
Abstract
The utility and commercial potential of genetically engineered (GE) plants would benefit from the use of site-specific recombination systems that enable efficient excision of the marker genes used to identify transformants. Although wheat is one of the most important food crops in the world, GE varieties have yet to be put into commercial production. To develop the Bxb1 recombination system (derived from the Mycobacterium smegmati bacteriophage Bxb1) for site-specific marker gene removal in transgenic wheat, we used biolistics to introduce into the wheat genome a codon optimized Bxb1 recombinase gene (BxbNom) under the control of the maize ubiquitin-1 promoter (Ubi1). Recombinase activity was monitored using a GUSPlus reporter gene activation assay. BxbNom recombinase-mediated excision of an att site-flanked stuffer DNA fragment activated β-glucuronidase reporter activity in callus, endosperm, and leaves in transient assays. The system also detected activity in leaves and endosperm of progeny of multiple independent transgenic wheat lines stably expressing BxbNom. Our results demonstrate that the Bxb1 recombinase is heritable in transgenic wheat plants and performs site-specific excision, providing a useful tool for generating marker-free GE plants. Establishment of wheat lines capable of efficiently excising unneeded marker genes removes one potential barrier to commercial deployment of GE wheat.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Plant biotechnology has the potential to produce crops with increased yield, resistance to key stresses like disease and drought, improved bioenergy production, and improved food products that provide valuable health benefits. Increases in crop yields can reduce land use, while still meeting food, feed, and fiber needs. Currently, genetically engineered (GE) crops under cultivation contain modified traits (e.g., herbicide or pest resistance) conferred by the presence of one or two transgenes of interest and are in use by many farmers worldwide (James 2011). Despite these benefits, biotechnology remains controversial (Herring 2008), and the deployment of genetically engineered plants in the field has elicited criticism from some quarters. Among the concerns that have been expressed are unintended effects on metabolism due to DNA insertion (Cellini et al. 2004), disruption of native genes at the sites of integration (König et al. 2004), the possibility of multiple insertions and/or DNA rearrangements of the integrating DNA (Cellini et al. 2004), and the potential for transgene flow into other crops or wild relatives growing in the surrounding environment (Gressel 2010). In addition, the retention of unneeded marker genes and their encoded proteins in transgenic plants raises concerns about their safety for human consumption (König et al. 2004).
New tools are being developed with the goal of making GE technology more precise and more acceptable to the public. Removal of selectable marker genes from plants destined for commercial production would address concerns about their safety and the potential for the transfer of these transgenes to related plants in the environment. Removal of the selectable marker also would allow reuse of the same selection regime for subsequent rounds of gene transfer. Site-specific recombination was among the first of several methods used to generate transgenic plants without selection transgenes (Dale and Ow 1991; Russell et al. 1992). Recently, a number of marker deletion strategies with various recombinase systems have been successfully applied to food crop species (Ballester et al. 2007; Cao et al. 2006; Chawla et al. 2006; Cuellar et al. 2006; Djukanovic et al. 2008; Gils et al. 2008; Hoa et al. 2002; Hu et al. 2008; Kerbach et al. 2005; Lyznik et al. 1996; Radhakrishnan and Srivastava 2005; Sreekala et al. 2005; Srivastava and Ow 2003; Srivastava et al. 1999; Zhang et al. 2003).
Cre-lox is a well-known site-specific recombination system that has been successfully utilized for marker gene deletion (Russell et al. 1992), site-specific gene integration (Albert et al. 1995; Day et al. 2000; Srivastava and Ow 2002, 2004), chromosomal translocation (Qin et al. 1994), and other genomic applications. While this system is quite versatile for genomic manipulations, it and other small tyrosine family recombinases have the disadvantage that the reactions they catalyze are readily reversible, making targeted integration applications less efficient (Albert et al. 1995; Srivastava and Ow 2004; Thomson et al. 2003; Zhao et al. 2003).
Because they have uni-directional modes of action, site-specific recombinase systems of the large serine family, such as Bxb1-att and phiC31-att, are potentially more powerful than the small tyrosine family recombinases as tools for the genomic manipulation of plants and other organisms. Large serine recombinases act on two unique recognition sequences, known as the attachment sites attB and attP, to yield the product sites known as attL and attR (Wang et al. 2011). Depending on the relative orientation of the attB and attP sites, the reaction can result in excision, inversion, or integration of sequences between the attachment sites and is not reversible unless an additional protein, an excisionase, is present. Several recombinase systems of this type including phiC31 (Thomason et al. 2001; Thomson et al. 2010), Bxb1, TP901-1, and U153 (Thomson and Ow 2006; Yau et al. 2011) have been shown to function in eukaryotic cells.
Bxb1 is a 500 amino acid protein that binds minimal recognition (attachment) sites attP and attB that are 39 bp and 34 bp, respectively, and enzymatically executes recombination (Ghosh et al. 2003). In vitro studies on the Bxb1 system have shown that it can catalyze site-specific recombination in the absence of other proteins or high-energy cofactors (Kim et al. 2003). The first plant study on the Bxb1–att system demonstrated its functionality in tobacco protoplasts (Yau et al. 2011) and Arabidopsis (Thomson et al. 2012). The Bxb1 system has also been shown to function in mammalian tissue culture (Keravala et al. 2006; Russell et al. 2006).
