
Abstract
MicroRNAs (miRNAs) are small ∼20–24 nt species of non-coding RNAs that modulate plant gene expression by means of gene silencing through sequence-specific inhibition of target mRNAs. MiRNAs derive from pol-II transcription of non-coding genes that are precisely processed in nuclear Dicing bodies by a microprocessor complex (dicer-like1–serrate–hyponastic leaves 1: DCL1-SE-HYL1), which recognizes stem-loop secondary-structure features of primary precursor miRNA transcripts (pri-miRNA). The proper processing of the pri-miRNAs results in a double-stranded small RNA that will eventually exit the nucleus and be loaded predominantly onto the effector complex Argonaute1 (Ago1). The single-stranded mature miRNA will guide AGO1, leading to cleavage or translational arrest of complementary mRNAs. MiRNA steady-state levels and activity are regulated not only by transcription rate of precursor transcripts, but also by direct degradation mediated by small RNA degrading nuclease1 (SDN1). miRNAs are retailored by 3′ editing through 2-O-methylation, uridylation and adenlylation, involving Hua enhancer1 (HEN1), HEN1 suppressor1 (HESO1) and probably the exosome—a phenomenon that has been elucidated only scarcely to date in Arabidopsis. MiRNA activity is involved not only in plant development, but also in signaling, abiotic stresses such as drought, heat and metal toxicity, pathogen interaction and symbiotic relationship regulation, among others. The engineering of miRNAs is paving the way to next-generation plant biotechnology by means of over-expression of natural miRNAs, generation of artificial microRNAs and inhibition of miRNA activity by target mimicry. This review highlights the importance of miRNAs in plant sciences by describing the latest updates in this research field.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plant miRNA biology has flourished during the last decade. The first discoveries in the field described a role for miRNAs in plant development through the inhibition of transcription factors (TF) (Rhoades et al. 2002; Park et al. 2002; Palatnik et al. 2003). In turn, the spectrum of action of these regulatory molecules expanded to abiotic stress and pathogen interaction (Jones-Rhoades et al. 2006; Rajagopalan et al. 2006). While the biogenesis of miRNAs has been explored in detail, the relevance of miRNA turnover is only starting to be elucidated. In recent years there has been a burst of publications in miRNA biology. This review, albeit discussing several seminal reports, focuses mainly on the most recent publications in the field, highlighting innovative advances and introducing future challenges in next-generation plant miRNA research.
MIRNA Gene Structure and Biogenesis
There is evidence that the miRNA pathway evolved before multicellularity. The unicellular alga Chlamydomonas reinhardtii generates miRNAs with similar characteristics to those of higher plants (Molnár et al. 2007). There are highly conserved miRNAs in the plant kingdom that emerged very early in plant evolution. The current model suggests that miRNAs arise from the inverted duplication of their target genes, which would generate perfect hairpin RNAs. Eventually, site-mutation leads to imperfect stem-loops, which are recognizable by the specialized dicer-like1 (DCL1) enzyme complex (Axtell and Bowman 2008). Transposable elements may have been responsible for the amplification of MIRNA genes during evolution. There is a link between the accumulation of miRNA-like hairpins and long terminal repeat retrotransposons (LTR-RT), suggesting that a selective pressure of the miRNA pathway may influence LTR-RTs directly by inhibiting their activity. LTR-RTs subject to mutation could generate miRNA-like hairpins that would eventually become MIRNA genes, and in turn would regulate LTR-RT activity (Zhou et al. 2013a). Plant genomes are paved with miRNA transcriptional units.
MIRNA genes usually encode an independent transcriptional unit and are transcribed by RNA polymerase II (Pol II). Primary miRNA transcripts (pri-miRNA) are 5′-capped and polyadenylated and contain imperfect foldback structures (Xie et al. 2005). Three features differentiate MIRNA genes from protein-coding genes at the promoter level. First, although with a binding peak at the transcription start site (TSS) for high transcription, the decline of the pol II signal is more intense downstream, at approximately 500 bp from the TSS in MIRNA genes. Second, the free energy change occurring between the immediate upstream and downstream of the TSS is much milder than in protein-coding genes, which is consistent with omission of translation. Third, the composition of the cis-elements of the promoter differs in frequency and composition of motifs; the TATA box is more enriched in MIRNA genes, and the G-Box is over-represented (Zhao et al. 2013). Although MIRNA genes often encode a unique RNA transcript, there are a few documented polycistronic miRNAs in plants—a phenomenon that is much more common in animals. For example, two functionally related miRNAs, miR842 and miR846 targeting jacalins, have been described as arising from the same transcriptional unit. With the addition of a new layer of regulation, its processing may result in three isoforms. miRNA 846 is expressed only in one isoform, whereas in the other forms a part of miR846 is included in an intron that is regulated by ABA. Alternative splicing of the intron results in a disruption of the mature miR846 and a loss of functionality of these isoforms (Jia and Rock 2013).
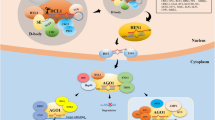
Transcription of MIRNA genes is dependent on the formation of a pre-initiation complex, in which Pol II binds to Mediator, recruits cyclin H-dependent CDKD kinases and basal TF IIH (Fig. 1). The CDKD and CAK-activating kinases phosporylate the carboxy-terminal domain of Pol II (CTD) at a Ser5 position, which is required for proper capping of nascent precursor RNA and recruitment of CBP80/ABA Hypersensitive1 and the CBP20 cap-binding complex (CBC). NOT2 interacts with CBC, Pol II, SE, and the Piwi/Ago/Zwille domain of DCL1 to promote the transcription of MIRNA genes and facilitates efficient DCL1 recruitment (Wang et al. 2013). The dicer-like1–serrate–hyponastic leaves 1 (DCL1–SE–HYL1) complex recognizes the stem-loop conformation of the precursor while interacting with the CBC (Hajheidari et al. 2013). With the help of SE, HYL1 is dephosphorylated by C-terminal domain phosphatase-like-1 (CPL1) and probably CPL2, which are required for accurate activity of HYL1. In the absence of CPL1, accurate processing and strand selection from miRNA duplexes are compromised (Manavella et al. 2012a).

Biogenesis of plant microRNAs: Pol II binds to Mediator, recruits CDKD kinases and basal TF IIH. CDKD and CDKF phosporylate Pol II for proper capping of nascent precursor RNA and recruiting of CBP80 and CBP20. NOT2 interacts with CBC, Pol II, SE, and DCL1.The dicer-like1–serrate–hyponastic leaves 1 (DCL1–SE–HYL1) complex recognizes the stem-loop. HYL1 is dephosphorylated by CPL1 and probably CPL2. Pre-miRNAs are recruited to dicing bodies. Tough (TGH) also cooperates with the DCL1-SE-HYL1 complex. When DCL1 binds to pre-miRNAs, its interaction with SE is no longer needed, and DCL1 proceeds with the cleavage of pri-miRNAs. miRNA duplexes composed of a miRNA guide and a miRNA passenger strand are protected by HEN1 methylation. After exiting the nucleus, miRNA duplexes are recruited to an AGO1 effector complex and miRNA* is degraded. After miRNA loading, target transcripts are often silenced through direct cleavage or translational repression
The activity of DCL1 is modulated by its helicase domain, which confers dependence of the pre-miRNA processing on ATP. This attenuation in DCL1 functioning is needed for precise processing of certain substrates, such as ath-pre-miR166b (Liu et al. 2012). This miRNA family has been explored to elucidate how defined secondary structures of precursors affect miRNA abundance. Multibranched terminal loops in some pre-miR166 species suppress miRNA accumulation considerably. The reason for this phenomenon is that, although DCL1 processes these pre-miRNAs “canonically” (at ∼15 bp from a loop region), in terminal branched precursors it is able to process the pre-miRNA from base-to-loop, or from loop-to-base—the latter generating an abortive miRNA (Zhu et al. 2013). DCL1 has two dsRNA-binding domains (dsRNA-BD), D1 and D2. D1, the N-terminal dsRNA-BD, plays a major role in binding to precursor miRNA, whereas the C-terminal dsRNA-BD, D2, is involved in protein–protein interactions with HYL1. Pri-miRNAs are recruited to dicing bodies, where DCL1 and HYL1 colocalize for accurate processing (Liu et al. 2013). The recently discovered MOS2 is a nuclear protein with G-patch and KOW RNA-binding domains that interacts with pri-miRNAs. MOS2 does not localize in dicing bodies nor does it interact with the DCL1-SE-HYL1 complex, although its impairment results in a reduced HYL1 activity and localization at the D-bodies (Wu et al. 2013b). The RNA-binding protein Tough (TGH) also cooperates with the DCL1-SE-HYL1 complex by binding to pri and pre-miRNAs and contributing to pri-miRNA HYL1 interaction (Ren et al. 2012b). A pre-mRNA processing factor, stabilized1 (STA1) indirectly affects DCL1 transcript levels and directly regulates the accumulation of intron-containing miRNAs by modulating splicing (Chaabane et al. 2013). SE induces DCL1 activity in an ionic strength-dependent scenario by its N-terminal binding domain interacting with RNA and the ZnF domain interacting with DCL1. When DCL1 binds to pre-miRNAs, its interaction with SE is no longer needed, and DCL1 proceeds with the cleavage of pri-miRNAs (Iwata et al. 2013). The interaction between DCL1 and SE is fundamental for the efficient enzymatic activity of DCL1. Scaffold proteins (Rack1) interact with SE, influencing the accumulation and processing of several miRNAs. Rack1 is also part of the AGO1 effector complex, with absence of Rack1 leading to over-accumulation of miRNA targets due to a deteriorated processing of pri-miRNAs (Speth et al. 2013). The structural constraints of the single-stranded secondary structure nature and requirements of pri-miRNAs have been studied extensively. In terms of conformational flexibility at the thermal equilibrium of the molecule, pri-miRNAs are characterized by plasticity and not by robustness (Rodrigo and Elena 2013).
