
Abstract
Selection and validation of appropriate reference genes should be the first step to consider in experiments based on quantitative real-time polymerase chain reaction (qRT-PCR). In this study, ten candidate genes were investigated for their stability as suitable reference genes in qRT-PCR data normalization using a diverse set of 12 Coffea cDNAs from plants from three different species/genotypes exposed to single or multiple abiotic stresses (drought and chilling, alone or in combination). Primer amplification efficiencies were calculated for all of the selected genes and varied according to each individual genotype. The expression of each gene was measured by qRT-PCR to evaluate its stability. A multiple analytical approach was followed, based on consensus merged data from four different complementary statistics, namely geNorm, BestKeeper, NormFinder, and coefficient of variation, which produced comparable but not identical results. According to this approach, the most suitable sets of reference genes for data normalization in the five experimental datasets are (1) total assay: GAPHD, Cycl, and UBQ10; (2) genotype: GAPDH, UBQ10, Ap47, and EF-1A; (3) cold stress: UBQ10, GAPDH, ACT, and EF-1A; (4) drought stress: GAPDH, ACT, EF1A, and Apt; and (5) multiple stress: UBQ10, GAPDH, ACT, and elf-4A. Normalization of gene expression using these selected genes was validated by examination of the expression of the photosynthetic-related ApoA2 gene in samples from non-stressed and stressed plants. Our results are useful to assist studies on Coffea physiology with the aim of breeding for increased tolerance to abiotic stress conditions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Coffee is a major crop of the world’s agricultural sector, supporting the economy of many countries from tropical regions. To assure its viability and sustainability, the use of genotypes with the ability to cope with environmental constraints is mandatory. Previous reports have highlighted that, within the Coffea genus, stress responses to energy overpressure related to cold, drought, and high irradiance conditions implicate acclimation changes that promote the tolerance of the photosynthetic apparatus (Ramalho et al. 1999, 2000, 2003; Batista-Santos et al. 2011) due to improved membrane stability and to the reinforcementof the anti-oxidative system (Fortunato et al. 2010; Lima et al. 2002; Partelli et al. 2009; Pinheiro et al. 2004; Ramalho et al. 1998). These differences to survive in environmental limiting areas make breeding for inter-species hybrids a promising strategy to explore in order to obtain plants adapted to a variety of climates and ecological units with regular productions and high yields. To accomplish such goal, it is fundamental to acquire knowledge on the physiological basis of the distinct adaptation mechanisms and to identify molecular cues associated with abiotic stress tolerance.
Understanding the complex relationship between the traits affecting a given relevant physiological response can be tackled by tracing the changes in the expression of responsible genes, which, although sometimes apparently small, often have high relevance to the knowledge of the underlying biological mechanisms. Quantitative real-time PCR (qRT-PCR) is presently the method of choice to monitor gene expression profiles in multiple samples involving a limited number of genes of interest, and to validate the results from high-throughput gene expression techniques, such as microarrays or next-generation sequencing-based methods, whose analyses are restricted to a limited number of experimental samples (Czechowski et al. 2005). qRT-PCR is an extremely accurate technique due to its high sensitivity, specificity and reproducibility, wide dynamic range (over 107-fold), high throughput capacity, and independence of post-PCR processing (Bustin et al. 2005). Despite that, qRT-PCR reliability depends on careful experimental designs that can assure adequate legitimacy of the biological significance of the mRNA quantifications. True gene-specific variation can only be obtained after correction of the expression data of target genes to remove non-biological sample-to-sample variations that may derive from intrinsic features of the qRT-PCR technique at a number of stages throughout the experimental protocol (Bustin et al. 2005; Phillips et al. 2009). Accurate normalization of the transcript expression level of a target gene to the expression level of internal reference genes has been considered the most appropriate solution to validate qRT-PCR results (Bustin et al. 2005; Vandesompele et al. 2009).
Suitable reference genes must be stably expressed in a wide variety of tissues or cell types and must be unaffected by the experimental conditions and treatments introduced. Traditionally, housekeeping genes have been chosen to be used as reference genes (e.g., Li et al. 2010; Chen et al. 2011). Housekeeping genes are coding genes that play a general role in basic cellular processes such as cell structure maintenance and basic primary cellular metabolism, and are assumed not to be influenced by external factors. However, increasing evidences show that the mRNA level of commonly used housekeeping genes is often not constant among individuals, tissues, or experimental conditions (Barber et al. 2005), so universally valid reference genes that can potentially account for all sources of variability are unlikely to exist. The reasons for the expressional variability of housekeeping genes include their possible participation in other cellular processes in addition to basic cell metabolism, or the occurrence and participation of different isoforms encoded by several genes in multigenic families (Hochstrasser 2000; Katsuyoshi et al. 1996).
