
Abstract
Simple sequence repeats (SSRs) were used to assess genetic diversity and study genetic relatedness in a large collection of Malus germplasm. A total of 164 accessions from the Malus core collection, maintained at the University of Illinois, were genotyped using apple SSR markers. Each of the accessions was genotyped using a single robust SSR marker from each of the 17 different linkage groups in Malus. Data were subjected to principal component analysis, and a dendrogram was constructed to establish genetic relatedness. As expected, this diverse core collection showed high allelic diversity; moreover, this allelic diversity was higher than that previously reported. Cluster analysis revealed the presence of four distinct clusters of accessions in this collection.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Availability of diverse Malus germplasm is critical for pursuing successful apple breeding efforts, as it increases genetic diversity and allows for development of new apple cultivars with enhanced and/or desirable traits. This also aids in diversifying the gene pool and preserving those unique genetic traits available in this material.
When characterizing plant germplasm, incidence of multiple clones of genetic material as well as mislabeling of accessions may occur, which are both costly and undesirable (Garkava-Gustavsson et al. 2008). Moreover, proper identification and characterization of plant germplasm will protect intellectual property as well as aid in identifying parents carrying genes of interest for breeding efforts (Goulão et al. 2001; Dávila et al. 1998). By selecting diverse parents and increasing genetic diversity through germplasm collections, progress can be made in apple plant breeding efforts towards developing new cultivars with economically valuable traits including those with enhanced fruit quality and disease and pest resistance.
As in vivo maintenance and management of Malus germplasm are labor-intensive, costly, and require commitment of land resources for germplasm conservation efforts, determining genetic identity and genetic relatedness among accessions also impacts efficiency and utilization of such germplasm collections in breeding programs (Kresovich and McFerson 1992; Russell et al. 1997). The constraints of management of the Malus germplasm collection have led to development of strategies for germplasm evaluation. One of these strategies is the development of a core collection consisting of accessions having high levels of genetic diversity that could serve as representatives of the entire genetic diversity available within a collection (Frankel 1984; Brown 1989; Marshall 1990; Brown 1995). Developing core subsets of a population enhances the efficiency of screening and evaluation of desirable target traits. To increase the usefulness of core collections, the genetic information must be clearly identified and documented (Hokanson et al. 1998).
The use of DNA-based molecular markers has been highly critical and valuable for pursuing studies to assess genetic diversity, determine genetic relatedness, and identify genes of interest (Han and Korban 2010; Chen et al. 2010; Baraket et al. 2011). Markers such as simple sequence repeats (SSRs) are highly polymorphic, reproducible, and are distributed throughout the genome, rendering these markers ideal for pursuing genetic diversity studies. This has proven successful for various other fruit crops such as apricot (Wang et al. 2011), cherry (Clarke and Tobutt 2009), and strawberry (Govan et al. 2008), among others. Therefore, the use of DNA-based markers to characterize the Malus germplasm is a highly valuable and reliable tool (Garkava-Gustavsson et al. 2008; Zhang et al. 2011).
Many apple SSR markers have been developed, and most have proven highly valuable for identifying and characterizing limited numbers of apple germplasm, identifying loci associated with target genes, and for map-based cloning efforts (Liebhard et al. 2002; Naik et al. 2006; Gasic et al. 2009; Han and Korban 2010; Zhang et al. 2011). The overall goal of this study is to assess genetic relatedness and diversity in a large collection of Malus germplasm, selected by the US Apple Crop Germplasm Committee as a core collection based on known and/or reported phenotypic and genetic traits, using SSR markers.
Materials and Methods
Plant Material and DNA Extraction
Young expanding leaves were collected from 164 Malus accessions maintained within the Malus core collection available at the University of Illinois (Table 1). This core collection, selected by the US Apple Crop Germplasm Committee based on either known or reported phenotypic and genotypic traits, is a subset of Malus accessions representing the genetic diversity of the entire Malus germplasm available at the clonal repository of the Plant Genetic Resources Unit in Geneva, NY, USA. It is comprised of wild accessions, old and new cultivars, as well as advanced selections.
DNA was extracted from leaf tissues of all of the above accessions following the protocol described by Kobayashi et al. (1998), but with a slight modification by extending the incubation period for an additional 20 min at 65°C. Moreover, after adding isopropanol, microfuge tubes were kept at −20°C for no less than 4 h.
