in the map. All the identified genes were randomly distributed on all the 8 chromosomes of chickpea. Chromosome 6 contains maximum numbers (8) of genes (7 transporters and 1 channel) while only 2 genes (both transporters) were present on each of chromosome 4 and 8. Almost 44% of members (15 out of 34) were present on chromosome 2 and 6 collectively (Fig. 6).

Chromosomal distribution of potassium channel and transporter genes. Thirty-four potassium transport-related genes are located on all eight chickpea chromosomes. The remaining two genes (CarKUP/HAK/KT4 and CarKUP/HAK/KT5) are located on scaffolds and not shown in this figure. The scale at the left side of the chromosomal bar indicates the position on the chromosome (mega base pairs; Mb). Duplicated genes are represented by a black line
Comparative Phylogenetic Analysis of K Transport System in Diverse Plant Species
Protein sequences of potassium transporters and channels belonging to different species like Arabidopsis thaliana, Glycine max, Medicago truncatula, Oryza sativa, and C. arietinum were used to find out the evolutionary and phylogenetic relationship (Figs. 3 and 4). For both the transporter channels, the sequences from leguminous plants (C. arietinum, G. max, and M. truncatula) appeared as close paraphyletic groups as compared to non-leguminous plants (O. sativa and A. thaliana). More specifically, members from C. arietinum shared same clades as M. truncatula.
In phylogenetic analysis of K+ channels, shaker and TPK/KCO appeared as separate groups (Fig. 4). Shaker channels are divided into five functional groups as AKT1-like inward rectifying channels, KAT1-like inward rectifying channels, silent channels, weak inward rectifying channels, and outward rectifying channels (Fig. 4.
Members of shaker family were subdivided into AKT, KAT, and GORK subfamilies. Members of KT (CarAKT1, CarAKT1.1, CarAKT1.2, and CarAKT2) family showed a close relationship with their counterparts in A. thaliana and O. sativa. CarAKT1.1 and CarKT1.2 showed a paralogous relation in C. arietinum, while they showed an orthologous and co-orthologous relation with AtAKT1 of A. thaliana. CarKAT1.1 and CarKAT1.2, members of KAT family, showed a close relationship with their counterpart (AtKAT1 and AtKAT2) in A. thaliana. Evolutionary analysis showed that these were duplicated through segmental duplication since 54.96 million years ago. CarTPK1.1 and CarTPK1.2, members of TPK family, duplicated as a result of segmental duplication before 45.64 million years ago, showed a paralogous relation within C. arietinum, while they showed an orthologous and co-orthologous relation with their counterparts in A. thaliana (AtTPK1) and rice (OsTPK1 and OsTPK2), respectively. Only one member of GORK subfamily was found which showed a close relation with GORK and SKOR in A. thaliana (Fig. 4).
According to the results of phylogenetic and evolutionary analysis, members of K+ transporter family were further subdivided into three subgroups: HKT proteins, KUP/HAK/KT proteins, and KEA proteins. There are two members of HKT subgroup (CarHKT1.1 and CarHKT1.2) in C. arietinum, duplicated 193.57 million years ago via tandem duplication, which showed a paralogous relation within the species. Both members showed a co-orthologous relation with their counterparts in A. thaliana (AtHKT1;1) and rice (OsHKT1.4 and OsHKT1.5).
The KUP/HAK/KT proteins in C. arietinum showed a close relation with respective members in A. thaliana and rice (Fig. 1). The sequence identity between KUP/HAK/KT protein sequences of C. arietinum and A. thaliana ranges from less than 40% to more than 75%. Car/KUP/HAK/KT6 and Car/KUP/HAK/KT8 showed a paralogous relationship in C. arietinum and exhibited tandem duplication. CarKUP/HAK/KT6 showed an orthologous relation with its counterpart, AtKUP/HAK/KT6, in A. thaliana with 71.6% identity, which is the highest identity rate as compared to the identity between the paralogous. CarKUP/HAK/KT8 showed the highest identity (77.2%) with its ortholog, AtKUP/HAK/KT8, in A. thaliana. CarKUP/HAK/KT3 and Car KUP/HAK/KT4 showed a paralogous relationship in C. arietinum, which were produced because of either tandem or segmental duplication before 148.6 million years ago. CarKUP/HAK/KT3 showed an ortholog relationship with its counterpart (AtKUP/HAK/KT3) in A. thaliana. CarKT4 showed a close relation with its counterpart in A. thaliana with 73.3% identity. CarKUP/HAK/KT5.1 and CarKUP/HAK/KT5.2 were emerged as a result of either tandem or segmental duplication since 158.93 million years ago. CarKUP/HAK/KT5.1 showed an orthologous relationship with AtKUP/HAK/KT5.2. CarKUP/HAK/KT10.1 and CarKUP/HAK/KT10.2, which were duplicated via segmental duplication before 47.86 million years ago, showed a paralogous relationship in C. arietinum. They contain a co-orthologous relation with their counterparts in A. thaliana (AtKUP/HAK/KT9, AtKUP/HAK/KT10.1, and AtKUP/HAK/KT11) and rice (OsKT11 and OsKT12). CarKUP/HAK/KT10.1 showed a high identity rate (77.5%) with its counterpart (AtKUP/HAK/KT10) in A. thaliana. CarKUP/HAK/KT12.1 and CarKUP/HAK/KT12.2 showed a paralogous relation in C. arietinum, which were produced as a result of segmental duplication before 39.54 million years ago. CarKUP/HAK/KT12.1 and CarKUP/HAK/KT12.2 showed a co-orthologous relationship with AtKUP/HAK/KT12, their A. thaliana counterpart. CarKUP/HAK/KT7.1 and CarKUP/HAK/KT7.2 originated as a result of segmental duplication since 41.27 million years ago. They showed a close association with their counterpart AtKUP/HAK/KT7.1 in A. thaliana. CarKUP/HAK/KT7.1 showed 77.1% identity with AtKUP/HAK/KT7.