Whilst the pathways from most pri-miRNA to mature miRNA, whether evolutionary ancient or young, involve the same proteins, the processing mechanism of precursors may differ. There are specific structural determinants in the precursor sequence that lead to divergence in the processing event even in miRNAs of the same family or alternative miRNAs from the same precursor. These processing mechanisms can be divided into four non-mutually exclusive categories: (1) a loop-to-base mechanism (Fig. 2a), guided by an upper stem releasing the mature miRNA after two consecutive cuts, in precursors with a conserved terminal region length of ∼42 nt and a small loop. (2) A long loop-to-base pathway (Fig. 2b), where the mature miRNA is released after four sequential cuts in pre-miRNAs with long conserved stems segments that can generate multiple sRNAs. (3) A pathway where the processing progress from base-to-loop via the recognition of a bulge followed by a cut at ∼15 nt from the lower stem (Fig. 2c), in precursors with a sharp transition from single-stranded RNA to lower stem. (4) Finally, a long base-to-loop pathway (Fig. 2d), similar to the latter, but in which the first cut is followed by two or three slices until the miRNA is released, and which might generate alternative small RNAs at low levels (Bologna et al. 2013).

Processing pathways of plant miRNA precursors. a Short loop-to-base pathway, guided by an upper stem releasing the mature miRNA after two consecutive cuts in precursors with a conserved terminal region length of ∼42 nt and a small loop. b Long loop-to-base pathway, where the mature miRNA is released after four sequential cuts in pre-miRNAs with long conserved stem segments that can generate multiple sRNAs. c Short base-to-loop pathway where the processing progresses from base-to-loop via the recognition of a bulge followed by a cut at ∼15 nt from the lower stem. d Long base-to-loop pathway, similar to the latter, but in which the first cut is followed by two or three slices until the miRNA is released
After exiting the nucleus, miRNA duplexes composed of a miRNA guide strand and a miRNA passenger strand (miRNA*) are recruited preferentially to AGO1, which favors 5′ terminal uridine, the most typical in plant miRNAs, whereas AGO2 and AGO4 recruit mainly sRNAs with a 5′ terminal adenosine and AGO5 mostly 5′ cytosine harboring sRNAs (Mi et al. 2008). It has been suggested that additional positions within the miRNA and sequence combinations may influence AGO sorting. Positions 2, 6, 9 and 13, as well as uracil bases in certain positions for AGO2 and AGO5, have been proposed (Thieme et al. 2012). After miRNA loading, highly complementary target transcripts are often silenced through direct cleavage (slicing), destabilization or translational repression (Jones-Rhoades et al. 2006; Brodersen et al. 2008). The most typical plant miRNA interaction with targets results in cleavage. This fact is consistent with a recent study reporting that the Arabidopsis mutant ago1-25 requires the catalytic residues of AGO1 to complement morphological and functional defects, supporting the consensus idea that the slicer activity is critical for AGO1 function (Carbonell et al. 2012).
Mature miRNAs may present length or sequence variants, generally named isomiRs. There is growing consensus about the importance of these miRNA versions (Jeong et al. 2011; Neilsen et al. 2012). These variants have also been described to be incorporated actively into RISC, and to regulate mRNA targets in mammals (Cloonan et al. 2011). In plants, a recent report has complemented isomiR identification with parallel analysis of RNA ends (PARE) data, revealing that predicted targets are regulated differently by certain isomiR variants. Notably, ath-miR161.1 and ath-miR161.3, two isoforms with only 1 nt shift, were predicted to regulate four PPR genes. However, the PARE data showed that two genes had cleavage intermediates arising only from ath-miR161.3, indicating that almost identical miRNA variants can regulate target genes distinctively (Jeong et al. 2013). Given the growing importance of miRNA variants, several pipelines have just been generated to address and standardize the identification of isomiRs from multiple high-throughput small RNA sequencing libraries (de Oliveira et al. 2013; Sablok et al. 2013).
Natural variants at the miRNA* sequence can also affect miRNA accumulation. A polymorphism in the ath-miR164a gene has been depicted, where a single base substitution in the miRNA* sequence results in reduced miR164 accumulation, probably by interfering in the precursor processing and affecting leaf shape and shoot architecture in a natural Arabidopsis strain. In that report, by exploring multiple A. thaliana accessions, the authors suggest that this is not an exceptional phenomenon but a common contributor to phenotypic variation in plants (Todesco et al. 2012). As a whole, it is the different expression levels, distinctive processing pathways, potential AGO loading, as well as the length and sequence variation that determine the resulting functional specification of miRNAs (Jeong et al. 2013).
MiRNA Editing and Turnover
The enzyme responsible for miRNA degradation is the exoribonuclease SDN1 (Ramachandran and Chen 2008). 3′ end modification of miRNAs influences abundance via stabilization and by directing their degradation. 2-O-methyl incorporation to the miRNA/miRNA* duplex terminal 3′ nucleotides, performed by the nuclear HEN1, protects miRNAs from degradation by exonucleases (Yu et al. 2005). Without methylation, miRNAs may undergo uridylation, promoting their degradation. Uridylation of miRNAs is performed by HESO1, a nontemplated terminal nucleotidyl transferase, which is inhibited completely by 2′-O-methylation (Ren et al. 2012a; Zhao et al. 2012b). After being uridylated, miRNAs are degraded; the enzyme responsible for this process has not been elucidated, but the involvement of the nuclear exosome subunit RRP6, a processive nuclease, has been suggested (Rogers and Chen 2012). This phenomenon has been documented in the alga Chlamydomonas reinhardtii, in which RRP6 is responsible for 3′–5′ degradation of uridylated miRNAs (Ibrahim et al. 2010). Two other types of miRNA editing have been described, 3′ adenylation and 3′ cytidine incorporation. 3′ adenlylation is proposed as promoting stabilization of miRNAs, consisting of one or a small number of post-transcriptionally added adenylic-acid residues that are different in length from the polyadenylate tail added to plant RNAs for exosome-mediated degradation (Lu et al. 2009). Cytidine incorporation at 3′ ends might promote miRNA degradation (Zhang et al. 2013a). The importance of these miRNA editing enzymes is reflected in, for example, the increased abundance of miRNAs in the heso1-2 mutants, the augmentation of truncated miRNA forms in the heso1-2 hen1 double mutants, and the reduction of miRNA abundance concomitant with vast accumulation of morphological defects of HESO1 over-expression in the hen1 background (Ren et al. 2012a). Moreover, the conservation of this attribute of HEN1 was determined by sequencing small RNAs (sRNA) of hen1 mutants from Arabidopsis, rice (Oryza sativa), and maize (Zea mays), in which a widespread 3′ truncation prior to tailing was observed, along with AGO1-bound miRNAs being actively truncated and tailed (Zhai et al. 2013).