PCR Amplification and Capillary Electrophoresis
DNA fragments were subjected to PCR amplification using 17 robust SSR markers spanning all 17 linkage groups (LG) of Malus. These SSR markers included the following: Hi02C07, CH02C06, GD12, NZ05g8, CH05f06, CH03d07, CH04e05, CH01h10, CH01f03b, CH02c11, CH02d08, CH01f02, GD147, CH04c07, CH02c09, CH04f10, and CH01h01 (Table 2). These markers were previously evaluated and developed by the European Cooperative Program for Plant Genetic Resources and were found to be highly polymorphic (Evans et al. 2007). PCR reactions were performed in 96-well plates in a total volume of 10 μL containing 50 ng template DNA, 4.25 nuclease-free water, 0.3 mM MgCl2, 0.2× Green GoTaq® Flexi Buffer, 0.05 U GoTaq® DNA polymerase (Promega, Madison, WI, USA), 0.04 mM of each dNTP, 0.25 μL forward primer (10 μM), 0.25 μL reverse primer (10 μM), and 0.15 μL M13 fluorescent dye (10 μM).
PCR amplification was carried out using either a Thermo Fisher Scientific multi-block thermal cycler (Pittsburgh, PA, USA) or an MJ Research PTC-100 or PTC-200 (Ramsey, MN, USA). Amplifications were performed using the following conditions: initial denaturation at 94°C for 4 min, 5 cycles of 94°C for 1 min, 54°C for 1 min, 72°C for 1 min, 30 cycles of 94°C for 1 min, 52°C for 1 min, 72°C for 1 min, and a final extension at 72°C for 30 min. This was followed by holding at 4°C.
Individual PCR products were labeled with one of four M13 dyes, FAM, VIC, PET, and NED, along with a corresponding LIZ 600 size standard. PCR products were then pooled for electrophoresis. Amplified PCR products were separated at the W.M. Keck Center at the University of Illinois using an ABI 3730xl sequencer (Applied Biosystems, Inc., Foster City, CA, USA). Raw fragment size data were analyzed using GeneMapper™ Software ver. 4.0 (Applied Biosystems, Inc.), and all automated results were manually reviewed.
Statistical Analysis
Statistical analysis was performed using SAS® 9.2 software. The proc univariate was used to determine data normality. Proc corr was used to assess fragment length data correlations. Proc cluster and proc tree were used to create the dendrogram using options rsquare and simple for proc cluster, and options ftext = triplexu, hsize = 10, ftext = 0.2, vsize = 10, interval = 0.2, htext = 0.2, horizontal, vpages = 5, height = rsq, and inc = 1.4 for proc tree. Proc princomp was used to obtain principal components, and proc g3d was used to create a scatter plot of the accessions with the options reset = all border, tilt = 50, rotate = 30, color = color, and shape = shape.
Data for each SSR were assessed using the correlation procedure to determine correlations among these variables. Ward’s minimum variance method, a non-hierarchical cluster method, was used to generate clusters. The number of clusters was determined by the cubic clustering criterion, the pseudo-F, and the pseudo-T 2 along with R 2 values. Data were then subjected to a principal component analysis to create principal components (PCs) for the construction of a three-dimensional scatter plot (Johnson 1998).
Expected heterozygosity and observed heterozygosity were calculated using the program GENEPOP (Raymond and Rousset 1995; Rousset 2008). Polymorphic information content (PIC) (Botstein et al. 1980; Hearne et al. 1992) was calculated using the program Cervus (Kalinowski et al. 2007). Effective alleles per locus were calculated according to Morgante et al. (1994).
Of 17 primer pairs, 10 were selected for final analysis on the basis of reliable amplification and signal strength. These included the following markers: Hi02c07, GD147, CH04e05, CH04c07, CH03d07, CH02c09, CH01h10, CH04f10, CH02d08, and CH01f03b. This number of markers was similar to that reported in previous studies, which have used anywhere from eight markers (Benson et al. 2001; Király et al. 2009) to 14 markers (Guilford et al. 1997).