There are six members (named CarKEA1 to CarKEA6) of KEA family, which showed a close relation with their counterparts in O. sativa and A. thaliana. CarKEA1 and CarKEA2 showed a paralogous relationship in C. arietinum. Evolutionary analysis indicated that they were evolved 38.16 million years ago via segmental duplication. CarKEA1 showed a co-orthologous relationship with A. thaliana counterparts (AtKEA1 and AtKEA2) with a high identity rate (65.4%). CarKEA2 showed an orthologous relationship with a member of CPA family (OsKEA2) in O. sativa. They showed a 64.3% identity with each other. CarKEA6 and CarKEA4 were considered as paralogous with 37.4% identity. CarKEA4 showed an orthologous relationship with AtKEA4 while CarKEA6 showed an orthologous relationship with AtKEA6 with 74.7 and 72.1% identity, respectively. Phylogenetic and evolutionary analysis showed that the K+ transporters and channels of C. arietinum, A. thaliana, and O. sativa contain close relationship. Analysis of duplication events revealed that mostly the K+ transporters and channels were evolved as a result of segmental duplication. Whole-genome sequence analysis also demonstrated that the significant number (69%) of the annotated chickpea genes have a history of gene duplication after the divergence of the legumes from A. thaliana and grape (Varshney et al. 2013), which also supports our observation of gene duplication in chickpea.
Phylogenetic trees of K+ transporters were constructed individually to show clear distinction among subgroups of respective K+ transporter family. The phylogenetic tree of HKT transporters indicated the two subfamilies. Members of CarHKTs were placed in subfamily I (Fig. 3a). The KEA transporters were subdivided into four subgroups (Fig. 3b). CarKEAs shared paraphyletic groups with legumes and dicotyledonous plants. In most of the clades, CarKEAs were present in the same clade as of M. truncatula. The phylogenetic tree involving KUP/HAK/KT members from C. arietinum, G. max, M. truncatula, O. sativa, and A. thaliana shows the clear division of these genes into four groups (Fig. 3c). The KUP/HAK/KT proteins in C. arietinum shared the same clades with M. truncatula. Moreover, members of leguminous plants appeared as separate paraphyletic groups (Fig. 3c).
Promoter Analysis
The cis-acting regulatory elements (CREs) are DNA regions in the promoter, where a number of transcription factors can bind and regulate the transcription of downstream genes. Screening of the promoter region for the CREs can provide information about gene regulatory networks. To understand the functional and tissue-specific regulation of K+-transporting genes in C. arietinum, upstream regions (1000–1500 bp) of putative members were screened. A number of putative cis-elements were found in promoter sequences of both K+ channels and transporters (Tables S1 and S2). Since members of shaker family and HKTs are best characterized in model plant species, selected abiotic stress-related cis-elements were searched in promoter regions of shaker and HKT family members in C. arietinum (Table 2). Among the identified elements, the most common cis-acting elements were known to be involved in abiotic stress response such as NAC core motif, ABRE, MYB/MYC recognition site, W-box, GT-1 motif, and G-box. Moreover, regulatory elements involved in plant response to K+, CO2, and light signals were also identified (Table 2).
Expression Analysis
Expression of K+ channels and transporters is regulated by biotic and abiotic stresses (Becker et al. 2003; Gaymard et al. 1998; Lacombe et al. 2000; Leonhardt et al. 2004; Maathuis et al. 2003; Pilot et al. 2003a; Reintanz et al. 2002). Therefore, to predict the potential role of K+ transport-related genes in chickpea, relative real-time qRT-PCR was performed for the estimation of transcript abundance of respective genes in leaf tissues under different abiotic stresses. Out of eight shaker channel genes, the expression of three members (CarAKT1.2, CarKAT1.1, and CarGORK) was found to be differentially modulated by abiotic stresses (Fig. 7). The transcript abundance of CarAKT2 and CarKAT1.2 seems unregulated under these conditions. CarAKT1.2 (putatively involved in K+ influx) was highly up-regulated in response to drought and mannitol stress. However, the expression of CarKAT1.1 (putatively involved in K+ influx) was down-regulated in response to drought, heat, mannitol, and salt stress. The expression of CarGORK (putatively involved in K+ efflux) was increased four to six times in response to osmotic stress while there was a mild increase of expression in response to drought, heat, and salt stresses (Fig. 7). The mRNA expression of CarKUP/HAK/KT1 and CarKUP/HAK/KT5.1 was significantly modulated by drought and salt stresses. CarKUP/HAK/KT1 is down-regulated while CarKUP/HAK/KT5.1 is up-regulated in response to these stresses. On the other hand, CarKUP/HAK/KT8 is slightly up-regulated in response to osmotic stress. CarTPK3.2 is slightly up-regulated in response to heat stress while it is down-regulated in response to osmotic and salt stress (Fig. 7). CarHKT1.1 (a putative Na+ transporter) is positively modulated upon all stress conditions under study. Salt and osmotic stresses particularly up-regulate (12–16 times) the transcript abundance of CarHKT1.1. However, CarHKT1.2 was not expressed at all. CarKEA5 seems unresponsive to environmental conditions used in this research.