MiRNA Target Acquisition
The sequence-specific nature of miRNA is bent by affinity to target mRNAs. The efficiency of miRNA-guided target inhibition can be modulated by the strength of the affinity between miRNA and target mRNA. For instance, ath-miRNA396 interacts with a target gene GRF2 TF with a bulge within positions 7–8 of the miRNA. This bulge modulates the repression induced by miR396 and shapes the precise spatio-temporal pattern of GRF2 expression (Debernardi et al. 2012). The acquisition of novel targets by conserved miRNAs might be biologically relevant in different backgrounds. ath-miR396 was also found to regulate bHLH transcription factors (Debernardi et al. 2012), and it was reported recently that Medicago truncatula plants over-expressing miR396b showed reduced growth and mycorrhizal associations, concomitant with repression of MtGRFs and bHLH-like genes (Bazin et al. 2013). Moreover, sunflower plants exposed to heat stress exhibit an induction of miR396, followed by a repression of a validated targeted WRKY TF. This miRNA, previously mentioned to be strongly involved in plant development, has acquired a new role in sunflower, regulating early response to heat-stress by gaining a new target, the HaWRKY6 TF (Giacomelli et al. 2012).
Firstly associated mainly with animals, miRNA-guided translational arrest of target mRNA is widespread in plants. The sub-cellular confinement of this process is dependent on Altered Meristem Program1 (AMP1)—an integral membrane protein associated with AGO1 and the endoplasmic reticulum (ER). A recruitment of miRNA target transcripts to membrane fractions has been observed, as well as the induction of translational arrest of target mRNA on the ER by miRNAs (Li et al. 2013b).
Since the introduction of next generation sequencing (NGS) to miRNA biology, the anecdotic high accumulation of miRNA passenger strands in several sRNA genome-wide profiles has raised attention. Although the functionality of miRNA* was suggested, the first report showing biological significance and activity of a miRNA* was demonstrated by active association of AGO1 with ath-miRNA171a* and loading onto RISC, triggering the tissue-specific silencing of a member of the large SET domain protein family, SUVH8 (Manavella et al. 2013).
MiRNAs and Plant Development
Plant development is influenced strongly by miRNAs (Rubio-Somoza and Weigel 2011; Chen 2012a; Jin et al. 2013). The juvenile phase is determined by miR156 regulation of target Squamosa Promoter-Binding Protein-Like (SPL) genes (Wang et al. 2009; Wu et al. 2009). HYL1 controls expression levels of miR156-targeted SPL genes, probably by ensuring proper processing of pri-miR156. In hyl1-2 mutants, the juvenile phase is compromised (Li et al. 2012b). Both miR156 and SPL genes are expressed in the developing gynoecium, controlling its patterning through interference with signaling and auxin homeostasis, and therefore directing the development of the female reproductive tract in Arabidopsis (Xing et al. 2013).
Flower maturation in Arabidopsis requires the coordinated activity of miR159, miR319 and miR167. miR159 regulates MYB and miR319, TCP TF. MYB33 and TCP4 induce miR167a transcription, which inhibits ARF6/8 TF, affecting auxins, gibberellic acid and jasmonic acid. This highly coordinated cross-regulation, including cis- and trans-interactions, modulates successive steps required for consecutive hormone-dependent transitions, leading to floral development (Rubio-Somoza and Weigel 2013). The influence of miR319 on flower maturation might be conserved among plants. A recent report indicates that tomato plants over-expressing miR319 flower with fewer leaves, suggesting tomato flowering delay through inhibition of miR319-sensitive TCPs, like Lanceolata (Burko et al. 2013). Flowering of the herbaceous biennial plant Cardamine flexuosa requires an age-dependent vernalization by exposure to cold. miR156 and miR172 control the timing of sensitivity to vernalization through modulation of the flower-promoting MADS-box gene CfSOC1, and in turn cueing flowering (Zhou et al. 2013b). The same signaling network has been observed in Arabis alpine, the perennial relative of A. thaliana. Before vernalization, miR172 levels are low, and its target Apetala2 represses flowering. Simultaneously, miR156 levels decline chronically, resulting in an increased abundance of SPLs, which are associated with flowering in response to cold. This process can be altered by maneuvering the levels of miR156, which is the main controller of time-dependent flowering in response to vernalization (Bergonzi et al. 2013). The influence of miR156 on vegetative phase change may be altered by its abundance. miR156 levels augment by reduced photosynthesis and decrease by exogenous sugar exposition. The outcome of these changes in miR156 loads is attributable primarily to the sensitive MIR156a and MIR156c genes (Yang et al. 2013). In the unicellular moss Physcomitrella patens, miR156 also influences phase change in an opposite direction to that in flowering plants, by promoting a developmental switch from young filamentous protonemata to leafy gametophores (Cho et al. 2012).
Another miRNA related to development is the conserved miR408, targeting copper protein encoding genes. miR408 is required for vegetative development in Arabidopsis and is altered by several environmental conditions, including copper deficiency (Cd). During Cd, SPL7 binds and induces the MIR408 promoter. The constitutive expression of miR408 increases growth of A. thaliana seedlings (Zhang and Li 2013). Mobile miRNAs have also been described as modulators of plant development. Arabidopsis miR394 inhibits F box protein Leaf Curling Responsiveness (LCR). MiR394 is transcribed at the protoderm and exerts its action in the distal meristem, potentiating signaling from underneath the stem cells by the TF Wuschel and thus maintaining stem cell pluripotency. This interaction restricts the dynamic meristem area, giving the surface layer area a role as a stable reference point of meristem development (Knauer et al. 2013).
MiRNAs and Abiotic Stress
Drought stress alters a wide range of miRNAs. For instance, miR474 has been shown to be sensitive to regulation of osmolyte production by proline dehydrogenase inhibition. MiR528 and miR398 fine-tune antioxidant production by repressing peroxidase and copper-zinc superoxide dismutases (CSD), respectively. MiR159, miR169, miR160 and miR167 calibrate ABA response by MYB, a subunit of the nuclear factor Y (NFYA) and auxin response factors (ARFs) TFs regulation. In addition, miR160, miR390 and miR393 mediate auxin signaling and cross talk by repressing ARF and transport inhibitor response TFs (reviewed in Ding et al. 2013).
Metal toxicity modifies several miRNAs. After cadmiun exposure, a downregulation of miR160 and miR164b was observed (targeting ARFS and NAC domain containing proteins such as cup-shaped cotyledon CUC, respectively), as well as an upregulation of miR393 targeting F-box proteins and bHLH TFs. This latter response of miR393 has also been described recently in radish (Raphanus sativus L.), suggesting that the outcome to cadmiun exposure might be conserved during evolution (Xu et al. 2013). During manganese toxicity, Arabidopsis miR166, miR319, miR393 and miR398 levels are altered. Increased expression of miR408, miR528 and miR397b, and a downregulation of miR1318 have been observed in Arabidopsis plants exposed to aluminum stress (reviewed in Gupta et al. 2014). During N starvation, an increment in the expression of miR160 miR780, miR826, miR842, and miR846 and a repression of miR169, miR171, miR395, miR397, miR398, miR399, miR408, miR827, and miR857 have been detected in Arabidopsis (Liang et al. 2012). This response does not appear to be conserved among plants. In maize, miR160 is repressed during N starvation (Zhao et al. 2012a).