In this investigation, we have developed a transient activity assay to detect stably transformed wheat expressing functional Bxb1 or Cre site-specific recombinases. The recombinase activity assay utilizes a sensor construct comprising an inactive reporter containing a loxP/att-flanked stuffer region located within the intron of the maize ubiquitin-1 promoter (Ubi1). When the stuffer region is removed via recombination, the maize ubiquitin promoter produces a functional transcript that encodes the readily detectable GUSPlus reporter enzyme. We have utilized this strategy to investigate Bxb1 recombinase function in transgenic wheat plants.
Materials and Methods
DNA Constructs
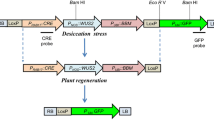
DNA vectors were constructed using standard recombinant DNA techniques (Sambrook 2001). Plasmid pUbi-BxbNom (Fig. 1a) was constructed by inserting a monocot codon optimized Bxb1 coding sequence with an added C-terminal nuclear localization signal (BxbNom, synthesized by GenScript, Piscataway, NJ; Online Resource 1) into the BamHI and SpeI restriction sites of the pUbi–BASK vector. The pUbi–BASK vector was derived from the pAHC20 vector containing the Ubi1 promoter (Christensen and Quail 1996) as follows. The EcoRI site within the Ubi1 intron was removed with site-directed mutagenesis. The bar coding sequence was excised with BamHI and KpnI and a synthetic BamHI–AscI–SpeI–KpnI oligonucleotide was ligated in its place. The expression cassette is terminated by the nos 3′ polyadenylation sequence.

Schematic representation of recombinase activity assay constructs. a Bxb1 recombinase (BxbNom) expression cassette. b Recombinase activity detection vector pGUNG–BxbBP. GUSPlus reporter gene expression is inhibited due to the terminating “stuffer” sequence. c Circular products of Bxb recombinase-mediated excision. The pGUNG–BxbBPexc expresses the GUSPlus reporter. The abbreviations are as follows: PUbi maize ubiquitin promoter, T 3′ Nos terminator, In5′ maize ubiquitin first intron 5′ fragment, tron3′ maize ubiquitin first intron 3′ fragment, GUSPlus β-glucuronidase gene sequence, db35S double enhanced CaMV 35S promoter, eGFP enhanced Green Fluorescent Protein gene. The thick black and gray arrows are loxP and att recombinase recognition sequences, respectively. The 782-bp Ubi::BxbNom PCR amplicon from wheat genomic DNA using primers w and x is shown as the dotted line in (a). The 1,550-bp fragment from the BxbNom coding region used as a probe for RNA blot hybridization is shown as a solid line in (a). The 1,175-bp Ubi::loxPattL::GusPlus PCR amplicon from pGUNG–BxbBPexc using primers y and z is shown as the dotted line in (c). Excision product for Cre-mediated recombination is not shown
Plasmid pGUNG–BxbBP (Fig. 1b) was constructed to include a loxP/att-flanked transcription terminator cassette embedded as a stuffer fragment within the maize Ubi1 promoter first intron (Christensen and Quail 1996). The presence of this terminator cassette blocks Ubi1-mediated expression of the Staphylococcus sp. GUSPlus (http://www.cambia.org/daisy/bioforge_gusplus/3705.html) reporter gene (Broothaerts et al. 2005). Bxb1 or Cre-mediated recombination is expected to remove the loxP/att-flanked terminator cassette from the intron and allow uninterrupted GUSPlus transcription from the Ubi1 promoter (Fig. 1c). The expression cassette is terminated by the nos 3′ polyadenylation sequence. Upstream and in the opposite orientation (i.e., the promoters are contiguous and facing opposite directions) is a double enhanced 35S promoter:eGFP:nos3′ expression cassette.
Plant Materials
Transformation was achieved by particle bombardment of immature embryos of hard white spring wheat ‘Bobwhite’ using methods slightly modified from those described previously (Blechl et al. 2007). Briefly, 0.6 μm gold particles (BioRad, Richmond, CA, USA) were coated as described (Weeks et al. 1993) with plasmids pUbi–BxbNom and pAHC20 (Ubi1:bar:Nos3′) (Christensen and Quail 1996) in 2:1 or 3:1 molar ratios. After bombardment, embryo-derived callus was cultured without selection for 2 weeks in the dark on MMS media (Okubara et al. 2002) containing 40 g/l maltose and 2 mg/l 2,4-D, followed by 2 weeks in the dark on the same media containing 2 or 3 mg/l bialaphos (Meiji Seika Kaisha, Tokyo, Japan). For shoot induction, embryogenic calli were cultured on MMS media containing 0.2 mg/l 2,4-D and 3 mg/l bialaphos for 2–8 weeks with transfers to fresh media every other week. Shoots that formed were rooted on media (Weeks et al. 1993) containing 3 mg/l bialaphos. Plantlets that formed roots on the selection media were transplanted to soil and acclimated to lower humidity over a period of 5–10 days in a growth chamber (23°C, 16 h light/8 h dark). After 5 days, small leaf samples were cut for isolation of DNA and PCR analysis, or in later experiments, for the recombination detection transient assay described below.
Transgenic wheat line, ‘Cre37’, which expresses the Cre recombinase from the maize Ubi1 promoter in the ‘Bobwhite’ background (Srivastava et al. 1999), was used as a positive control for pGUNG–BxbBP functionality.