Results and Discussion
The Malus core collection used in this study was originally selected as the best representative of available phenotypic and genetic diversity for various economic traits of interest for use in evaluations for various biotic and abiotic stresses as well as fruit quality traits (Forsline 1996).
All SSR primer pairs used in this study generated multiple fragments in the Malus germplasm core collection. Of 164 Malus accessions, 39 did not amplify at least one PCR product. Due to the nature of multivariate analysis, accessions with missing data were not used for either the dendrogram or the scatter plot. A total of 125 accessions were included in generating both the dendrogram and the scatter plot (Table 3). However, due to the nature of the analysis and the strength of the remaining data, all 164 accessions were included in calculating expected heterozygosity, observed heterozygosity, PIC, and effective alleles per locus.
A total of 283 fragments were amplified using the selected 10 SSR markers (Table 4). In general, each primer pair amplified several alleles in large numbers, while many alleles were either rare or unique, representing only 2–5% of the entire Malus core collection. Overall, a higher allelic diversity was observed in this Malus core collection than that reported in previous studies (Hokanson et al. 1998; Kitahara et al. 2005; Garkava-Gustavsson et al. 2008; Zhang et al. 2011). Discrepancies in allelic diversity could be attributed to differences in DNA isolation protocols and perhaps increased ability to detect 1–2 bp length differences utilizing modern fragment analysis technologies.
In this study, rare alleles (<5% of the total alleles per marker) have been detected at a frequency of 39% compared to frequencies of 53% (Garkava-Gustavsson et al. 2008) and 59% (Hokanson et al. 1998). These discrepancies could be attributed to the use of different SSRs, as only three markers are common among these three different studies. Another possible explanation is that rare alleles have been selected for in domesticated cultivars, whereas studies targeting wild germplasm are not selected for these rare alleles. Rare alleles are critical for maintaining genetic diversity as they are unique and they are likely to be involved in plant adaptation to environmental shifts (Richter et al. 1994; Bengtsson et al. 1995). Thus, these alleles are important components of this core collection.
In this study, 164 Malus accessions from diverse genetic backgrounds include Malus species, M. × domestica cultivars, and selections, while previous studies have focused on assessing allelism using smaller collections consisting primarily of M. × domestica cultivars. As modern cultivars are derived from a relatively narrow genetic base, it is not unexpected that these collections would be less genetically diverse. Only five founding clones were progenitors for 64% of a total of 439 cultivars in a co-ancestry study (Noiton and Alspach 1996), thus pointing to the highly shared genetic identity of modern apple cultivars.
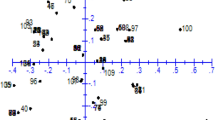
The cluster function of SAS 9.2 (proc cluster) produced a dendrogram composed of six distinct clusters (Fig. 1). These clusters were populated as follows: The first cluster consisted of 34 accessions, a second cluster consisted of 33 accessions, a third cluster consisted of 21 accessions, and a fourth cluster consisted of 14 accessions, while both fifth and sixth clusters consisted of 14 and nine accessions, respectively, for a total of 125 accessions. In addition, a scatter plot of these accessions (Fig. 2) was produced using the first, second, and third principal components, and accounting for 35% of the variance. Principal component one was comprised mainly of SSR markers CH01h10, CH03d07, GD147, and CH01f03b. Principal component two was mainly comprised of markers CH02c09, CH04e05, and CH01f03b1, whereas principal component 3 was mainly comprised of markers CH04e05, CH01f03b, and CH04f10.

A dendrogram of 125 Malus accessions clustered based on genotypic analysis of 10 robust SSR markers into six distinct clusters

A scatter plot of 125 Malus accessions analyzed based on principal components, accounting for 35% of variability detected in this material. Principal component one (Prin1) was comprised mainly of SSR markers CH01h10, CH03d07, GD147, and CH01f03b. Principal component two (Prin2) was mainly comprised of markers CH02c09, CH04e05, and CH01f03b1, while principal component 3 (Prin3) was mainly comprised of markers CH04e05, CH01f03b, and CH04f10
The expected heterozygosity ranged from 0.821 to 0.954, with a mean value of 0.887, while observed heterozygosity ranged from 0.600 to 0.795, with a mean value of 0.715 (Table 2). PIC values ranged from 0.802 to 0.949 with a mean value of 0.875 (Table 2). The range of effective alleles per locus was wide, ranging from 5.507 to 20.489, with an average of 9.791 (Table 2).