Furthermore, miRNAs may be regulated by heat stress. Ath-miR400 is an intronic miRNA from the At1g32583 gene transcriptional unit. In Arabidopsis plants, heat exposure induces the intron where miR400 is located, and an alternative splicing event excises a portion of pri-miR400, resulting in accumulation of precursor transcripts and a decrease of the mature miR400 forms (Yan et al. 2012b). MiR400 gene targets are members of the pentatricopeptide repeat-containing proteins (PPR). Although this family has been studied extensively, the specific impacts of miR400 abundance on its gene targets under heat stress have not been explored. A rapid induction of miR398 levels has also been observed under heat, concomitant with inhibition of mRNA abundance of its target genes CSD 1 and 2 and a copper chaperone for the CSDs (CCS). This modulation is orchestrated under a regulatory circuit derived from the inhibition of miR398 levels under oxidative stress, preventing the impairment of CSD2 inhibition to cope with ROS species (Guan et al. 2013). However, an over-accumulation of this miRNA under oxidative stress was observed during late virus infection (Manacorda et al. 2013), indicating that the regulation of miR398 levels is not restricted only to ROS detection, and is more complex than expected.
In addition, plant miRNAs response to cold stress has been described for a thermosensitive male-sterile wheat line (Tang et al. 2012), Chinese white poplar Populus tomentosa (Chen et al. 2012b) and in an inter-fertile relative of citrus trifoliate orange Poncirus trifoliata known to be freezing-tolerant (Zhang et al. 2014). Several miRNAs were found to be altered differently during cold conditions, suggesting plant-specific pathways to co-opt this stress response. However, miR167, controlling ARF TFs, and miR396, regulating GRF TFs, both related to developmental process, were commonly induced as a response to cold exposure in these unrelated plants.
Finally, in Arabidopsis seedlings, growth arrest is a typical osmotic stress aftermath, as a strategy to survive this disturbing condition. MiR172b actively represses the AP2-like gene Schnarchzapfen (SNZ). This gene controls the ABI3 (B3 type)/ABI5 (bZIP type) TFs pathway, which switches plant metabolism leading from the heterotrophic to an autotrophic state. During osmotic stress, ABA signaling represses miR172b, compromising SNZ regulation of cotyledon greening during seedling growth (Zou et al. 2013).
MiRNAs and Biotic Stress
Not long ago, the modification of several miRNAs during viral infection was reported for the first time. This alteration during Tobamoviridae, Potyviridae, and Potexviridae infection was correlated to viral symptoms and a developmental shift (Bazzini et al. 2007). There is increasing evidence suggesting that this phenomenon might be a general response of the host plant to virus infection. Whether this alteration is a side-effect of the host miRNA pathway saturation by viral transcripts or a coordinated response of the plant to co-opt with viruses is under debate (reviewed in Zvereva and Poogin 2012; Balmer and Mauch-Mani 2013). Nevertheless, the interaction between viruses and the RNAi pathway is far more complex and multidimensional. It has been proposed that, although the overlapping responses against viruses might be studied and analyzed in detail in the short-term, in the long-term there is a need to fit together the various responses in order to generate a unified theory of plant–virus interactions (Palukaitis et al. 2013).
It is widely known that RNA silencing protects plants against most viruses and that successful infection depends on suppression and evasion of antiviral silencing, based mostly on the action of viral silencing suppressors (VSR) as a mechanism to hijack the host defense response (reviewed in Pumplin and Voinnet 2013). VSR are structurally and functionally diverse. Several VSR have been described to inhibit or alter the miRNA pathway (Chapman et al. 2004). For instance, the tombusvirus P19 protein is able to bind and sequester most 21 nt sRNA species, preventing them from being incorporated into the silencing pathway. Strikingly, miR168 does not bind efficiently to p19, and tombusviruses induce MIR168 expression. This miRNA is loaded onto AGO1, targeting AGO1 mRNA precisely in a feedback loop. As a net result, AGO1 levels are strongly reduced, therefore inhibiting the miRNA pathway (Varallyay et al. 2010; Varallyay and Havelda 2013).
A different type of interaction has been elucidatedrecently in relation to virus infection, involving the modulation of plant immune receptors through miRNAs. nta-miR6019 and nta-miR6020 target and inhibit the Toll and Interleukin-1 immune receptor NB-LRR (N) from tobacco that confers resistance to tobacco mosaic virus (TMV). The cleavage of N by nta-miR6019 and nta-miR6020 triggers the RdRD6- and DCL4-dependent generation of multiple secondary siRNAs, suggesting that, in turn, several related immune receptors might be regulated in a coordinated response. Nta-miR6019 and nta-miR6020 are repressed and N is induced during pathogen response, indicating that this mechanism controls and restricts costly production of immune receptors only when required by a pathogen-induced interaction (Li et al. 2012a). This mechanism was shown to be conserved in tomato, in which miR482 and miR2118 target cleavage of mRNA sequences also for NB-LRRs disease resistance proteins with coiled-coil domains at their N terminus (Shivaprasad et al. 2012). In addition, a miRNA identified from apple, Md-miRLn11, targets an NB-LRRs coding gene (Md-NBS), in particular during pathogen infection (Ma et al. 2014). Moreover, in a new study describing the sRNA profile of spruce, grape and poplar, a large fraction of 21 nt siRNAs was originated from NBS-LRR receptors, further suggesting to be originated by phased processing of miRNA cleaved NBS-LRR transcripts (Källman et al. 2013). In rice, miR7695 is regulated by elicitors from the blast fungus Magnaporthe oryzae and inhibits an alternatively spliced transcript, Natural resistance-associated macrophage protein 6 (OsNramp6). Over-expression of miR7695 results in resistance to the blast fungus (Campo et al. 2013). miRNAs have also been involved in defense pathways against insect pests. Myzus persicae produces less progeny in Arabidopsis plants deficient in the miRNA pathway. This miRNA compromised plants, when exposed to aphid infestation, increasing production of camalexin and therefore inhibiting the production of aphid progeny (Kettles et al. 2013). Finally, there is evidence that miRNAs might be involved in soybean nodulation. Over-expression of gma-miR172 increases the expression of symbiotic leghemoglobin and non-symbiotic hemoglobin and boosts nodule number in a complex regulatory circuit, linking miR156 regulation of miR172 expression and the level of AP2 TF (Yan et al. 2013).
MiRNAs and Phased Small RNAs
The nature of the mechanism encompassing the production of secondary phased siRNAs (phasiRNA) derived from miRNA-guided cleavage of intermediary transcripts has been studied extensively in recent years. PhasiRNAs are triggered by 22 nt long miRNAs (Cuperus et al. 2010; Chen et al. 2010), or by the presence of asymmetrically positioned bulged bases in the miRNA:miRNA* duplex. PhasiRNA production is programmed during the early steps of miRNA loading onto AGO1, before disassembly of the RNA duplex and selection of the miRNA guide strand (Manavella et al. 2012b). A subset of this secondary siRNAs, denominated tasiRNAs, are loaded onto AGO effectors and direct the cleavage in trans of mRNA targets with complementary sequences (Peragine et al. 2005; Vazquez et al. 2004; Allen et al. 2005).
The presence of phasiRNA generating miRNAs is highly dynamic and widespread in eudicots. A new report infers that these types of miRNAs might have originated by diversification of a widely conserved ancient miR390. miR390-dependent phasiRNA production does not rely on length or duplex asymmetry, but on a “two-hit trigger” strategy, resulting in secondary siRNAs arising from regions flanked by dual miR390 target sites (Axtell et al 2006). miR390 would have originated the miR7122 super family, which has pronounced identity with miR173, the first described 22 nt miRNA that generates phasiRNAs derived from PPR target genes. This scenario is supported by the fact that even though the diverse miRNA target transcripts present high sequence divergence among eudicots, the MIR173-like genes, such as miR7122, miR1509, and fve-PPRtri1/2, show pronounced identity (Xia et al. 2013). The evolutionary pathway of miRNAs and phasiRNAs presents intriguing aspects of convergence and divergence in the plant kingdom. As an example, a unique role of the ancient miR156 has been observed in Physcomitrella patens but not in flowering plants. In the moss, miR156 induces the accumulation of particular phasiRNAs reliant on the conserved miR390, by targeting a phasiRNA primary transcript (Cho et al. 2012).