RNA Blot Analysis
Wheat total leaf RNA was isolated using TRI Reagent (Sigma-Aldrich, St. Louis, MO, USA) with chloroform washes. Isopropanol and sodium acetate were used to pellet the RNA. The pellet was rinsed with 70 % ethanol and resuspended in RNase-free water. Five micrograms of each RNA sample was separated on a 1 % agarose gel containing 2 % formaldehyde and transferred onto Hybond N+ (Amersham Biosciences, Piscataway, NJ, USA) membrane using 20× SSC buffer (Sambrook 2001). The RNA was cross-linked to the blot using a UV Stratalinker 2400 (Agilent Technologies, Santa Clara, CA, USA). For the probe, a 1,550-bp BxbNom fragment was amplified with primers BxbNom F60 5′-GGATCCATGGGGAGAGCTCTCG-3′ and BxbNom R60 5′-CACACTTTCCGCTTTTTCTTAGGC-3′ (Fig. 1a). The BxbNom DNA was radioactively labeled with αP32-dCTP using the NEBlot kit (New England Biolabs, Ipswich, MA, USA) and purified through ProbeQuant G-50 microcolumns from Amersham Biosciences. Blot hybridizations were performed using the Sigma PerfectHyb™ Plus hybridization buffer (Sigma-Aldrich) as recommended by the manufacturer. Hybridized blots were washed to 1× SSC 0.1 % SDS at 50°C. The radioactive signal present on the blot was detected using a Molecular Dynamics Storm 820 Phosphoimager™ (Sunnyvale, CA, USA). IMAGEQUANT software (Molecular Dynamics) was used to view and export the blot image.
Molecular Analysis
Genomic DNA from leaves of T0 and T1 plants was isolated and used for PCR amplification with the Extract-N-Amp Plant PCR kit (Sigma, St. Louis, MO, USA) as specified by the manufacturer. For detection of the BxbNom transgene, forward primer w 5′-CCTGCCTTCATACGCTATTTATTTGC-3′ from the Ubi1 gene promoter and reverse primer x 5′-CCTTGTGGTTCCCTACCTTGGAGTTG-3′ from the BxbNom gene were used to amplify a 782-bp fragment (Fig. 1a). The amplification was performed for 35 cycles with a 1-min denaturation at 96°C, a 1-min annealing at 60°C, and a 1-min extension at 72°C followed by a final elongation step at 72°C for 15 min. As an internal control for wheat genomic DNA purity, a 228-bp fragment was amplified from the wheat high molecular weight glutenin Glu-D1-2b gene in the same reaction tubes, using the primers YF1-Dy10 5′-AGCAGCTCCGAGATGTTAGC-3′ and YR1-Dy10 5′-TGGCCTGGATAATATGACCC-3′ (Altenbach et al. 2002). Fragments were separated on 1.2 % agarose gels in TAE buffer (Sambrook 2001).
For detection of the 1,175-bp pGUNG–BxbBPexc fragment (Fig. 1c) after bombardment of transgenic and wild-type leaves with pGUNG–BxBP, DNA isolated as above was subjected to PCR amplification using the “Phire Hot Start II DNA Polymerase” kit (New England Biolabs). Primers y (5′-AACCAGATCTCCCCCAAATC-3′) and z (5′-CCATGGCTGCAGAAGTAACA-3′) (Fig. 1c) were mixed with 2 μl of leaf DNA in a total reaction volume of 50 μl. Amplification conditions were as follows: 95°C, 5 min, one cycle; 95°C 30 s, 62°C 22 s, 72°C 10 s, 32 cycles; 72°C 5 min, one cycle; 18°C hold.
Development of a Bxb1 and Cre Recombination Detection Transient Assay
Wheat ‘Bobwhite’ tissues used for the detection transient assay were embryos and endosperm excised from caryopses about 3 weeks after anthesis, callus derived from the scutellum of embryos excised from caryopses 10 days after anthesis and cultured on MMS media with 2 mg/l 2,4-D for 1 week, and leaves of various ages (also see below). The tissues were arranged to cover at least 2.5 cm in the center of 9-cm-diameter Petri plate containing either MMS media supplemented with 0.5 M sucrose (embryos, endosperm, calli) or sterile filter paper (leaves). The tissues were bombarded with 1 μM gold particles (Seashell, La Jolla, CA, USA) coated as specified by the manufacturer with a total of 11.25 μg of DNA from plasmids pGUNG–BxbBP or pAHC15 (Ubi:GUS) (Christensen and Quail 1996) or pGUNG–BxbBP + pUbi–BxbNom. The rupture disc pressure was 1,100 psi and the distance between the macrocarrier and the target was 9 cm. After approximately 16 h, the tissues were viewed under a Leica MZ16F stereomicroscope (Leica Microsystems, Bannockburn, IL, USA) with an attached Retiga 2000R FAST Cooled Color 12-bit digital camera (Q Imaging, Pleasanton, CA, USA) and an XCite 120 Fluorescence Illumination System (EXFO Life Sciences, Mississauga, ON, Canada). A 450–490 nm excitation and 515 nm barrier/long pass fluorescence filter set was used for visualization of the GFP fluorescence activity and then the tissues were histochemically stained for GUS activity with 1 mM X-Gluc (Gold Biotechnologies, St. Louis, MO, USA) essentially as previously described (Jefferson 1987). Bombarded tissues were submerged in 1–2 ml of the staining solution, vacuum-infiltrated for 5–20 min, and incubated at 37°C overnight. Following staining, the leaves were treated with several changes of 70 % or 95 % ethanol to remove the chlorophyll.