Similarities were found both by pedigree analysis and by previously reported genetic relatedness studies. As expected, “McIntosh” sports “Kimball McIntosh,” “Marshall McIntosh,” and “Wijcik McIntosh” were genetically identical for all markers, as similarly reported by Hokanson et al. (1998). “Golden Delicious” is a parent of “Gala” (Kouassi et al. 2009) and both closely clustered together.
Multiple groups, including “McIntosh” sports and “Cortland,” “Ein Shemer” and “Virginia Gold,” “Koningszuur” and “Spokane Beauty,” “Murray” and “Viking,” as well as “Northern Spy” and “Rhode Island Greening” are clustered together in this study, and this is similar to findings reported by Hokanson et al. (1998). “PRI 1484-1,” “PRI 1773-6,” and “E36-7” are clustered at high proximity to each other, and this is similar to findings of Hokanson et al. (2001). Moreover, accessions “PRI 1918-1” and “PRI 2482-100,” “PRI 2050-2,” and “PRI 1346-2,” as well as “Demir” and M. kirghisorum clustered similarly to those reported by Hokanson et al. (2001).
Previously, Hokanson et al. (1998) have reported that “Murray” is distantly clustered from other “McIntosh” accessions. However, in this study, “Murray” is clustered in close proximity to other “McIntosh” accessions, which is to be expected as “Murray” is to known to be a “McIntosh” seedling. In other findings in this study, “Ein Shemer” is separated from both “Golden Delicious” and “Gala”; “Northern Spy” is separated from “Jonafree”; “Irish Peach” is separated from “Keepsake”; and “Korichnoe Polosatoje” is separated from “Winter Majetin.” Previously, Hokanson et al. (1998) have reported instead that the above groups of accessions are clustered together. Among other discrepancies, M. × soulardii and “Arnold Crab” are found in different clusters by Hokanson et al. (2001), while these have clustered similarly in this study. This is also the case with “Hansen’s #2” and Malus halliana (PI 589972), “Novosibirski Sweet” and “White Angel,” as well as “Robert’s Crab” and “Inuringo.” Such discrepancies could be due to use of either different or possible mislabeling in one of these collections. A solution to mislabeling has been proposed via the use of common DNA samples from a single source as control across different studies, which could then be used to check for trueness-to-type (Evans et al. 2009).
Another possible explanation for the above observed differences could be attributed to the different statistical analyses used in these two studies. While the unweighted pair-group method has been used by Hokanson et al. (1998), Ward’s minimum variance method is used in this study. The unweighted pair-group method is a hierarchical method of creating clusters, which results in a nested dendrogram. Additionally, the unweighted pair-group method assumes equal rates of evolution between lineages, whereas Ward’s minimum variance method is nonhierarchical, and it is designed to obtain clusters with the least amount of within-group variance and the most amount of between-group variance, thus allowing for better separation of distinct groups. It is also likely that these observed differences may be attributed to differences in DNA separation methodologies used in these two studies. Hokanson et al. (1998) have used the ABI 377 or 373 DNA sequencing system (Applied Biosystems, Inc.) for gel-based separation, whereas the ABI 3730xl sequencer (Applied Biosystems, Inc.) for separation via capillary electrophoresis is used in this study. It is likely that capillary electrophoresis separation has resulted in different DNA fragment sizes than is possible with using gel-based separation.
Although there were clusters of accessions with common origin, or research station if the accession was a selection, a sizeable portion of the accessions did not cluster in a meaningful way. Decreased clustering by origin and taxonomy has been exhibited previously as a result of adding species and derived hybrids to smaller cultivar collections (Hokanson et al. 2001; Dunemann et al. 1994).
In this study, “Kansas K14” and an unlabeled accession (planted adjacent to “Kansas K14”) were grouped together with an R 2 value of 1.00, thus suggesting that the unlabeled accession was in fact a duplicate clone of “Kansas K14.” Additionally, two accessions, M. sieversii “KAZ 93-24-01” and M. orientalis “RUS 98 07-01,” unexpectedly grouped together with an R 2 value of 1.00. These two accessions originated from different collections; thus, it is likely that these accessions are likely to have been mislabeled at either the original collection or subsequently in the core collection used in this study.