Target Mimicry
Since the discovery of miRNAs, due to its active induction of target inhibition, it has been widely postulated that miRNAs are indeed regulators of messenger RNAs. An opposite hypothesis postulating that miRNA targets regulate miRNA availability, termed “target mimicry”, is supported strongly by a class of long non-coding RNAs discovered in Arabidopsis. Induced by Phosphate Starvation 1 (IPS1) is targeted by ath-miR399, the miRNA-target pair presents a site that contains a central bulge protecting IPS1 from cleavage. IPS1 thereby reduces the amount of miR399 available to repress its target mRNA PHOSPHATE 2—an E2 ubiquitin conjugase-related protein that negatively affects shoot Pi content and Pi remobilization (Franco-Zorrilla et al. 2007). In recent years this hypothesis has been expanded to animals and humans, holding that target mRNAs are not only mere inert substrates of miRNA action, but dynamic regulators of miRNA availability, and proposing mRNAs, transcribed pseudo-genes, and long non-coding RNAs as competing endogenous RNA(ceRNA) communicating to each other using microRNA as response elements (Salmena et al. 2011). This inverse rationale offers a novel mode of miRNA regulation and a redefinition of the rules governing miRNA biology, reinstalling mRNAs at the central stage of regulatory networks (Cesana and Daley 2013). It is worth noting that the intrinsic concept at the core of this expanded hypothesis was anticipated in the target mimicry principle (Rubio-Somoza et al. 2011).
Target mimicry (Fig. 3) as a natural phenomenon, has been extensively validated by a novel study reporting the existence of several widespread long non-coding RNAs that are target mimics in plants. Twenty conserved endogenous miRNA target mimics (eTMs) were found in Arabidopsis and rice, with marked sequence conservation along the target binding site. Among them, two eTMs of miR160 and miR166 were proven to be functional target mimics regulating plant development (Wu et al. 2013a). Target mimic has been exploited for the generation of knock-down libraries of Arabidopsis miRNAs, by emulating natural long-noncoding RNAs such as IPS1. Strong morphological defects were observed when several conserved miRNA families were inhibited. Milder effects observed when young miRNAs were inhibited suggest that they do not influence basic developmental processes (Todesco et al. 2010). An improvement of this technology comprises the expression of a short tandem target mimic (STTM), composed of two miRNA target sites with a CTA tri-nucleotide bulge corresponding to positions 10 and 11 from the 5′ mature miRNAs to avoid cleavage, and separated by an optimal 48–88 nt size spacer. This STTM results in an increased inhibition of miRNAs (Yan et al. 2012a).

Engineering of plant miRNAs. a Natural miRNAs (e.g. miRX) originate by processing of a gene precursor miRNA (pre-miRX), resulting in a duplex RNA (miRX/miRX*) that will be incorporated into AGO1 and induce cleavage of complementary targets (mRNA-X). b Artificial miRNAs (e.g., amiRX) consist of modified natural miRNA precursors (pre-amiRX) that redirect miRNA silencing activity to newly defined targets (mRNA-Y). c Over-expression of natural miRNAs is based on the transformation of plants with strong promoters directing transcription of miRNA units (e.g., 35S:pre-miRX), resulting in over-accumulation of miRNAs and depletion of target mRNA. d Target mimicry consists of the expression of artificial targets of natural miRNAs (mim-X) that sequester miRNAs and prevent natural targets (mRNA-X) from degradation
Engineering miRNAs
Different molecular engineering approaches have been developed to exploit plant miRNA biology. Artificial miRNAs (amiRNAs), in which plant pri-miRNAs are modified at the miRNA/miRNA* duplex to re-direct their action to silencing of newly defined target genes (Fig. 3b; Schwab et al. 2005), are perhaps the most representative case. One of the first reported uses of amiRNAs was the generation of virus-resistant transgenic plants targeting viral silencing suppressors (Niu et al. 2006). A concern with this strategy related to virus escape to amiRNAs during mixed infections was raised recently. In amiRNA-mediated turnip mosaic virus (TuMV) resistant plants, pre-infection with another virus has been found to jeopardize the resistance phenotype to TuMV (Martínez et al. 2013). To circumvent this problem, a design solution that improves resistance by combining several amiRNAs targeting highly conserved viral genomic regions to successfully maintain the resistance phenotype has been suggested (Lafforgue et al. 2013). Recently, for research purposes, a genome-wide family-specific amiRNA library was generated, encompassing 22,000 amiRNAs targeting defined functional protein classes for genetic screens of the functionality of the complete gene landscape in Arabidopsis (Hauser et al. 2013). To improve the screening of amiRNA target inhibition, an epitope-tagged protein-based amiRNA platform (ETPamir) has been developed. When ETPamir are co-expressed in protoplasts with amiRNA candidates, it permits the parallel quantification of target genes and proteins as a measure of amiRNA efficacy (Li et al. 2013a).
A different strategy has been deployed by exploiting the generation of phasiRNAs of target genes by 22 nt miRNA-guided cleavage of gene target transcripts fused to an upstream miR173 target site. This target site is sufficient to efficiently trigger silencing of endogenous genes; it may be used to target multiple genes and can be applied to other plant species besides Arabidopsis by co-expressing ath-miR173 (de Felippes et al. 2012). A version of this strategy was evaluated and compared to different silencing approaches based on expression of hairpin RNA constructs (hpRNA) and regular 21 nt amiRNAs. The 22 nt phasiRNA-triggering miRNAs were successful in silencing every tissue, including roots and seed coats—two regions where silencing efficiency of regular amiRNAs and hpRNA is compromised (McHale et al. 2013).
First used to understand miRNA function, over-expression of natural miRNAs (Fig. 3c) has now been re-oriented to the generation of biotechnologically relevant traits in plants. In switchgrass (Panicum virgatum L.)—a perennial warm season bunchgrass used primarily for soil conservation and forage production, over-expression of miR156 and a strong silencing of its SPL gene targets result in several morphological alterations leading to enhanced biomass production (Fu et al. 2012). The popular ornamental tuberous gloxinia (Sinningia speciosa) has been engineered to over-express miR159, which resulted in a delayed flowering state concomitant with a repression of its MYB TF gene target (SsGAMYB). When miR159 was inhibited by means of target mimicry, gloxinia plants presented an early flowering phenotype (Li et al. 2013c). Over-expression of osa-miR397 in rice induced a substantial depletion of its laccase-like gene target (OsLAC) involved in sensitivity to brassinosteroids. Rice over-expresser lines increased grain size and promoted panicle branching, resulting in improved yield (Zhang et al. 2013b). Despite the lack of reports related to miRNA engineering to reduce the amount of N-based fertilizer, it has been suggested that such reduction could be attained by expression of MIRNA genes upregulated during N starvation, such as miR156, along with inhibition of repressed miRNAs amid N starvation, such as miR827. This might reduce production costs and environmental damages (Fischer et al. 2013). Manipulation of miR160 can alter nodule primordium formation in soybean. When miR160 is over-expressed in soy, silencing of ARF TFs, hyposensitivity to cytokinin, and hypersensitivity to auxin in roots are observed, which reduces nodule development (Turner et al. 2013).
Concluding Remarks and Future Challenges
In the miRNA biogenesis sphere, several important new players, such as CPL1, THG, MOS2 and STA1, have been discovered in recent years. In addition, the elucidation of a much more complex than previously thought panorama in the processing pathway of pri-miRNAs in the elegant report of Bologna et al. (2013) is worth mentioning.
One notable future challenge is the elucidation of the miRNA degradation process during biotic or abiotic stress. Even though SDNs were described in Ramachandran and Chen (2008), there is not a single report detailing SDN1 steady-state levels or alterations during plant development, pathogen interaction or environmental stress. It is also not clear if any protein member of the exosome contributes directly to miRNA degradation. It is interesting to point out that we know a lot more about miRNA biogenesis than about turnover.