Relative qRT-PCR assay of potassium channel/transporter genes under heat, drought, salt, and osmotic stresses. The default expression value for each gene was one in non-treated plants
Discussion
Potassium plays an important role in different cellular processes like enzyme activation, stabilization of protein synthesis, neutralization of protein charges, and maintenance of pH in plants. It also acts as a key player in the regulation of water potential, turgor pressure, cell elongation, opening and closing of stomata, and membrane polarization (Maathuis 2009). A complex set of systems is involved in acquisition and translocation of K+ from soil to different parts of the plants (Dreyer and Uozumi 2011; Mäser et al. 2001; Ward et al. 2009). Current genome-wide analysis demonstrated eight shaker genes present in chickpea. This number is almost consistent with those of Vitis vinifera (9 genes) (Cuéllar et al. 2010), A. thaliana (9 genes) (Lebaudy et al. 2007), and O. sativa (11 genes) (Amrutha et al. 2007). However, in comparison to two outward rectifying channel genes (AtGORK and AtSKOR) in A. thaliana, only one outward rectifying channel gene (CarGORK) was identified in C. arietinum. It predicts important role of CarGORK in long-distance potassium transport from roots to shoots and stomatal movements (Hosy et al. 2003). Domain analysis of shaker family in chickpea indicated similar domain pattern as the members of shaker family in A. thaliana. These results are reinforced by phylogenetic analysis. Potassium channels in C. arietinum share close homology with dicots (M. truncatula, G. max, and A. thaliana) and divergence from monocots (O. sativa). Among dicots, K+ channels from C. arietinum are more closely related to K+ channels from M. truncatula as compared to those from G. max and A. thaliana (Fig. 4). Moreover, five-group distribution of shaker channels is also conserved (Fig. 4; Pilot et al. 2003b). These results indicate that K+ channels from C. arietinum are expected to be functionally conserved as well.
Genome-wide analysis of A. thaliana revealed that it contains five members of TPK family (TPKs) and only one member of Kir-like family (AtKCO). Members of TPK family contain 4TM/2P topology while Kir-like channels are 2TM/1P. Current analysis identified five members of TPK family in C. arietinum with 4TM/2P topology, while no member of Kir-like family was identified. TPK channels have also been identified in a number of plant species other than A. thaliana, i.e., Oryza sativa, Nicotiana tabacum, Hordeum vulgare, and Solanum tuberosum (Czempinski et al. 1999; Hamamoto et al. 2008; Isayenkov et al. 2011). Plant Kir-like channels were initially considered as a separate group, although they show a great similarity to TPK channels. Nonetheless, to date, they have been only discovered in A. thaliana (Gomez-Porras et al. 2012; Marcel et al. 2010). Apparently, they emerged because of recent evolutionary events. The phylogenetic analysis indicates that they are originated after the duplication of TPK channels followed by deletion of one P-loop (Voelker et al. 2010). Domain analysis indicated that Kir-like channels contain the same pattern as their homologs in O. sativa and A. thaliana. The GYGD motif that acts as a hallmark of potassium channels is highly conserved among all the members of TPK family in chickpea. A highly conserved motif, “RSXpS/pTXP” where p represents the phosphorylation of serine/threonine residues, has been reported at the N-terminal of TPKs. Phosphorylation of serine/threonine plays a crucial role in interaction with the 14-3-3 proteins (for review, see Sharma et al. 2013). In C. arietinum, serine/threonine residue of “RSXpS/pTXP” motif was found highly conserved among all members except CarTPK1.1, which indicated that it might not be regulated by 14-3-3 proteins.
Individual members of KUP/HAK/KT family have been studied in number of plant species other than A. thaliana, i.e., Lotus japonicas (Desbrosses et al. 2004), V. vinifera (Davies et al. 2006), Piper nigrum, Solanum lycopersicum (Wang et al. 2002), M. crystallinum (Su et al. 2002), H. vulgare (Rubio et al. 2000; Senn et al. 2001; Vallejo et al. 2005), and seagrass (Garciadeblas et al. 2002). Fifteen members of KUP/HAK/KT family were found in the genome of chickpea, which contain the same domain pattern as their orthologues in A. thaliana. However, they possess 12 TM domains and an additional K_trans-domain, which play a crucial role in K+ transport. It suggests a potentially distinct role of CarKUP/HAK/KTs in potassium transport. According to phylogenetic analysis, CarKUP/HAK/KTs were divided into four groups (Fig. 3c), as described for other plants (Bañuelos et al. 2002; Rubio et al. 2000). Some transporters in group I (AtHAK5, OsHAK1, and OsHAK5) are functionally well characterized and are known to mediate K+ uptake in roots (Bañuelos et al. 2002; Rubio et al. 2000). Knockout mutants of OsHAK5 displayed decreased net short-term K+ influx rate by 80% in 0.1 mM K+ solution (Yang et al. 2014). AtHAK5 is the only gene mediating K+ uptake at concentrations below 0.01 mM. At the concentration between 0.01 and 0.05 mM K+, AtHAK5 and AtAKT1 are the only contributors to K+ acquisition. At higher K+ concentrations, unknown systems come into operation and participate together with AtAKT1 in low-affinity K+ uptake (Rubio et al. 2010). CarKUP/HAK/KT5.2 shares phylogenetic cluster with OsHAK5 (69% similarity) and AtHAK5 (71% similarity) in the phylogenetic analysis. Therefore, this gene is predicted to perform likewise functions and might help plant growth in K+-limiting conditions. AtKUP7 is considered a potential alternative system involved in K+ uptake besides AtAKT1 channel and AtHAK5 transporters (Nieves-Cordones et al. 2016).There are two homologs of AtKUP7 in C. arietinum as CarKUP/HAK/KT7.1 (87% similarity) and CarKUP/HAK/KT7.2 (84% similarity). Therefore, it is predicted that these proteins also share functional properties with AtKUP7.