Taking into account “Kansas K14” and its duplicate unlabeled accession along with other duplicate pairs as well as those genetically identical “McIntosh” accessions, a total of 121 unique genotypes have been identified from 125 Malus accessions that were genetically characterized using the final set of markers. Using a set of ten robust SSR markers, it was possible to differentiate all accessions. All other additional markers were useful for confirmation of these findings. Moreover, two accessions with identical PI numbers (PI 633920 M. sieversii “KAZ 96 09-05”), but at different locations (K1-17-15 and K1-17-7), were not genetically similar as they were clustered in different groups, clusters 3 and 6, respectively.
Although six clusters generated by the scatter plot overlapped and were not clearly distinguishable, it should be noted that the scatter plot was a three-dimensional graphical representation of these data and accounted for only 35% of the existing variability. As only three of the PCs were used for visualization, it was assumed that clusters would be clearly differentiated in the ten-dimensional space required to observe most of the variability (Johnson 1998).
A relatively high allelic diversity has been reported in studies that included wild Malus species (Hokanson et al. 2001; Richards 2009; Zhang et al. 2011). Using a different set of SSRs on a slightly smaller number of similar accessions, Hokanson et al. (2001) have reported slightly higher numbers of effective alleles than that found in this study. In contrast, two previous studies evaluating only apple cultivars have identified significantly lower numbers of effective alleles (Hokanson et al. 1998; Garkava-Gustavsson et al. 2008). This is an expected finding due to lower levels of genetic diversity present in modern cultivars (Noiton and Alspach 1996).
Overall, levels of heterozygosity detected in this study were sometimes different than those reported previously. Although Hokanson et al. (1998, 2001) have used a similar collection, they reported lower levels of heterozygosity than that detected in this study. This could be attributed to the use of different marker sets, especially since markers used in this study had higher PIC values than those used in earlier studies (Table 2). In contrast, three studies focusing on apple cultivars (Liebhard et al. 2002; Kitahara et al. 2005; Garkava-Gustavsson et al. 2008) displayed higher levels of heterozygosity than that reported in this study. During cross-hybridization and selection efforts in crop improvement, higher levels of heterozygosity are generated (Lamboy and Alpha 1998). As the Malus core collection used in this study included wild Malus species, heterozygosity levels were lower than what would generally be observed in collections containing only domesticated apples.
As expected, this diverse core collection of Malus germplasm showed high allelic diversity. Although fewer rare alleles were found than in previous studies, 39% of the alleles detected in this collection were only present in five or fewer accessions, indicating alleles to maintain genetic diversity (Richter et al. 1994; Bengtsson et al. 1995). Genetic relatedness, as determined by cluster analysis, showed both similarities and dissimilarities to previously studies. One unlabeled accession was discovered to be a replicate of another accession, “Kansas K14,” and two accessions thought to be duplicate were not genetically identical. The set of ten SSR markers were sufficient to differentiate all accessions in the core collection except for three “McIntosh” sport mutations, the determined “Kansas K14” replicate, and a duplicate accession. Heterozygosity was lower than studies focusing on M. × domestica cultivars, likely due to increased heterozygosity as a result of selection (Lamboy and Alpha 1998).