For the earliest detection of Bxb1 activity in plants transformed with pUbi–BxbNom, T0 leaves from plantlets 5–10 days after removal from tissue culture were bombarded with pGUNG–BxbBP and stained for GUSPlus expression as above. Although such tissues clearly contained cells with histochemical staining (Online Resource 2a), they typically also contained areas of dead cells in the centers of the bombardment zone. In subsequent experiments with T1 seedlings that had been shown to contain pUbi–BxbNom transgenes by PCR amplification, we noted that leaves from plants acclimated to growth in the greenhouse withstood bombardment better and the results of the transient assay were clearer (Online Resource 2b). Reporter activity could readily be visualized, even in the cells in the center of the bombardment zones, and the stained cells were larger and more numerous (Online Resource 2b).
Other tissues tested included embryogenic calli, endosperm, and embryos of wheat. All supported expression of the Ubi:GUS positive control (Fig. 2 and Online Resource 3). Wild-type wheat callus and leaves did not exhibit reporter gene activity following bombardment with the pGUNG–BxbBP construct (Figs. 2 and 5), but the embryos and occasionally the dorsal (embryo) sides of endosperm exhibited GUS positive cells indicative of leaky expression in these tissues of the interrupted (un-excised) GUSPlus cassette of pGUNG–BxbBP (Online Resource 3). Thus, only embryogenic callus, leaves, and the ventral (crease) side of endosperm tissues were useful for recombinase activity assays.

Detection of site-specific recombinase activity with the pGUNG-BxbBP vector. a Transient expression in bombarded non-transgenic ‘Bobwhite’ or ‘Cre37’ wheat leaves. Reporter gene expression for β-glucuronidase activity (GUS) or blue light illumination (GFP) is shown. Leaves were bombarded with pAHC15 (positive control) or pGUNG–BxbBP. Detection of GUS activity in tissues bombarded with the pGUNG–BxbBP vector indicates the site-specific removal of the “stuffer” region allowing GUSPlus gene expression. GFP fluorescence indicates the presence of pGUNG–BxbBP DNA. b Transient expression in bombarded ‘Bobwhite’ wheat callus (top row) and the ventral side of endosperm (bottom row). Tissues were bombarded with pAHC15 (positive control), pGUNG–BxbBP alone, or both pGUNG–BxbBP and pUbi–BxbNom. Samples were stained for β-glucuronidase activity and imaged for green fluorescence activity
Results
Detection of Recombinase Activity via a Transient Assay Utilizing the pGUNG–BxbBP Sensor Construct
To develop a unidirectional recombinase system that functions in wheat, we devised a transient assay for the detection of site-specific recombination activity. The approach utilizes biolistics to introduce into wheat cells a sensor plasmid (pGUNG–BxbBP; Fig. 1b) that expresses the GUSPlus reporter gene only after it has undergone recombinase-mediated excision. In the absence of a functional recombinase, the GUSPlus reporter is not expressed due to presence of a lox/att-flanked stuffer fragment containing transcription termination sequences. Introduction of pGUNG–BxbBP into cells containing the BxbNom or Cre recombinase is expected to result in the deletion of lox/att-flanked termination region (Fig. 1c) and activation of the GUSPlus gene expression, which can be detected by histochemical staining. The pGUNG–BxbBP plasmid also contains an expression cassette for GFP visualization as an internal control verifying that the cells received and expressed the introduced DNA.
To ascertain whether the assay would work to successfully detect recombinase activity, leaves of wild-type plants and the transgenic wheat line ‘Cre37’ that expresses the Cre recombinase were bombarded with pGUNG–BxbBP. The wheat Cre37 line contains a single copy of the Cre gene expressed under the control of the Ubi1 promoter (Srivastava et al. 1999). GUS activity was readily detectable in wild-type leaves after bombardment with a positive control plasmid pAHC15 (Ubi1::GUS) (Fig. 2a), but not after bombardment with pGUNG–BxbBP, even though GFP expression indicated that the cells had received the detection plasmid. In contrast, the Cre37 line exhibited GUS activity after bombardment with pGUNG–BxbBP as a result of Cre-mediated removal of the loxP-flanked stuffer sequence (Fig. 2a). This result demonstrates that the pGUNG–BxbBP construct was a competent substrate for recombinase-mediated excision of the stuffer region allowing activation of GUSPlus expression.
In other preliminary experiments to develop the recombinase detection transient assay, embryos, embryogenic callus, and endosperm from non-transformed ‘Bobwhite’ were bombarded with pAHC15 or pGUNG–BxbBP or co-bombarded with pGUNG–BxbBP + pUbi–BxbNom and stained for GUS activity. The pAHC15 plasmid containing the Ubi1::GUS cassette served as the positive control and all the tissues bombarded with it showed GUS activity detected as blue spots after histochemical staining (Fig. 2b; Online Resource 3). Bombardment with pGUNG–BxbBP did not result in detectable GUS activity in wild-type wheat leaves (Fig. 2a), callus, or the ventral (crease) side of endosperm (Fig. 2b; Online Resource 3b). Based on these results, the functionality of the Bxb1–att recombination system in wheat cells was tested by co-bombardment with both the pGUNG–BxbBP and pUbi–BxbNom constructs into callus tissue and the ventral side of endosperm tissue. The GFP fluorescence and GUSPlus staining observed 16 h after bombardment, compared to the positive and negative controls, is shown in Fig. 2b. These results clearly demonstrate that the codon optimized BxbNom gene performed recombination that activated expression of the GUSPlus reporter gene in wheat cells.