Two members of HKT family (CarHKT1.1 and CarHKT1.2) were found in chickpea. These members of HKT family belong to Trk superfamily that has a topology similar to K+ channels and is primarily involved in K+/Na+ transport. Multiple sequence alignment showed that “G” residue is substituted by “S” in both members; therefore, they may be involved in Na+ transport. In phylogenetic analysis, both members were seen in close relationship with OsHKT1.4 and OsHKT1.5, which further supports their involvement in Na+ transport (Platten et al. 2006).
It has already been established that glycosylation may affect fundamental properties of membrane proteins such as stability, folding, ligand binding, and localization (Strasser 2016). N-Glycosylation has been observed as a typical characteristic in numerous eukaryotic membrane proteins (van Geest and Lolkema 2000). In Arabidopsis thaliana, a Na+ transporter AtHKT1;1 has been shown to contain one functional glycosylation site at a position of N429, which is located at the extracellular side of the protein (Kato et al. 2001). In the loss of site mutant N429Q, the AtHKT1;1 expressed in Xenopus oocytes retained selectivity for Na+ and hence remained functional resembling wild-type AtHKT1;1 (Kato et al. 2001). In contrary, disruption or inhibition of N-glycosylation could lead to protein misfolding resulting proteasomal degradation (Beers et al. 2013). It can also change transporter membrane density and hence disrupt turnover rates (Dauterive et al. 1996). In this context, the presence of putative multiple glycosylation sites in the chickpea potassium transporters provides an opportunity to explore their role through functional characterization.
Previous reports have demonstrated that potassium transport-related genes were involved in plant response to abiotic stresses (Hosy et al. 2003; Lebaudy et al. 2007; Lebaudy et al. 2008; Nieves-Cordones et al. 2016; Sharma et al. 2013). We studied the expression pattern of shaker channel genes and representatives from TPK, KUP/HAK/KT, KEAs, and HKTs families. The expression was studied in response to four environmental stresses (for details, see “Materials and Methods”). Five shaker channel genes (CarAKT1.2, CarKAT1.1, CarKAT1.2, CarAKT2, and CarGORK) out of eight were expressed in leaves under normal conditions (Fig. 7). These results are in accordance with previous studies, which report tissue-specific expression of shaker channel genes (reviewed by (Lebaudy et al. 2007)). Moreover, gene expression was also recorded for CarKUP/HAK/KT1, CarKUP/HAK/KT5.2, CarKUP/HAK/KT7.1, CarTPK3.2, CarKEA5, and CarHKT1.1. Gene expression profiles in response to various abiotic stresses revealed that CarAKT1.2, CarAKT2, CarHAK/KUP/KT7.1, and CArKEA5 were unresponsive to stresses. However, differential expression was observed for CarAKT1.2, CarGORK, CarKAT1.1, CarKUP/HAK/KT1, CarKUP/HAK/KT5.2, CarTPK3.2, and CarHKT1.1. Homologs of these genes were reported to participate in plant stress response in different plants (Becker et al. 2003; Liu et al. 2006; Voelker et al. 2010). Osakabe et al. 2013; Hamamoto et al. 2015; Ahmad et al. 2016). Moreover, Cis-elements involved in abiotic stress adaption (Table 2) have been reported in the promoter regions of potassium transport-related genes (Hyun et al. 2014), and potassium homeostasis is important for stress tolerance (Assaha et al. 2017). Therefore, differentially expressing K+ transport-related genes are likely to function in plant adaptation to environmental stresses.
Conclusion
Potassium is one of the three major nutrients (NPK) for plants, yet it is given least importance both at farmer and researcher ends. Chickpea is also cultivated on nutrient poor soils. A number of studies have demonstrated the importance of potassium transport systems for plant growth and development. Therefore, a detailed investigation is required to get more information about the molecular mechanisms of K+ homeostasis in chickpea and their impact on plant growth, development, and stress response. The current study was carried out to identify the K+-transporting genes in C. arietinum. On the basis of their sequence and structural identity with K+-transporting genes in A. thaliana, total 36 members were found and divided as K+ transporters (23 members) and channels (13 members). In silico analyses indicated the presence of important residues. Moreover, a detailed phylogenetic analysis and gene structure analysis provided information about evolutionary conservation of these genes in monocots/dicots and legumes/non-legumes. Gene promoter and expression analysis indicated the potential involvement of potassium transport-related genes in abiotic stress response of chickpea. Current study provided a first insight of K+ transport system in C. arietinum, which will be helpful in exploring the functioning of these genes.