References
Baraket G, Chatti K, Saddoud O, Ben Abdelkarim A, Mars M, Trifi M, Hannachi A (2011) Comparative assessment of SSR and AFLP markers for evaluation of genetic diversity and conservation of fig, Ficus carica L., genetic resources in Tunisia. Plant Mol Biol Rep 29:171–184
Bengtsson B, Weibull P, Ghatnekar L (1995) The loss of alleles by sampling: a study of the common outbreeding grass Festuca ovina over three geographical scales. Hereditas 122:221–238
Benson L, Lamboy W, Zimmerman R (2001) Molecular identification of Malus hupehensis (tea crabapple) accessions using simple sequence repeats. HortScience 36:961–966
Botstein D, White R, Skolnick M, Davis R (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Human Genet 32:314–331
Brown A (1989) The case for core collections. In: Brown AHD, Frankel OH, Marshall DR, Williams JT (eds) The use of plant genetic resources. Cambridge University Press, Cambridge, pp 136–156
Brown A (1995) The core collection at the crossroads. In: Hodgkin T, Brown AHD, Hintum TJL, Morales EAV (eds) Core collections of plant genetic resources. Wiley, Chichester, pp 3–19
Chen X, Xia Z, Fu Y, Lu C, Wang W (2010) Constructing a genetic linkage map using an F1 population of non-inbred parents in Cassava (Manihot esculenta Crantz). Plant Mol Biol Rep 28:676–683
Clarke J, Tobutt K (2009) A standard set of accessions, microsatellites and genotypes for harmonising the fingerprinting of cherry collections for the ECPGR. Acta Hort 814:615–618
Dávila J, Sánchez de la Hoz M, Loarce Y, Ferrer E (1998) The use of random amplified microsatellite polymorphic DNA and coefficients of parentage to determine genetic relationships in barley. Genome 41:477–486
Dunemann F, Kahnau R, Schmidt H (1994) Genetic relationships in Malus evaluated by RAPD ‘fingerprinting’ of cultivars and wild species. Plant Breed 113:150–159
Evans K, Fernández F, Laurens F, Feugey L, Van de Weg E (2007) Harmonizing fingerprinting protocols to allow comparisons between germplasm collections. Eucarpia XII Fruit Selection Symposium, September 16–20, 2007, Zaragoza, Spain, pp 57–58
Evans K, Fernández F, Govan C (2009) Harmonising fingerprinting protocols to allow comparisons between germplasm collections—Pyrus. Acta Hort 814:103–106
Forsline P (1996) Core subsets in the USDA/NPGS with Apple as an example. Proceedings of the 2nd Workshop on Clonal Genetic Resources, Ottawa, Ontario, January 23–24, 1996. pp 172–175
Frankel O (1984) Genetic perspectives of germplasm conservation. In: Arber WK, Llimensee K, Peacock WJ, Starlinger P (eds) Genetic manipulation: Impact on man and society. Cambridge University Press, Cambridge, pp 161–170
Garkava-Gustavsson L, Brantestam A, Sehic J, Nybom H (2008) Molecular characterisation of indigenous Swedish apple cultivars based on SSR and S-allele analysis. Hereditas 145:99–112
Gasic K, Han Y, Kertbundit S, Shulaev V, Iezzoni A, Stover E, Bell R, Wisniewski M, Korban SS (2009) Characteristics and transferability of new apple EST-derived SSRs to other Rosaceae species. Mol Breed 23:397–411
Goulão L, Cabrita L, Oliveira C, Leitao J (2001) Comparing RAPD and AFLPTM analysis in discrimination and estimation of genetic similarities among apple (Malus domestica Borkh.) cultivars—RAPD and AFLP analysis of apples. Euphytica 119:259–270
Govan C, Simpson D, Johnson A, Tobutt K, Sargent D (2008) A reliable multiplexed microsatellite set for genotyping Fragaria and its use in a survey of 60 F. × ananassa cultivars. Mol Breed 22:649–661
Guilford P, Prakash S, Zhu J, Rikkerink E, Gardiner S, Bassett H (1997) Microsatellites in Malus xdomestica (apple): abundance, polymorphism and cultivar identification. Theor Appl Genet 94:249–254
Han Y, Korban SS (2010) Strategies for map-based cloning in apple. Crit Rev Plant Sci 29:265–284
Hearne C, Ghosh S, Todd J (1992) Microsatellites for linkage analysis of genetic traits. Trends Genet 8:288–294
Hokanson S, Szewc-McFadden A, Lamboy W, McFerson J (1998) Microsatellite (SSR) markers reveal genetic identities, genetic diversity and relationships in a Malus xdomestica Borkh. core subset collection. Theor Appl Genet 97:671–683