Generation of BxbNom Expressing Wheat Transformants
In five separate biolistic transformation experiments, ‘Bobwhite’ was co-transformed with pAHC20 (Ubi1::bar) and pUbi–BxbNom (Fig. 1a). Genomic DNA was isolated from leaves of plantlets regenerated under bialaphos selection and assayed by PCR amplification with primers designed to reveal the presence of pUbi–BxbNom. Thirteen T0 plantlets were found to carry pUbi–BxbNom (Table 1). Of these, all but one transmitted the transgene to their progeny as determined by PCR of genomic DNA from T1 plantlets (Table 1).
For six of the 12 heritable BxbNom transgenic lines, more than 10 progeny were tested for inheritance. Three of these lines exhibited segregation that was consistent with transgene integration at a single locus (3:1 segregation) (Table 1). PCR results for genomic DNA from 13 T1 progeny from one such line, Bxb146-110, are shown in Fig. 3a. For events Bxb145-12, Bxb144-19, and Bxb146-14, transmission of the transgene to the progeny was lower than expected (Table 1). For the other six events, only a few progeny were tested, and at least half inherited the transgene (Table 1).

Segregation of the Ubi1:BxbNom transgene and recombinase activity in a family of 13 T1 plants from event Bxb146-110. a PCR amplification of genomic DNA from 13 T1 progeny (numbered lanes) and non-transformed ‘Bobwhite’ (BW). Expected products for BxbNom and Dy10 amplification are denoted by arrows. Lane marked M contains DNA size markers. b Excision activated GUSPlus reporter gene activity detected in the T0 Bxb146-110 leaves and the leaves of the same 13 T1 progeny as in (a). A GFP image of leaves from T1 progeny number 13 is also shown bottom right
To ascertain whether the BxbNom transgene was transcribed in the transgenic wheat plants, RNA was isolated from the leaves of T1 or T2 plants of six of the events that showed inheritance of the BxbNom transgene. Five of the lines contained the predicted 1.9-kb transcript (Fig. 4) that hybridized to the BxbNom coding region probe (Fig. 1a). A sixth line (Bxb125-17) did not exhibit detectable levels of transcript (Fig. 4), which was consistent with the lack of recombinase activity observed in the recombinase detection assay (see below, Fig. 5a). Among the lines with detectable transcripts, their levels varied approximately 2–5-fold, with Bxb145-35 having the highest level (Fig. 4).

Blot hybridization analysis of RNA from T2 (Bxb125-52) or T1 leaves of selected Bxb1 transgenic wheat lines and non-transformed ‘Bobwhite’. The nylon membrane was probed with the BxbNom coding sequence (Fig. 1a). Size markers in kilobases (kb) are shown. The image of the ethidium bromide stained gel ribosomal RNA (rRNA) is shown in the bottom panel to illustrate RNA loading

Bxb1 recombinase activity detected in leaves of nine independent transgenic wheat lines. Wheat leaves from plants stably transformed with BxbNom were bombarded with the pGUNG–BxbBP recombinase activity sensor vector and stained for β-glucuronidase activity (a) or subjected to DNA extraction and PCR analyses (b). All plants except ‘Bobwhite’ contained the Ubi:BxbNom transgene fragment as detected by PCR of genomic DNA (see Table 1). a Leaves of line Bxb125-53 were from T3 generation plants; leaves of lines Bxb125-17, Bxb126-98, and Bxb145-35 were from T1 generation plants, while leaves of the other lines were from T0 plants. b Amplification products from T3 (Bxb125-53) or T1 leaf DNA of five transgenic events or non-transformed ‘Bobwhite’ bombarded with pGUNG–BxbBP (BW+GGBxBP), and unbombarded ‘Bobwhite’, after PCR with primers y and z (Fig. 1c). Size markers in kilobases (kb) are shown to the left. The B lane shows no products were produced in the absence of DNA template; the P lane contains the product of the excised plasmid DNA (Fig. 1c) after amplification with the same primers
Bxb Recombinase Activity in Transgenic Wheat
To assess whether the Bxb1 recombinase encoded by the BxbNom transgene was functional for marker gene excision in wheat, we bombarded the detection plasmid into leaves from transgenic plants at various stages of development. We found we could detect GUSPlus activity encoded by the excised version of pGUNG–BxbBP in leaves of T0 plantlets as early as 5–10 days after transfer from tissue culture to soil (the same stage used for isolation of genomic DNA for PCR). However, leaves from plants allowed to grow in the greenhouse to at least the five-leaf stage tolerated bombardment better and yielded larger numbers of bigger spots (Online Resource 2). Applying the transient assay to T0 leaves of the 13 independent transgenic pUbi–BxbNom lines revealed that 10 had functional Bxb recombinase activity, as shown by histochemical detection of β-glucuronidase activity (Table 1; examples in Fig. 5a). Furthermore, GUSPlus activity due to Bxb1-mediated excision was detected in leaves of transgenic T1 progeny of eight lines that had recombinase activity in T0 leaves (Table 1; examples in Figs. 3b and 5a). Figure 3b shows the results of the recombinase detection transient assay in leaves of progeny of line Bxb146-110. As can be seen, there is a one-to-one correspondence between inheritance of the BxbNom transgene (Fig. 3a) and recombinase activity (Fig. 3b) in the 13 progeny. For event Bxb125-53, Bxb1 recombinase activity was detected in each generation from T0 to T3 (results for T3 pictured in Fig. 5a).