Regarding miRNA target acquisition, there is an important question that has not yet been conclusively addressed. In animals, miRNAs regulate gene targets by “seed” binding, which targets only the first 8 nt of the mature miRNA. Thus, the typical interaction in animal miRNAs and its numerous targets results in a mild modulation of multiple mRNAs. In plants, most of the experimentally validated targets share extended sequence complementary to the miRNA (Jones-Rhoades and Bartel 2004; Schwab et al. 2005). Thus, in general, plant miRNAs regulate only a few targets via a strong interaction (Ding et al. 2012). The extended complementarities and typical straightforward validation by mRNA accumulation may have biased the identification of more subtle modulated targets. In a recent report, this animal-like interaction has been analyzed by a transient sensor system at both the RNA and protein levels, and has been ruled out for several conserved miRNAs in plants (Liu et al. 2014). Nevertheless, a new study of the same group describes the discovery and validation of a non-canonical target harboring a 6 nt bulge at the 5′ complementary region of the miRNA (Brousse et al. 2014). This completely unconventional and unexpected target highlights that it is not yet definitely shown if new types of interaction between miRNAs and targets are possible in plants, and, more importantly, if they could be biologically relevant.
Every biotechnology breakthrough derived from miRNA engineering will be subjected to restrictions before an important question is answered: what are the biosafety and environmental risks associated with the use of small RNAs for plant genetic improvement? (reviewed in Auer and Frederick 2009; Auer 2011). This concern has deepened since the publication of an article describing a rice miRNA in animal samples and the possibility of cross-kingdom inhibition of mammalian targets (Zhang et al. 2011), although the implications of this report have been diminished in recent years by the identification of some technical concerns in the report and the difficulties in replicating the results by an independent group (corrigendum to Zhang et al. 2011: Dickinson et al. 2013; Chen et al 2013).
Overall, the expansion of knowledge in the miRNA field is overwhelming. While miRNA incumbency was first believed to be exceptional and anecdotic, it turned out to be central to plant biology, encompassing and modulating every aspect of plant life investigated thus far.
References
Allen E, Xie Z, Gustafson AM, Carrington JC (2005) microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121:207–221
Auer C (2011) Small RNAs for crop improvement: applications and considerations for ecological risk assessments. In: Erdmann VA, Barciszewski J (eds) Non-coding RNAs in plants. Springer, Berlin, pp 461–484
Auer C, Frederick R (2009) Crop improvement using small RNAs: applications and predictive ecological risk assessments. Trends Biotechnol 27:644–651
Axtell MJ, Bowman JL (2008) Evolution of plant microRNAs and their targets. Trends Plant Sci 13:343–349
Axtell MJ, Jan C, Rajagopalan R, Bartel DP (2006) A two-hit trigger for siRNA biogenesis in plants. Cell 127:565–577
Balmer D, Mauch-Mani B (2013) Small yet mighty microRNAs in plant-microbe interactions. MicroRNA 2(1):73–80
Bazin J, Khan GA, Combier J-P et al (2013) miR396 affects mycorrhization and root meristem activity in the legume Medicago truncatula. Plant J 74:920–934
Bazzini AA, Hopp HE, Beachy RN, Asurmendi S (2007) Infection and coaccumulation of tobacco mosaic virus proteins alter microRNA levels, correlating with symptom and plant development. Proc Natl Acad Sci USA 104:12157–12162
Bergonzi S, Albani MC, Loren V, van Themaat E et al (2013) Mechanisms of age-dependent response to winter temperature in perennial flowering of Arabis alpina. Science 340:1094–1097
Bologna NG, Schapire AL, Zhai J et al (2013) Multiple RNA recognition patterns during microRNA biogenesis in plants. Genome Res 23:1675–1689
Brodersen P, Sakvarelidze-Achard L, Bruun- M (2008) Widespread translational inhibition by plant miRNAs and siRNAs. Science 320:1185–1190
Brousse C, Liu Q, Beauclair L, et al. (2014) A non-canonical plant microRNA target site. Nucleic Acids Res. doi: 10.1093/nar/gku157
Burko Y, Shleizer-Burko S, Yanai O et al (2013) A role for APETALA1/fruitfull transcription factors in tomato leaf development. Plant Cell 25:2070–2083
Campo S, Peris-Peris C, Siré C et al (2013) Identification of a novel microRNA (miRNA) from rice that targets an alternatively spliced transcript of the Nramp6 (Natural resistance-associated macrophage protein 6) gene involved in pathogen resistance. New Phytol 199:212–227
Carbonell A, Fahlgren N, Garcia-Ruiz H et al (2012) Functional analysis of three Arabidopsis ARGONAUTES using slicer-defective mutants. Plant Cell 24:3613–3629
Cesana M, Daley GQ (2013) Deciphering the rules of ceRNA networks. Proc Natl Acad Sci USA 110:7112–7113
Chaabane S, Liu R, Chinnusamy V et al (2013) STA1, an Arabidopsis pre-mRNA processing factor 6 homolog, is a new player involved in miRNA biogenesis. Nucleic Acids Res 41:1984–1997
Chapman EJ, Prokhnevsky AI, Gopinath K et al (2004) Viral RNA silencing suppressors inhibit the microRNA pathway at an intermediate step. Genes Dev 18:1179–1186
Chen X (2012) Small RNAs in development—insights from plants. Curr Opin Genet Dev 22:361–7
Chen H-M, Chen L-T, Patel K et al (2010) 22-nucleotide RNAs trigger secondary siRNA biogenesis in plants. Proc Natl Acad Sci USA 107:15269–15274
Chen L, Zhang Y, Ren Y et al (2012) Genome-wide identification of cold-responsive and new microRNAs in Populus tomentosa by high-throughput sequencing. Biochem Biophys Res Commun 417:892–896
Chen X, Zen K, Zhang C-Y (2013) Reply to Lack of detectable oral bioavailability of plant microRNAs after feeding in mice. Nat Biotechnol 31:967–969
Cho SH, Coruh C, Axtell MJ (2012) miR156 and miR390 regulate tasiRNA accumulation and developmental timing in Physcomitrella patens. Plant Cell 24:4837–4849
Cloonan N, Wani S, Xu Q et al (2011) MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol 12:R126
Cuperus J, Carbonell A, Fahlgren NH (2010) Unique functionality of 22-nt miRNAs in triggering RDR6-dependent siRNA biogenesis from target transcripts in Arabidopsis. Nat Struct Mol Biol 17:997–1003
De Felippes FF, Wang J, Weigel D (2012) MIGS: miRNA-induced gene silencing. Plant J 70:541–547
De Oliveira LFV, Christoff AP, Margis R (2013) isomiRID: a framework to identify microRNA isoforms. Bioinformatics 29:2521–2523
Debernardi JM, Rodriguez RE, Mecchia MA, Palatnik JF (2012) Functional specialization of the plant miR396 regulatory network through distinct microRNA-target interactions. PLoS Genet 8:e1002419
Dickinson B, Zhang Y, Petrick JS et al (2013) Lack of detectable oral bioavailability of plant microRNAs after feeding in mice. Nat Biotechnol 31:965–967
Ding J, Li D, Ohler U et al (2012) Genome-wide search for miRNA-target interactions in Arabidopsis thaliana with an integrated approach. BMC Genomics 13(Suppl 3):S3
Ding Y, Tao Y, Zhu C (2013) Emerging roles of microRNAs in the mediation of drought stress response in plants. J Exp Bot 64:3077–3086
Fischer JJ, Beatty PH, Good AG, Muench DG (2013) Manipulation of microRNA expression to improve nitrogen use efficiency. Plant Sci 210:70–81
Franco-zorrilla M, Puga I, Todesco M et al (2007) Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet 39:1033–1037
Fu C, Sunkar R, Zhou C et al (2012) Overexpression of miR156 in switchgrass (Panicum virgatum L.) results in various morphological alterations and leads to improved biomass production. Plant Biotechnol J 10:443–452