References
Ahmad I, Mian A, Maathuis FJM (2016) Overexpression of the rice AKT1 potassium channel affects potassium nutrition and rice drought tolerance. J Exp Bot 67:2689–2698. https://doi.org/10.1093/jxb/erw103
Almeida P, Katschnig D, de Boer A (2013) HKT transporters—state of the art. Int J Mol Sci 14:20359–20385. https://doi.org/10.3390/ijms141020359
Amrutha RN, Sekhar PN, Varshney RK, Kishor PBK (2007) Genome-wide analysis and identification of genes related to potassium transporter families in rice (Oryza sativa L.). Plant Sci 172:708–721. https://doi.org/10.1016/j.plantsci.2006.11.019
Assaha DVM, Ueda A, Saneoka H, al-Yahyai R, Yaish MW (2017) The role of Na+ and K+ transporters in salt stress adaptation in glycophytes. Front Physiol 8. https://doi.org/10.3389/fphys.2017.00509
Bañuelos MA, Garciadeblas B, Cubero B, Rodríguez-Navarro A (2002) Inventory and functional characterization of the HAK potassium transporters of rice. Plant Physiol 130:784–795. https://doi.org/10.1104/pp.007781
Becker D, Hoth S, Ache P, Wenkel S, Roelfsema MRG, Meyerhoff O, Hartung W, Hedrich R (2003) Regulation of the ABA-sensitive Arabidopsis potassium channel gene GORK in response to water stress. FEBS Lett 554:119–126. https://doi.org/10.1016/S0014-5793(03)01118-9
Beers MF, Zhao M, Tomer Y, Russo SJ, Zhang P, Gonzales LW, Guttentag SH, Mulugeta S (2013) Disruption of N-linked glycosylation promotes proteasomal degradation of the human ATP-binding cassette transporter ABCA3. Am J Physiol Cell Mol Physiol 305:L970–L980. https://doi.org/10.1152/ajplung.00184.2013
Clarkson DT, Hanson JB (1980) The mineral nutrition of higher plants. Annu Rev Plant Physiol Plant Mol Biol 31:239–298
Crooks GE (2004) WebLogo: a sequence logo generator. Genome Res 14:1188–1190. https://doi.org/10.1101/gr.849004
Cuéllar T, Pascaud F, Verdeil J-L et al (2010) A grapevine shaker inward K(+) channel activated by the calcineurin B-like calcium sensor 1-protein kinase CIPK23 network is expressed in grape berries under drought stress conditions. Plant J 61:58–69. https://doi.org/10.1111/j.1365-313X.2009.04029.x
Czempinski K, Gaedeke N, Zimmermann S, Müller-Röber B (1999) Molecular mechanisms and regulation of plant ion channels. J Exp Bot 50:955–966. https://doi.org/10.1093/jxb/50.Special_Issue.955
Dauterive R, Laroux S, Bunn RC, Chaisson A, Sanson T, Reed BC (1996) C-terminal mutations that alter the turnover number for 3-O-methylglucose transport by GLUT1 and GLUT4. J Biol Chem 271:11414–11421. https://doi.org/10.1074/jbc.271.19.11414
Davies C, Shin R, Liu W, Thomas MR, Schachtman DP (2006) Transporters expressed during grape berry (Vitis vinifera L.) development are associated with an increase in berry size and berry potassium accumulation. J Exp Bot 57:3209–3216. https://doi.org/10.1093/jxb/erl091
Desbrosses G, Kopka C, Ott T, Udvardi MK (2004) Lotus japonicus LjKUP is induced late during nodule development and encodes a potassium transporter of the plasma membrane. Mol Plant-Microbe Interact 17:789–797. https://doi.org/10.1094/MPMI.2004.17.7.789
Dreyer I, Uozumi N (2011) Potassium channels in plant cells. FEBS J 278:4293–4303. https://doi.org/10.1111/j.1742-4658.2011.08371.x
Garciadeblas B, Benito B, Rodríguez-Navarro A (2002) Molecular cloning and functional expression in bacteria of the potassium transporters CnHAK1 and CnHAK2 of the seagrass Cymodocea nodosa. Plant Mol Biol 50:623–633
Gaymard F, Pilot G, Lacombe B, Bouchez D, Bruneau D, Boucherez J, Michaux-Ferrière N, Thibaud JB, Sentenac H (1998) Identification and disruption of a plant shaker-like outward channel involved in K+ release into the xylem sap. Cell 94:647–655
Gomez-Porras JL, Riaño-Pachón DM, Benito B, Haro R, Sklodowski K, Rodríguez-Navarro A, Dreyer I (2012) Phylogenetic analysis of K+ transporters in bryophytes, lycophytes, and flowering plants indicates a specialization of vascular plants. Front Plant Sci 3:167. https://doi.org/10.3389/fpls.2012.00167
Gupta AS, Berkowitz GA, Pier PA (1989) Maintenance of photosynthesis at low leaf water potential in wheat: role of potassium status and irrigation history. Plant Physiol 89:1358–1365
Hamamoto S, Marui J, Matsuoka K, Higashi K, Igarashi K, Nakagawa T, Kuroda T, Mori Y, Murata Y, Nakanishi Y, Maeshima M, Yabe I, Uozumi N (2008) Characterization of a tobacco TPK-type K+ channel as a novel tonoplast K+ channel using yeast tonoplasts. J Biol Chem 283:1911–1920. https://doi.org/10.1074/jbc.M708213200
Hamamoto S, Horie T, Hauser F, Deinlein U, Schroeder JI, Uozumi N (2015) HKT transporters mediate salt stress resistance in plants: from structure and function to the field. Curr Opin Biotechnol 32:113–120. https://doi.org/10.1016/j.copbio.2014.11.025
Hedrich R (2012) Ion channels in plants. Physiol Rev 92:1777–1811. https://doi.org/10.1152/physrev.00038.2011
Hosy E, Vavasseur A, Mouline K, Dreyer I, Gaymard F, Poree F, Boucherez J, Lebaudy A, Bouchez D, Very AA, Simonneau T, Thibaud JB, Sentenac H (2003) The Arabidopsis outward K+ channel GORK is involved in regulation of stomatal movements and plant transpiration. Proc Natl Acad Sci U S A 100:5549–5554. https://doi.org/10.1073/pnas.0733970100