To confirm that the GUSPlus reporter activity detected in the recombination assay was the result of Bxb-mediated excision, DNA from transgenic and control leaves bombarded with pGUNG–BxbBP was isolated and subjected to PCR amplification with primers designed to detect the excision product (Fig. 1c). Fragments of the expected size, 1,175 bp, were present in amplified DNA extracts from each of five different transgenic plants, but absent in non-transgenic ‘Bobwhite’ bombarded with pGUNG–BxbBP (Fig. 5b). These results confirm at the DNA level that Bxb-mediated excision had occurred in wheat cells expressing BxbNom.
Discussion
In this report, we have shown that the uni-directional Bxb1 recombinase system is active in wheat, one of the most important food crops that thus far has not benefited from commercial release of GE varieties. Bxb1 functions in wheat leaves and endosperm to precisely excise DNA between its attB and attP recognition sites. Expression of the BxbNom gene is heritable. These results provide another tool—along with Cre (Srivastava et al. 1999) and phiC31 (Rubtsova et al. 2008)—for recombinase-mediated genetic engineering to remove selectable marker genes from GE wheat. The transgenic wheat lines generated here can be used in genetic crosses to introduce Bxb1 activity into other wheat cultivars.
Previous publications reported that phenotypic abnormalities can occur in transgenic plants constitutively expressing site-specific recombinase genes. Widespread expression of Cre in tomato, petunia, and tobacco generated misshapen leaves (Coppoolse et al. 2003) while the Gin recombinase elicited lesions in tobacco (Maeser and Kahmann 1991). In contrast, our transgenic wheat plants constitutively expressing the BxbNom recombinase under the control of the maize ubiquitin promoter did not exhibit abnormal regeneration, growth, development, or fertility. These results are similar to those reported for wheat expressing Cre (Srivastava et al. 1999) and phiC31 (Rubtsova et al. 2008). Whether wheat’s tolerance of active recombinases is due to its large genome size and/or the redundancy of genes in hexaploid varieties or to some other reason remains to be determined.
Deployment of the Bxb1/att (Thomson et al. 2012) or the phiC31/att (Kempe et al. 2010; Thomson et al. 2010) irreversible site-specific recombinase systems into plants such as Arabidopsis and wheat raises the possibility of developing sophisticated genome engineering applications that require irreversible integration capabilities. Because of the unidirectional nature of these enzymes, a dual recombinase-mediated cassette exchange (RMCE) strategy enabling targeted and oriented gene stacking within the genome becomes feasible (Wang et al. 2011). Various genetic one-way switches could be triggered through the use of tightly controlled organ-specific promoters expressing the Bxb1 recombinase, allowing activation or repression of target genes.
In conclusion, we have shown that the expression of a functional codon-optimized version of the Bxb1 recombinase gene can be maintained over at least two generations in wheat. The BxbNom recombinase mediates site-specific excision and can be utilized in further applications for precise genome modifications and crop enhancement. In enabling the creation of GE plants free of marker genes and proteins, site-specific recombination systems such as Bxb1 could make GE wheat more acceptable to farmers, government regulators, and consumers, and facilitate the use of genetic engineering to improve this major food crop.
Abbreviations
- GUSPlus :
-
Staphylococcus sp. β-glucuronidase gene
- eGFP:
-
Enhanced green fluorescent protein
References
Albert H, Dale EC, Lee E, Ow DW (1995) Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J 7(4):649–659. doi:10.1046/j.1365-313X.1995.7040649.x
Altenbach SB, Kothari KM, Lieu D (2002) Environmental conditions during wheat grain development alter temporal regulation of major gluten protein genes. Cereal Chem J 79(2):279–285. doi:10.1094/cchem.2002.79.2.279
Ballester A, Cervera M, Peña L (2007) Efficient production of transgenic citrus plants using isopentenyl transferase positive selection and removal of the marker gene by site-specific recombination. Plant Cell Rep 26(1):39–45. doi:10.1007/s00299-006-0197-3
Blechl A, Lin J, Nguyen S, Chan R, Anderson OD, Dupont FM (2007) Transgenic wheats with elevated levels of Dx5 and/or Dy10 high-molecular-weight glutenin subunits yield doughs with increased mixing strength and tolerance. J Cereal Sci 45(2):172–183. doi:10.1016/j.jcs.2006.07.009
Broothaerts W, Mitchell HJ, Weir B, Kaines S, Smith LM, Yang W, Mayer JE, Roa-Rodriguez C, Jefferson RA (2005) Gene transfer to plants by diverse species of bacteria. Nature 433(7026):629–633. doi:10.1038/nature03309
Cao M-X, Huang J-Q, Yao Q-H, Liu S-J, Wang C-L, Wei Z-M (2006) Site-specific DNA excision in transgenic rice with a cell-permeable Cre recombinase. Mol Biotechnol 32(1):55–63. doi:10.1385/mb:32:1:055