Giacomelli JI, Weigel D, Chan RL, Manavella PA (2012) Role of recently evolved miRNA regulation of sunflower HaWRKY6 in response to temperature damage. New Phytol 195:766–773
Guan Q, Lu X, Zeng H et al (2013) Heat stress induction of miR398 triggers a regulatory loop that is critical for thermotolerance in Arabidopsis. Plant J 74:840–851
Gupta OP, Sharma P, Gupta RK, Sharma I (2014) MicroRNA mediated regulation of metal toxicity in plants: present status and future perspectives. Plant Mol Biol 84:1–18
Hajheidari M, Koncz C, Eick D (2013) Emerging roles for RNA polymerase II CTD in Arabidopsis. Trends Plant Sci 18:633–643
Hauser F, Chen W, Deinlein U et al (2013) A genomic-scale artificial microRNA library as a tool to investigate the functionally redundant gene space in Arabidopsis. Plant Cell 25:2848–2863
Ibrahim F, Rymarquis LA, Kim E-J et al (2010) Uridylation of mature miRNAs and siRNAs by the MUT68 nucleotidyltransferase promotes their degradation in Chlamydomonas. Proc Natl Acad Sci USA 107:3906–3911
Iwata Y, Takahashi M, Fedoroff NV, Hamdan SM (2013) Dissecting the interactions of SERRATE with RNA and DICER-LIKE 1 in Arabidopsis microRNA precursor processing. Nucleic Acids Res 41:9129–9140
Jeong D-H, Park S, Zhai J et al (2011) Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 23:4185–4207
Jeong D-H, Thatcher SR, Brown RSH et al (2013) Comprehensive investigation of microRNAs enhanced by analysis of sequence variants, expression patterns, ARGONAUTE loading, and target cleavage. Plant Physiol 162:1225–1245
Jia F, Rock CD (2013) MIR846 and MIR842 comprise a cistronic MIRNA pair that is regulated by abscisic acid by alternative splicing in roots of Arabidopsis. Plant Mol Biol 81:447–460
Jin D, Wang Y, Zhao Y, Chen M (2013) MicroRNAs and their cross-talks in plant development. J Genet Genomics 40:161–70
Jones-Rhoades MW, Bartel DP (2004) Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell 14:787–799
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol 57:19–53
Källman T, Chen J, Gyllenstrand N, Lagercrantz U (2013) A significant fraction of 21-nucleotide small RNA originates from phased degradation of resistance genes in several perennial species. Plant Physiol 162:741–754
Kettles GJ, Drurey C, Schoonbeek H et al (2013) Resistance of Arabidopsis thaliana to the green peach aphid, Myzus persicae, involves camalexin and is regulated by microRNAs. New Phytol 198:1178–1190
Knauer S, Holt AL, Rubio-Somoza I et al (2013) A protodermal miR394 signal defines a region of stem cell competence in the Arabidopsis shoot meristem. Dev Cell 24:125–132
Lafforgue G, Martínez F, Niu Q-W et al (2013) Improving the effectiveness of artificial microRNA (amiR)-mediated resistance against Turnip mosaic virus by combining two amiRs or by targeting highly conserved viral genomic regions. J Virol 87:8254–8256
Li F, Pignatta D, Bendix C et al (2012a) MicroRNA regulation of plant innate immune receptors. Proc Natl Acad Sci USA 109:1790–1795
Li S, Yang X, Wu F, He Y (2012b) HYL1 controls the miR156-mediated juvenile phase of vegetative growth. J Exp Bot 63:2787–98
Li J-F, Chung HS, Niu Y et al (2013a) Comprehensive protein-based artificial microRNA screens for effective gene silencing in plants. Plant Cell 25:1507–1522
Li S, Liu L, Zhuang X et al (2013b) MicroRNAs inhibit the translation of target mRNAs on the endoplasmic reticulum in Arabidopsis. Cell 153:562–574
Li X, Bian H, Song D et al (2013c) Flowering time control in ornamental gloxinia (Sinningia speciosa) by manipulation of miR159 expression. Ann Bot 111:791–799
Liang G, He H, Yu D (2012) Identification of nitrogen starvation-responsive microRNAs in Arabidopsis thaliana. PLoS One 7:e48951
Liu C, Axtell MJ, Fedoroff NV (2012) The helicase and RNaseIIIa domains of Arabidopsis Dicer-Like1 modulate catalytic parameters during microRNA biogenesis. Plant Physiol 159:748–758
Liu Q, Yan Q, Liu Y et al (2013) Complementation of HYPONASTIC LEAVES1 by double-strand RNA-binding domains of DICER-LIKE1 in nuclear dicing bodies. Plant Physiol 163:108–117
Liu Q, Wang F, Axtell MJ (2014) Analysis of complementarity requirements for plant microRNA targeting using a Nicotiana benthamiana quantitative transient assay. Plant Cell. doi: 10.1105/tpc.113.120972
Lu S, Sun Y, Chiang VL (2009) Adenylation of plant miRNAs. Nucleic Acids Res 37:1878–1885
Ma C, Lu Y, Bai S et al (2014) Cloning and characterization of miRNAs and their targets, including a novel miRNA-targeted NBS-LRR protein class gene in apple (Golden Delicious). Mol Plant 7:218–230
Manacorda CA, Mansilla C, Debat HJ et al (2013) Salicylic acid determines differential senescence produced by two Turnip mosaic virus strains involving reactive oxygen species and early transcriptomic changes. Mol Plant Microbe Interact 26:1486–1498
Manavella PA, Hagmann J, Ott F et al (2012a) Fast-forward genetics identifies plant CPL phosphatases as regulators of miRNA processing factor HYL1. Cell 151:859–870
Manavella PA, Koenig D, Weigel D (2012b) Plant secondary siRNA production determined by microRNA-duplex structure. Proc Natl Acad Sci USA 109:2461–2466
Manavella PA, Koenig D, Rubio-Somoza I et al (2013) Tissue-specific silencing of Arabidopsis SU(VAR)3-9 HOMOLOG8 by miR171a. Plant Physiol 161:805–812
Martínez F, Elena SF, Daròs J-A (2013) Fate of artificial microRNA-mediated resistance to plant viruses in mixed infections. Phytopathology 103:870–876
McHale M, Eamens AL, Finnegan EJ, Waterhouse PM (2013) A 22-nt artificial microRNA mediates widespread RNA silencing in Arabidopsis. Plant J 76:519–529
Mi S, Cai T, Hu Y et al (2008) Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 133:116–127
Molnár A, Schwach F, Studholme DJ et al (2007) miRNAs control gene expression in the single-cell alga Chlamydomonas reinhardtii. Nature 447:1126–1129
Neilsen CT, Goodall GJ, Bracken CP (2012) IsomiRs–the overlooked repertoire in the dynamic microRNAome. Trends Genet 28:544–549
Niu Q-W, Lin S-S, Reyes JL et al (2006) Expression of artificial microRNAs in transgenic Arabidopsis thaliana confers virus resistance. Nat Biotechnol 24:1420–1428
Palatnik JF, Allen E, Wu X, et al. (2003) Control of leaf morphogenesis by microRNAs. Nature 425:257–263
Palukaitis P, Groen SC, Carr JP (2013) The Rumsfeld paradox: some of the things we know that we don’t know about plant virus infection. Curr Opin Plant Biol 16:513–519
Park W, Li J, Song R et al (2002) CARPEL FACTORY, a dicer homolog, and HEN1, a novel protein, Act in microRNA metabolism in Arabidopsis thaliana. Curr Biol 12:1484–1495
Peragine A, Yoshikawa M, Park MY, Poethig RS (2005) A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis. A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis. Genes Dev 19:2164–2175
Pumplin N, Voinnet O (2013) RNA silencing suppression by plant pathogens: defence, counter-defence and counter-counter-defence. Nat Rev Microbiol 11:745–760
Rajagopalan R, Vaucheret H, Trejo J, Bartel D (2006) A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev 20:3407–3425
Ramachandran V, Chen X (2008) Degradation of microRNAs by a family of exoribonucleases in Arabidopsis. Science 321:490–1492
Ren G, Chen X, Yu B (2012a) Uridylation of miRNAs by hen1 suppressor1 in Arabidopsis. Curr Biol 22:695–700
Ren G, Xie M, Dou Y et al (2012b) Regulation of miRNA abundance by RNA binding protein TOUGH in Arabidopsis. Proc Natl Acad Sci USA 109:12817–12821
Rhoades MW, Reinhart BJ, Lim LP et al (2002) Prediction of Plant MicroRNA Targets. Cell 110:513–520