Hyun TK, Rim Y, Kim E, Kim J-S (2014) Genome-wide and molecular evolution analyses of the KT/HAK/KUP family in tomato (Solanum lycopersicum L.). Genes Genomics 36:365–374. https://doi.org/10.1007/s13258-014-0174-0
Isayenkov S, Isner J-C, Maathuis FJM (2011) Rice two-pore K+ channels are expressed in different types of vacuoles. Plant Cell 23:756–768. https://doi.org/10.1105/tpc.110.081463
Jain M, Misra G, Patel RK, Priya P, Jhanwar S, Khan AW, Shah N, Singh VK, Garg R, Jeena G, Yadav M, Kant C, Sharma P, Yadav G, Bhatia S, Tyagi AK, Chattopadhyay D (2013) A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J 74:715–729. https://doi.org/10.1111/tpj.12173
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282
Kato Y, Sakaguchi M, Mori Y, Saito K, Nakamura T, Bakker EP, Sato Y, Goshima S, Uozumi N (2001) Evidence in support of a four transmembrane-pore-transmembrane topology model for the Arabidopsis thaliana Na+/K+ translocating AtHKT1 protein, a member of the superfamily of K+ transporters. Proc Natl Acad Sci U S A 98:6488–6493. https://doi.org/10.1073/pnas.101556598
Koizumi N, Ujino T, Sano H, Chrispeels MJ (1999) Overexpression of a gene that encodes the first enzyme in the biosynthesis of asparagine-linked glycans makes plants resistant to tunicamycin and obviates the tunicamycin-induced unfolded protein response. Plant Physiol 121:353–361
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7 . 0 for bigger datasets brief communication. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Lacombe B, Pilot G, Michard E, Gaymard F, Sentenac H, Thibaud JB (2000) A shaker-like K(+) channel with weak rectification is expressed in both source and sink phloem tissues of Arabidopsis. Plant Cell 12:837–851
Latz A, Becker D, Hekman M, Müller T, Beyhl D, Marten I, Eing C, Fischer A, Dunkel M, Bertl A, Rapp UR, Hedrich R (2007) TPK1, a Ca(2+)-regulated Arabidopsis vacuole two-pore K(+) channel is activated by 14-3-3 proteins. Plant J 52:449–459. https://doi.org/10.1111/j.1365-313X.2007.03255.x
Lebaudy A, Véry A-A, Sentenac H (2007) K+ channel activity in plants: genes, regulations and functions. FEBS Lett 581:2357–2366. https://doi.org/10.1016/j.febslet.2007.03.058
Lebaudy A, Vavasseur A, Hosy E, Dreyer I, Leonhardt N, Thibaud JB, Véry AA, Simonneau T, Sentenac H (2008) Plant adaptation to fluctuating environment and biomass production are strongly dependent on guard cell potassium channels. Proc Natl Acad Sci U S A 105:5271–5276. https://doi.org/10.1073/pnas.0709732105
Leigh RA (2001) Potassium homeostasis and membrane transport. J Plant Nutr Soil Sci 164:193–198. https://doi.org/10.1002/1522-2624(200104)164:2<193::AID-JPLN193>3.0.CO;2-7
Leonhardt N, Kwak JMJ, Robert N, Waner D, Leonhardt G, Schroeder JI (2004) Microarray expression analyses of Arabidopsis guard cells and isolation of a recessive abscisic acid hypersensitive protein phosphatase 2C mutant. Plant Cell 16:596–615. https://doi.org/10.1105/tpc.019000.2
Lerouge P, Cabanes-Macheteau M, Rayon C, Fischette-Lainé AC, Gomord V, Faye L (1998) N-glycoprotein biosynthesis in plants: recent developments and future trends. Plant Mol Biol 38:31–48. https://doi.org/10.1023/A:1006012005654
Li W, Liu B, Yu L, Feng D, Wang H, Wang J (2009) Phylogenetic analysis, structural evolution and functional divergence of the 12-oxo-phytodienoate acid reductase gene family in plants. BMC Evol Biol 9:90. https://doi.org/10.1186/1471-2148-9-90
Li W-Y, Wang X, Li R, Li WQ, Chen KM (2014) Genome-wide analysis of the NADK gene family in plants. PLoS One 9:e101051. https://doi.org/10.1371/journal.pone.0101051
Liu K, Li L, Luan S (2006) Intracellular K+ sensing of SKOR, a Shaker-type K+ channel from Arabidopsis. Plant J 46:260–268. https://doi.org/10.1111/j.1365-313X.2006.02689.x
Maathuis FJM (2009) Physiological functions of mineral macronutrients. Curr Opin Plant Biol 12:250–258. https://doi.org/10.1016/j.pbi.2009.04.003
Maathuis FJM, Filatov V, Herzyk P, C. Krijger G, B. Axelsen K, Chen S, Green BJ, Li Y, Madagan KL, Sánchez-Fernández R, Forde BG, Palmgren MG, Rea PA, Williams LE, Sanders D, Amtmann A (2003) Transcriptome analysis of root transporters reveals participation of multiple gene families in the response to cation stress. Plant J 35:675–692. https://doi.org/10.1046/j.1365-313X.2003.01839.x
Marcel D, Müller T, Hedrich R, Geiger D (2010) K+ transport characteristics of the plasma membrane tandem-pore channel TPK4 and pore chimeras with its vacuolar homologs. FEBS Lett 584:2433–2439. https://doi.org/10.1016/j.febslet.2010.04.038
Mäser P, Thomine S, Schroeder JI, Ward JM, Hirschi K, Sze H, Talke IN, Amtmann A, Maathuis FJ, Sanders D, Harper JF, Tchieu J, Gribskov M, Persans MW, Salt DE, Kim SA, Guerinot ML (2001) Phylogenetic relationships within cation transporter families of Arabidopsis. Plant Physiol 126:1646–1667. https://doi.org/10.1104/pp.126.4.1646