Cellini F, Chesson A, Colquhoun I, Constable A, Davies HV, Engel KH, Gatehouse AMR, Kärenlampi S, Kok EJ, Leguay JJ, Lehesranta S, Noteborn HPJM, Pedersen J, Smith M (2004) Unintended effects and their detection in genetically modified crops. (Food Chem Toxicol 42(7):1089–1125. doi:10.1016/j.fct.2004.02.003
Chawla R, Ariza-Nieto M, Wilson AJ, Moore SK, Srivastava V (2006) Transgene expression produced by biolistic-mediated, site-specific gene integration is consistently inherited by the subsequent generations. Plant Biotechnol J 4(2):209–218. doi:10.1111/j.1467-7652.2005.00173.x
Christensen AH, Quail PH (1996) Ubiquitin promoter-based vectors for high-level expression of selectable and/or screenable marker genes in monocotyledonous plants. Transgenic Res 5(3):213–218. doi:10.1007/bf01969712
Coppoolse ER, de Vroomen MJ, Roelofs D, Smit J, van Gennip F, Hersmus BJ, Nijkamp HJ, van Haaren MJ (2003) Cre recombinase expression can result in phenotypic aberrations in plants. Plant Mol Biol 51(2):263–279. doi:10.1023/a:1021174726070
Cuellar W, Gaudin A, Solórzano D, Casas A, Ñopo L, Chudalayandi P, Medrano G, Kreuze J, Ghislain M (2006) Self-excision of the antibiotic resistance gene nptII using a heat inducible Cre-loxP system from transgenic potato. Plant Mol Biol 62(1):71–82. doi:10.1007/s11103-006-9004-3
Dale EC, Ow DW (1991) Gene transfer with subsequent removal of the selection gene from the host genome. Proc Natl Acad Sci 88(23):10558–10562
Day CD, Lee E, Kobayashi J, Holappa LD, Albert H, Ow DW (2000) Transgene integration into the same chromosome location can produce alleles that express at a predictable level, or alleles that are differentially silenced. Genes Dev 14(22):2869–2880. doi:10.1101/gad.849600
Djukanovic V, Lenderts B, Bidney D, Lyznik LA (2008) A Cre::FLP fusion protein recombines FRT or loxP sites in transgenic maize plants. Plant Biotechnol J 6(8):770–781. doi:10.1111/j.1467-7652.2008.00357.x
Ghosh P, Kim AI, Hatfull GF (2003) The orientation of mycobacteriophage Bxb1 integration is solely dependent on the central dinucleotide of attP and attB. Molecular Cell 12(5):1101–1111. doi:10.1016/s1097-2765(03)00444-1
Gils M, Marillonnet S, Werner S, Grützner R, Giritch A, Engler C, Schachschneider R, Klimyuk V, Gleba Y (2008) A novel hybrid seed system for plants. Plant Biotechnol J 6(3):226–235. doi:10.1111/j.1467-7652.2007.00318.x
Gressel J (2010) Needs for and environmental risks from transgenic crops in the developing world. New Biotechnol 27(5):522–527. doi:10.1016/j.nbt.2010.05.015
Herring RJ (2008) Opposition to transgenic technologies: ideology, interests and collective action frames. Nat Rev Genet 9(6):458–463. doi:10.1038/nrg2338
Hoa TTC, Bong BB, Huq E, Hodges TK (2002) Cre/lox site-specific recombination controls the excision of a transgene from the rice genome. Theor Appl Genet 104(4):518–525. doi:10.1007/s001220100748
Hu Q, Kononowicz-Hodges H, Nelson-Vasilchik K, Viola D, Zeng P, Liu H, Kausch AP, Chandlee JM, Hodges TK, Luo H (2008) FLP recombinase-mediated site-specific recombination in rice. Plant Biotechnol J 6(2):176–188. doi:10.1111/j.1467-7652.2007.00310.x
James C (2011) Global status of commercialized biotech/GM crops: 2011. ISAAA Briefs 43:1–29
Jefferson R (1987) Assaying chimeric genes in plants: the GUS gene fusion system. Plant Mol Biol Rep 5(4):387–405. doi:10.1007/bf02667740
Kempe K, Rubtsova M, Berger C, Kumlehn J, Schollmeier C, Gils M (2010) Transgene excision from wheat chromosomes by phage phiC31 integrase. Plant Mol Biol 72(6):673–687. doi:10.1007/s11103-010-9606-7
Keravala A, Groth A, Jarrahian S, Thyagarajan B, Hoyt J, Kirby P, Calos M (2006) A diversity of serine phage integrases mediate site-specific recombination in mammalian cells. Mole Genet Genom 276(2):135–146. doi:10.1007/s00438-006-0129-5
Kerbach S, Lörz H, Becker D (2005) Site-specific recombination in Zea mays. Theor Appl Genet 111(8):1608–1616. doi:10.1007/s00122-005-0092-2
Kim AI, Ghosh P, Aaron MA, Bibb LA, Jain S, Hatfull GF (2003) Mycobacteriophage Bxb1 integrates into the Mycobacterium smegmatis groEL1 gene. Mol Microbiol 50(2):463–473. doi:10.1046/j.1365-2958.2003.03723.x
König A, Cockburn A, Crevel RWR, Debruyne E, Grafstroem R, Hammerling U, Kimber I, Knudsen I, Kuiper HA, Peijnenburg AACM, Penninks AH, Poulsen M, Schauzu M, Wal JM (2004) Assessment of the safety of foods derived from genetically modified (GM) crops. Food Chem Toxicol 42(7):1047–1088. doi:10.1016/j.fct.2004.02.019
Lyznik LA, Rao KV, Hodges TK (1996) FLP-mediated recombination of FRT sites in the maize genome. Nucleic Acids Res 24(19):3784–3789. doi:10.1093/nar/24.19.3784
Maeser S, Kahmann R (1991) The Gin recombinase of phage Mu can catalyse site-specific recombination in plant protoplasts. Mol Gen Genet 230(1):170–176. doi:10.1007/bf00290665