Rodrigo G, Elena SF (2013) MicroRNA precursors are not structurally robust but plastic. Genome Biol Evol 5:181–186
Rogers K, Chen X (2012) microRNA biogenesis and turnover in plants. Cold Spring Harb Symp Quant Biol 77:183–194
Rubio-Somoza I, Weigel D (2011) MicroRNA networks and developmental plasticity in plants. Trends Plant Sci 16:258–264
Rubio-Somoza I, Weigel D (2013) Coordination of flower maturation by a regulatory circuit of three microRNAs. PLoS Genet 9:e1003374
Rubio-Somoza I, Weigel D, Franco-Zorilla J-M et al (2011) ceRNAs: miRNA target mimic mimics. Cell 147:1431–1432
Sablok G, Milev I, Minkov G et al (2013) isomiRex: web-based identification of microRNAs, isomiR variations and differential expression using next-generation sequencing datasets. FEBS Lett 587:2629–2634
Salmena L, Poliseno L, Tay Y et al (2011) A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146:353–358
Schwab R, Palatnik JF, Riester M et al (2005) Specific effects of microRNAs on the plant transcriptome. Dev Cell 8:517–527
Shivaprasad PV, Chen H-M, Patel K et al (2012) A microRNA superfamily regulates nucleotide binding site-leucine-rich repeats and other mRNAs. Plant Cell 24:859–874
Speth C, Willing E-M, Rausch S et al (2013) RACK1 scaffold proteins influence miRNA abundance in Arabidopsis. Plant J 76:433–445
Tang Z, Zhang L, Xu C et al (2012) Uncovering small RNA-mediated responses to cold stress in a wheat thermosensitive genic male-sterile line by deep sequencing. Plant Physiol 159:721–738
Thieme CJ, Schudoma C, May P, Walther D (2012) Give it AGO: The search for miRNA-Argonaute sorting signals in Arabidopsis thaliana indicates a relevance of sequence positions other than the 5′-position alone. Front Plant Sci 3:272
Todesco M, Rubio-Somoza I, Paz-Ares J, Weigel D (2010) A collection of target mimics for comprehensive analysis of microRNA function in Arabidopsis thaliana. PLoS Genet 6:e1001031
Todesco M, Balasubramanian S, Cao J et al (2012) Natural variation in biogenesis efficiency of individual Arabidopsis thaliana microRNAs. Curr Biol 22:166–170
Turner M, Nizampatnam NR, Baron M et al (2013) Ectopic expression of miR160 results in auxin hypersensitivity, cytokinin hyposensitivity, and inhibition of symbiotic nodule development in soybean. Plant Physiol 162:2042–2055
Varallyay E, Havelda Z (2013) Unrelated viral suppressors of RNA silencing mediate the control of ARGONAUTE1 level. Mol Plant Pathol 14:567–575
Varallyay E, Valoczi A, Agyi A et al (2010) Plant virus-mediated induction of miR168 is associated with repression of ARGONAUTE1 accumulation. EMBO J 29:3507–3519
Vazquez F, Vaucheret H, Rajagopalan R et al (2004) Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol Cell 16:69–79
Wang J, Czech B, Weigel D (2009) miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 138:738–749
Wang L, Song X, Gu L et al (2013) NOT2 proteins promote polymerase II-dependent transcription and interact with multiple MicroRNA biogenesis factors in Arabidopsis. Plant Cell 25:715–727
Wu G, Park MY, Conway SR et al (2009) The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 138:750–759
Wu H-J, Wang Z-M, Wang M, Wang X-J (2013a) Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol 161:1875–1884
Wu X, Shi Y, Li J et al (2013b) A role for the RNA-binding protein MOS2 in microRNA maturation in Arabidopsis. Cell Res 23:645–657
Xia R, Meyers BC, Liu Z et al (2013) MicroRNA superfamilies descended from miR390 and their roles in secondary small interfering RNA Biogenesis in Eudicots. Plant Cell 25:1555–1572
Xie Z, Allen E, Fahlgren N et al (2005) Expression of Arabidopsis MIRNA genes. Plant Physiol 138:2145–2154
Xing S, Salinas M, Garcia-Molina A et al (2013) SPL8 and miR156-targeted SPL genes redundantly regulate Arabidopsis gynoecium differential patterning. Plant J 75:566–577
Xu L, Wang Y, Zhai L et al (2013) Genome-wide identification and characterization of cadmium-responsive microRNAs and their target genes in radish (Raphanus sativus L.) roots. J Exp Bot 64:4271–4287
Yan J, Gu Y, Jia X et al (2012a) Effective small RNA destruction by the expression of a short tandem target mimic in Arabidopsis. Plant Cell 24:415–427
Yan K, Liu P, Wu C-A et al (2012b) Stress-induced alternative splicing provides a mechanism for the regulation of microRNA processing in Arabidopsis thaliana. Mol Cell 48:521–531
Yan Z, Hossain MS, Wang J et al (2013) miR172 regulates soybean nodulation. Mol Plant Microbe Interact 26:1371–1377
Yang L, Xu M, Koo Y et al (2013) Sugar promotes vegetative phase change in Arabidopsis thaliana by repressing the expression of MIR156A and MIR156C. Elife 2:e00260
Yu B, Yang Z, Li J et al (2005) Methylation as a crucial step in plant microRNA biogenesis. Science 307:932–935
Zhai J, Zhao Y, Simon SA et al (2013) Plant microRNAs display differential 3′ truncation and tailing modifications that are ARGONAUTE1 dependent and conserved across species. Plant Cell 25:2417–2428
Zhang H, Li L (2013) SQUAMOSA promoter binding protein-like7 regulated microRNA408 is required for vegetative development in Arabidopsis. Plant J 74:98–109
Zhang L, Hou D, Chen X et al (2011) Exogenous plant MIR168a specifically targets mammalian LDLRAP1: evidence of cross-kingdom regulation by microRNA. Cell Res 22:107–126
Zhang J, Zhang S, Li S et al (2013a) A genome-wide survey of microRNA truncation and 3’ nucleotide addition events in larch (Larix leptolepis). Planta 237:1047–1056
Zhang Y-C, Yu Y, Wang C-Y et al (2013b) Overexpression of microRNA OsmiR397 improves rice yield by increasing grain size and promoting panicle branching. Nat Biotechnol 31:848–852
Zhang X-N, Li X, Liu J-H (2014) Identification of conserved and novel cold-responsive microRNAs in trifoliate orange (Poncirus trifoliata (L.) Raf.) using high-throughput sequencing. Plant Mol Biol Report 32:328–341
Zhao M, Tai H, Sun S et al (2012a) Cloning and characterization of maize miRNAs involved in responses to nitrogen deficiency. PLoS One 7:e29669
Zhao Y, Yu Y, Zhai J et al (2012b) The Arabidopsis nucleotidyl transferase HESO1 uridylates unmethylated small RNAs to trigger their degradation. Curr Biol 22:689–694
Zhao X, Zhang H, Li L (2013) Identification and analysis of the proximal promoters of microRNA genes in Arabidopsis. Genomics 101:187–194
Zhou Z, Wang Z, Li W et al (2013a) Comprehensive analyses of microRNA gene evolution in paleopolyploid soybean genome. Plant J 76:332–344
Zhou C-M, Zhang T-Q, Wang X et al (2013b) Molecular basis of age-dependent vernalization in Cardamine flexuosa. Science 340:1097–1100
Zhu H, Zhou Y, Castillo-González C et al (2013) Bidirectional processing of pri-miRNAs with branched terminal loops by Arabidopsis Dicer-like1. Nat Struct Mol Biol 20:1106–1115
Zou Y, Wang Y, Wang L et al (2013) miR172b controls the transition to autotrophic development inhibited by ABA in Arabidopsis. PLoS One 8:e64770
Zvereva AS, Pooggin MM (2012) Silencing and innate immunity in plant defense against viral and non-viral pathogens. Viruses 4:2578–2597
Acknowledgments
This work was supported by the National Program of Plant Protection, Instituto Nacional de Tecnología Agropecuaria (INTA). The authors would like apologize to colleagues whose work could not be fully cited owing to space constraints.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Debat, H.J., Ducasse, D.A. Plant microRNAs: Recent Advances and Future Challenges. Plant Mol Biol Rep 32, 1257–1269 (2014). https://doi.org/10.1007/s11105-014-0727-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-014-0727-z