Moshelion M, Becker D, Czempinski K, Mueller-Roeber B, Attali B, Hedrich R, Moran N (2002) Diurnal and circadian regulation of putative potassium channels in a leaf moving organ. Plant Physiol 128:634–642. https://doi.org/10.1104/pp.010549
Munro AW, Ritchie GY, Lamb AJ, Douglas RM, Booth IR (1991) The cloning and DNA sequence of the gene for the glutathione-regulated potassium-efflux system KefC of Escherichia coli. Mol Microbiol 5:607–616
Nieves-Cordones M, Martínez V, Benito B, Rubio F (2016) Comparison between Arabidopsis and rice for main pathways of K+ and Na+ uptake by roots. Front Plant Sci 7. https://doi.org/10.3389/fpls.2016.00992
Osakabe Y, Yamaguchi-Shinozaki K, Shinozaki K, Tran L-SP (2013) Sensing the environment: key roles of membrane-localized kinases in plant perception and response to abiotic stress. J Exp Bot 64:445–458. https://doi.org/10.1093/jxb/ers354
Pilot G, Gaymard F, Mouline K, Chérel I, Sentenac H (2003a) Regulated expression of Arabidopsis shaker K+ channel genes involved in K+ uptake and distribution in the plant. Plant Mol Biol 51:773–787
Pilot G, Pratelli R, Gaymard F et al (2003b) Five-group distribution of the shaker-like K+ channel family in higher plants. J Mol Evol 56:418–434. https://doi.org/10.1007/s00239-002-2413-2
Platten JD, Cotsaftis O, Berthomieu P, Bohnert H, Davenport RJ, Fairbairn DJ, Horie T, Leigh RA, Lin HX, Luan S, Mäser P, Pantoja O, Rodríguez-Navarro A, Schachtman DP, Schroeder JI, Sentenac H, Uozumi N, Véry AA, Zhu JK, Dennis ES, Tester M (2006) Nomenclature for HKT transporters, key determinants of plant salinity tolerance. Trends Plant Sci 11:372–374. https://doi.org/10.1016/j.tplants.2006.06.001
Rasool S, Latef AAHA, Ahmad P (2015) Chickpea. In: Legumes under environmental stress. Wiley, Chichester, pp 67–79
Reintanz B, Szyroki A, Ivashikina N, Ache P, Godde M, Becker D, Palme K, Hedrich R (2002) AtKC1, a silent Arabidopsis potassium channel alpha-subunit modulates root hair K+ influx. Proc Natl Acad Sci U S A 99:4079–4084. https://doi.org/10.1073/pnas.052677799
Rubio F, Santa-Maria GE, Rodriguez-Navarro A (2000) Cloning of Arabidopsis and barley cDNAs encoding HAK potassium transporters in root and shoot cells. Physiol Plant 109:34–43. https://doi.org/10.1034/j.1399-3054.2000.100106.x
Rubio F, Alemán F, Nieves-Cordones M, Martínez V (2010) Studies on Arabidopsis athak5, atakt1 double mutants disclose the range of concentrations at which AtHAK5, AtAKT1 and unknown systems mediate K uptake. Physiol Plant 139:220–228. https://doi.org/10.1111/j.1399-3054.2010.01354.x
Schroeder JI, Ward JM, Gassmann W (1994) Perspectives on the physiology and structure of inward-rectifying K+ channels in higher plants: biophysical implications for K+ uptake. Annu Rev Biophys Biomol Struct 23:441–471. https://doi.org/10.1146/annurev.bb.23.060194.002301
Senn ME, Rubio F, Bañuelos MA, Rodríguez-Navarro A (2001) Comparative functional features of plant potassium HvHAK1 and HvHAK2 transporters. J Biol Chem 276:44563–44569. https://doi.org/10.1074/jbc.M108129200
Sharma T, Dreyer I, Riedelsberger J (2013) The role of K+ channels in uptake and redistribution of potassium in the model plant Arabidopsis thaliana. Front Plant Sci 4. https://doi.org/10.3389/fpls.2013.00224
Shi H, Ishitani M, Kim C, Zhu JK (2000) The Arabidopsis thaliana salt tolerance gene SOS1 encodes a putative Na+/H+ antiporter. Proc Natl Acad Sci U S A 97:6896–6901. https://doi.org/10.1073/pnas.120170197
Singh KB, Ocampo B, Robertson LD (1998) Diversity for abiotic and biotic stress resistance in the wild annual Cicer species. Genet Resour Crop Evol 45:9–17. https://doi.org/10.1023/A:1008620002136
Strasser R (2016) Plant protein glycosylation. Glycobiology 26:926–939. https://doi.org/10.1093/glycob/cww023
Su H, Golldack D, Zhao C, Bohnert HJ (2002) The expression of HAK-type K(+) transporters is regulated in response to salinity stress in common ice plant. Plant Physiol 129:1482–1493. https://doi.org/10.1104/pp.001149
Tholema N, Bruggen MV d, Maser P et al (2005) All four putative selectivity filter glycine residues in KtrB are essential for high affinity and selective K+ uptake by the KtrAB system from Vibrio alginolyticus. J Biol Chem 280:41146–41154. https://doi.org/10.1074/jbc.M507647200
Vallejo AJ, ML P, GE S-M (2005) Expression of potassium-transporter coding genes, and kinetics of rubidium uptake, along a longitudinal root axis. Plant Cell Environ 28:850–862. https://doi.org/10.1111/j.1365-3040.2005.01334.x
van Geest M, Lolkema JS (2000) Membrane topology and insertion of membrane proteins: search for topogenic signals. Microbiol Mol Biol Rev 64:13–33. https://doi.org/10.1128/MMBR.64.1.13-33.2000
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Tar'an B, Millan T, Zhang X, Ramsay LD, Iwata A, Wang Y, Nelson W, Farmer AD, Gaur PM, Soderlund C, Penmetsa RV, Xu C, Bharti AK, He W, Winter P, Zhao S, Hane JK, Carrasquilla-Garcia N, Condie JA, Upadhyaya HD, Luo MC, Thudi M, Gowda CLL, Singh NP, Lichtenzveig J, Gali KK, Rubio J, Nadarajan N, Dolezel J, Bansal KC, Xu X, Edwards D, Zhang G, Kahl G, Gil J, Singh KB, Datta SK, Jackson SA, Wang J, Cook DR (2013) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31:240–246. https://doi.org/10.1038/nbt.2491