Hokanson S, Lamboy W, Szewc-McFadden A, McFerson J (2001) Microsatellite (SSR) variation in a collection of Malus (apple) species and hybrids. Euphytica 118:281–294
Johnson D (1998) Principal component analysis, and cluster analysis. In: Johnson DE (ed) Applied multivariate methods for data analysts. Brooks/Cole, Pacific Grove, pp 319–396
Kalinowski S, Taper M, Marshall T (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106
Király I, Tóth M, Pedryc A, Halász J, Deák T (2009) Parent identification of Hungarian apple cultivars using SSR markers. Acta Hort 839:471–477
Kitahara K, Matsumoto S, Yamamoto T, Soejima J, Kimura T, Komatsu H (2005) Molecular characterization of apple cultivars in Japan by S-RNase analysis and SSR markers. J Amer Soc Hort Sci 130:885–892
Kobayashi N, Horikoshi T, Katsuyama H, Handa T, Takayanagi K (1998) A simple and efficient DNA extraction method for plants, especially woody plants. Plant Tiss Cult Biotech 4:76–80
Kouassi A, Durel C, Costa F, Tartarini S, van de Weg E, Evans K (2009) Estimation of genetic parameters and prediction of breeding values for apple fruit-quality traits using pedigreed plant material in Europe. Tree Genet Genomes 5:659–672
Kresovich S, McFerson J (1992) Assessment and management of plant genetic diversity: considerations of intra- and inter-specific variation. In: Kresovich S (ed.) The impact of progress in genetics on plant resources (germplasm) conservation and utilization. Field Crops Res 29:185–204
Lamboy W, Alpha C (1998) Using simple sequence repeats (SSRs) for DNA fingerprinting germplasm accessions of grape (Vitis L.) species. J Amer Soc Hort Sci 123:182–188
Liebhard R, Gianfranceschi L, Koller B, Ryder C, Tarchini R, Van de Weg E (2002) Development and characterisation of 140 new microsatellites in apple (Malus × domestica Borkh.). Mol Breed 10:217–241
Marshall D (1990) Crop genetic resources: current and emerging issues. In: Brown AHD, Clegg MH, Kahler AL, Weir BS (eds) Plant population genetics, breeding, and genetic resources. Wiley, Chichester, pp 367–388
Morgante M, Rafalski A, Biddle P, Tingey S, Olivieri A (1994) Genetic mapping and variability of seven soybean simple sequence repeat loci. Genome 37:763–769
Naik S, Hampson C, Gasic K, Bakkeren G, Korban S (2006) Development and linkage mapping of E-STS and RGA markers for functional gene homologues in apple. Genome 49:959–968
Noiton D, Alspach P (1996) Founding clones, inbreeding, coancestry, and status number of modern apple cultivars. J Amer Soc Hort Sci 121:773–782
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Heredity 86:248–249
Richards C (2009) Genetic diversity and population structure in Malus sieversii, a wild progenitor species of domesticated apple. Tree Genet Genomes 5:339–347
Richter T, Soltis P, Soltis D (1994) Genetic variation within and among populations of the narrow endemic, Delphinium viridescens (Ranunculaceae). Amer J Bot 81:1070–1076
Rousset F (2008) Genepop'007: a complete reimplementation of the Genepop software for Windows and Linux. Mol Ecol Res 8:103–106
Russell J, Fuller J, Macaulay M, Hatz B, Jahoor A, Powell W, Waugh R (1997) Direct comparison of levels of genetic variation among barley accessions detected by RFLPs, AFLPs, SSRs and RAPDs. Theor Appl Genet 95:714–722
Wang Y, Zhang J, Sun H, Ning N, Yang L (2011) Construction and evaluation of a primary core collection of apricot germplasm in China. Scient Hort 128:311–319
Zhang Q, Li L, Zhao Y, Korban S, Han Y (2011) Evaluation of genetic diversity in Chinese wild apple species along with apple cultivars using SSR markers. Plant Mol Biol Rep. doi:10.1007/s11105-011-0366-6
Acknowledgments
This research was supported by Pioneer Hi-Bred International, Inc., Illinois Plant Breeding Center, USDA-NIFA-SCRI grant AG 2009-51181-06023, University of Illinois Office of Research Project 875-325, and University of Illinois Office of Research Project 875-922.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Potts, S.M., Han, Y., Khan, M.A. et al. Genetic Diversity and Characterization of a Core Collection of Malus Germplasm Using Simple Sequence Repeats (SSRs). Plant Mol Biol Rep 30, 827–837 (2012). https://doi.org/10.1007/s11105-011-0399-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-011-0399-x