Okubara PO, Blechl AB, McCormick SM, Alexander NA, Dill-Macky RD-M, Hohn TH (2002) Engineering deoxynivalenol metabolism in wheat through the expression of a fungal trichothecene acetyltransferase gene. TAG Theor Appl Genet 106(1):74–83. doi:10.1007/s00122-002-1066-2
Qin M, Bayley C, Stockton T, Ow DW (1994) Cre recombinase-mediated site-specific recombination between plant chromosomes. Proc Natl Acad Sci 91(5):1706–1710
Radhakrishnan P, Srivastava V (2005) Utility of the FLP–FRT recombination system for genetic manipulation of rice. Plant Cell Rep 23(10):721–726. doi:10.1007/s00299-004-0876-x
Rubtsova M, Kempe K, Gils A, Ismagul A, Weyen J, Gils M (2008) Expression of active Streptomyces phage phiC31 integrase in transgenic wheat plants. Plant Cell Rep 27(12):1821–1831. doi:10.1007/s00299-008-0604-z
Russell SH, Hoopes JL, Odell JT (1992) Directed excision of a transgene from the plant genome. Mol Gen Genet 234(1):49–59. doi:10.1007/bf00272344
Russell JP, Chang DW, Tretiakova A, Padidam M (2006) Phage Bxb1 integrase mediates highly efficient site-specific recombination in mammalian cells. Biotechniques 40(4):460–464. doi:10.2144/000112150
Sambrook J (2001) Molecular cloning: a laboratory manual. vol Accessed from http://nla.gov.au/nla.cat-vn2284148. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
Sreekala C, Wu L, Gu K, Wang D, Tian D, Yin Z (2005) Excision of a selectable marker in transgenic rice (Oryza sativa L.) using a chemically regulated Cre/loxP system. Plant Cell Rep 24(2):86–94. doi:10.1007/s00299-004-0909-5
Srivastava V, Ow DW (2002) Biolistic mediated site-specific integration in rice. Mol Breed 8(4):345–349. doi:10.1023/a:1015229015022
Srivastava V, Ow DW (2003) Rare instances of Cre-mediated deletion product maintained in transgenic wheat. Plant Mol Biol 52(3):661–668. doi:10.1023/a:1024839617490
Srivastava V, Ow DW (2004) Marker-free site-specific gene integration in plants. Trends Biotechnol 22(12):627–629. doi:10.1016/j.tibtech.2004.10.002
Srivastava V, Anderson OD, Ow DW (1999) Single-copy transgenic wheat generated through the resolution of complex integration patterns. Proc Natl Acad Sci 96(20):11117–11121
Thomason LC, Calendar R, Ow DW (2001) Gene insertion and replacement in Schizosaccharomyces pombe mediated by the Streptomyces bacteriophage phiC31 site-specific recombination system. Mole Genet Genom 265(6):1031–1038. doi:10.1007/s004380100498
Thomson JG, Ow DW (2006) Site-specific recombination systems for the genetic manipulation of eukaryotic genomes. Genesis 44(10):465–476. doi:10.1002/dvg.20237
Thomson JG, Rucker EB, Piedrahita JA (2003) Mutational analysis of loxP sites for efficient Cre-mediated insertion into genomic DNA. Genesis 36(3):162–167. doi:10.1002/gene.10211
Thomson JG, Chan R, Thilmony R, Yau Y-Y, Ow DW (2010) PhiC31 recombination system demonstrates heritable germinal transmission of site-specific excision from the Arabidopsis genome. BMC Biotechnol 10:17. doi:10.1186/1472-6750-10-17
Thomson JG, Chan R, Smith J, Thilmony R, Yau Y-Y, Wang Y, Ow DW (2012) The Bxb1 recombination system demonstrates heritable transmission of site-specific excision in Arabidopsis. BMC Biotechnol 12(1):9. doi:10.1186/1472-6750-12-9
Wang Y, Yau Y-Y, Perkins-Balding D, Thomson JG (2011) Recombinase technology: applications and possibilities. Plant Cell Rep 30(3):267–285. doi:10.1007/s00299-010-0938-1
Weeks JT, Anderson OD, Blechl AE (1993) Rapid production of multiple independent lines of fertile transgenic wheat (Triticum aestivum). Plant Physiol 102(4):1077–1084. doi:10.1104/pp.102.4.1077
Yau Y-Y, Wang Y, Thomson JG, Ow DW (2011) Method for Bxb1-mediated site-specific integration in planta. In: Methods in molecular biology, vol 701. Plant chromosome engineering: methods and protocols. Springer, New York, pp 147–166. doi:10.1007/978-1-61737-957-4_8
Zhang W, Subbarao S, Addae P, Shen A, Armstrong C, Peschke V, Gilbertson L (2003) Cre/lox-mediated marker gene excision in transgenic maize (Zea mays L.) plants. Theor Appl Genet 107(7):1157–1168. doi:10.1007/s00122-003-1368-z
Zhao X, Coats I, Fu P, Gordon-Kamm B, Lyznik LA (2003) T-DNA recombination and replication in maize cells. Plant J 33(1):149–159. doi:10.1046/j.1365-313X.2003.016016.x
Acknowledgments
We thank Dr. V. Srivastava for seeds of the Cre-expressing transgenic wheat line ‘Cre37’ and Mara Guttman for excellent technical assistance. Mention of trade names or commercial products in this publication is solely to provide specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity employer.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PPT 4692 kb)
Rights and permissions
About this article
Cite this article
Blechl, A., Lin, J., Shao, M. et al. The Bxb1 Recombinase Mediates Site-Specific Deletion in Transgenic Wheat. Plant Mol Biol Rep 30, 1357–1366 (2012). https://doi.org/10.1007/s11105-012-0454-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-012-0454-2