Véry A-A, Sentenac H (2003) Molecular mechanisms and regulation of K+ transport in higher plants. Annu Rev Plant Biol 54:575–603. https://doi.org/10.1146/annurev.arplant.54.031902.134831
Véry A-A, Nieves-Cordones M, Daly M, Khan I, Fizames C, Sentenac H (2014) Molecular biology of K+ transport across the plant cell membrane: what do we learn from comparison between plant species? J Plant Physiol 171:748–769. https://doi.org/10.1016/j.jplph.2014.01.011
Voelker C, Gomez-Porras JL, Becker D, Hamamoto S, Uozumi N, Gambale F, Mueller-Roeber B, Czempinski K, Dreyer I (2010) Roles of tandem-pore K+ channels in plants—a puzzle still to be solved. Plant Biol 12:56–63. https://doi.org/10.1111/j.1438-8677.2010.00353.x
Walker DJ, R a L, Miller a J (1996) Potassium homeostasis in vacuolate plant cells. Proc Natl Acad Sci U S A 93:10510–10514
Wang Y-H, Garvin DF, Kochian LV (2002) Rapid induction of regulatory and transporter genes in response to phosphorus, potassium, and iron deficiencies in tomato roots. Evidence for cross talk and root/rhizosphere-mediated signals. Plant Physiol 130:1361–1370. https://doi.org/10.1104/pp.008854
Wang M, Zheng Q, Shen Q, Guo S (2013) The critical role of potassium in plant stress response. Int J Mol Sci 14:7370–7390. https://doi.org/10.3390/ijms14047370
Ward JM, Mäser P, Schroeder JI (2009) Plant ion channels: gene families, physiology, and functional genomics analyses. Annu Rev Physiol 71:59–82. https://doi.org/10.1146/annurev.physiol.010908.163204
Yang T, Zhang S, Hu Y, Wu F, Hu Q, Chen G, Cai J, Wu T, Moran N, Yu L, Xu G (2014) The role of a potassium transporter OsHAK5 in potassium acquisition and transport from roots to shoots in rice at low potassium supply levels. Plant Physiol 166:945–959. https://doi.org/10.1104/pp.114.246520
Funding
This study was funded by International Foundation for Science (IFS) (grant number no. C/5684-1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic Supplementary Material
Table S1
(DOCX 17 kb)
Table S2
(DOCX 21 kb)
Table S3
(DOCX 12 kb)
Figure S1
A) An unrooted phylogenetic tree of HKT family is generated via MEGA 7. The complete protein sequences of all members were aligned to generate the tree. Two subfamilies of HKTs (subfamily I and subfamily II) on the base of G or S residues are also shown in the tree. (B) Multiple sequence alignment of HKT family is shown. First P loop of various plant (Cicer arietinum L, Eucalyptus camaldulensis, Oryza sativa and Mesembryanthemum crystallinum) HKTs is compared with TrkH from Pseudomonas aeruginosa, P-loop of the Drosophila Shaker channel, Trk1from S. cerevisiae and to the KtrB from Vibrio alginolyticus, Alignment was done via ClustalX2. Conserved G residue is shown above the alignment. (JPG 202 kb)
Figure S2
(A) An unrooted phylogenetic tree of KUP/HAK/KT family is presented. The tree was generated using MEGA 7. The complete protein sequence of all members was aligned and TreeView program was used for visualization and analysis of graphical output. (B) Multiple sequence alignment of KUP/HAK/KT family is shown. Alignment was done via ClustalX2. Conserved motifs are shown above the alignment. Highly conserved residues are highlighted. (JPG 377 kb)
Figure S3
(A) An unrooted phylogenetic tree of KEA family is given. The tree was generated using Neighbor joining method via MEGA 7. TreeView program was used for visualization of graphical output. (B) Multiple sequence alignment of KEA family is shown. Alignment was done via ClustalX2. Conserved motifs are shown above the alignment. Highly conserved residues are highlighted. (JPG 348 kb)
Figure S4
(A) An unrooted phylogenetic tree of Shaker family is presented. The tree was generated using MEGA 7. The complete protein sequence of all members was aligned to generate the tree. Subfamilies of Shaker family are also shown in the tree. (B) Multiple sequence alignment of Shaker family is shown. Alignment was done via ClustalX2. TXXTXGYGD motif, a hallmark of K+ channels, is shown above the alignment . Highly conserved residues are highlighted in black. (JPG 447 kb)
Figure S5
(A) An unrooted phylogenetic tree of KCO family is given. The tree was generated using Neighbor joining method via MEGA 7. TreeView program was used for visualization and analysis of tree. (B)Multiple sequence alignment of KCO family is shown. Alignment was done via ClustalX2. Conserved motifs i.e. RSXpSX, necessary to interact with regulatory proteins, and TXXTXGYGD (a hallmark of K+ channels) are shown above the alignment. Highly conserved residues are highlighted in black. (JPG 220 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Azeem, F., Ahmad, B., Atif, R.M. et al. Genome-Wide Analysis of Potassium Transport-Related Genes in Chickpea (Cicer arietinum L.) and Their Role in Abiotic Stress Responses. Plant Mol Biol Rep 36, 451–468 (2018). https://doi.org/10.1007/s11105-018-1090-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-018-1090-2