Since most experiments are restricted to few different tissues of treatments, conducted under controlled conditions and varying one factor at the time, it is likely that one or more genes will be stably expressed in particular and limited experimental designs. Although useful to understand gene function in the laboratory, such circumstances never occur in the field, where interactions and unexpected factors usually affect the pattern and the overall degree of transcriptional responses, including for housekeeping genes. In particular, abiotic stresses generally occur simultaneously, and the plant responses and coping mechanisms may differ according to which stress or combination of stresses it is experiencing. Therefore, the molecular and metabolic responses to a given combination should not be directly extrapolated from the response exhibited to each stress applied separately (Mittler 2006). Hence, a careful evaluation of the behavior of candidate reference genes in every individual experimental setup is required since reference genes with a stable expression under a certain condition may not be suitable to normalize gene expression in another condition, particularly when multiple variables are involved.
On the other hand, as stated before, comparison of genotypes from different species displaying contrasting survival responses under stress is advantageous to understand the underlying physiological mechanisms and to plan effectual breeding approaches. Natural sequence variation in target genes is expected in genotypes belonging to different species and, even in reduced number, is likely to affect PCR efficiency. Although it is not widely acknowledged, there is an intimate association between quantification and amplification efficiency. Neglecting this aspect can be dramatically misleading when reference genes and target genes are selected and analyzed.
Taking these aspects in consideration, we provide a set of reference genes to be used in experiments of Coffea gene expression, involving cold stress, drought stress, taken individually or together (multiple stress), and including genotypes from different species. To our knowledge, this is the first study that evaluates the suitability of potential reference genes for gene expression analyses in coffee involving different species and multiple abiotic stresses. Additionally, we use a combination of different algorithms to effectively select appropriate reference genes.
Materials and Methods
Plant Material and Growth Conditions
The experiments were carried out using potted 18-month-old plants from the genotypes ‘Icatu’ (IAC 2944—Coffea canephora × Coffea arabica, widely considered as C. arabica due to the large backcross with C. arabica genotypes), C. canephora cv. ‘Apoatã’ (IAC 2258) and C. arabica cv. ‘Obatã’, which are frequently used in breeding programs. The assay was conducted in walk-in growth chambers (10000 EHHF, ARALAB, Portugal) with photoperiod set to 12 h, RH to 65–70%, and irradiance to ca. 750–850 μmol m−2 s−1 at the upper part of the canopy. Plants were divided into two groups: the control group was maintained fully irrigated (ca. 80% of the field capacity) and the drought stress group was maintained at ca. 15% of the field capacity. After the imposition of the irrigation treatments, plants were submitted successively to (1) a gradual temperature decrease (0.5°C per day) from 25/20°C to 13/8°C (day/night), over 24 days, (2) a 3-day chilling cycle (3 × 13/4°C), where the plants were subjected to 4°C during the night and in the first 4 h of the morning, followed by a rise to 13°C applied throughout the rest of the diurnal period. Subsequently, re-warming conditions (25/20°C) were applied for 7 days and, afterwards, re-watering took place. Leaf material for expression studies and physiological determinations was collected from the two top pairs of recent mature, fully expanded leaves from both plagiotropic and orthotropic branches, in the upper part of eight to ten plants per genotype. Leaf samples were collected after 2 h of diurnal light exposure, at 25/20°C from well-watered plants (no stress, NS) and drought-stressed plants (drought stress, DS), and after the chilling treatment from well-watered plants (cold stress, CS) and drought-stressed plants (multiple stress, MS). Similar leaves were collected for all samples and conditions and immediately ground independently in liquid nitrogen and stored at −80°C until RNA extraction.
Measurements of Physiological Parameters
Leaf net photosynthetic rates (P n) were measured under steady-state conditions (after ca. 2 h of light exposure), using a CO2/H2O open system portable infra-red gas analyzer (CIRAS I, PP Systems, UK) with external CO2 set to 380 μl CO2 l−1. Measurements of oxygen evolution, expressing the photosynthetic capacity, A max, were performed on 1.5-cm2 discs from the same leaves, under light (PPFD 750 μmol m−2 s−1) and CO2 (ca. 7%) saturating conditions, at 25°C, in a Clark-type leaf disc O2 electrode (LD2/2; Hansatech, Kings Lynn, UK). Saturating PPFD was provided by a Björkman lamp (Hansatech).Water potential determinations (Ψ w) were performed in leaves collected at predawn, at the end of night period, with a Schölander pressure chamber (Model 1000, PMS Instrumental Company), according to Schölander et al. (1965).
RNA Isolation and cDNA Synthesis
Total RNA for each of the 12 experiments [three genotypes, each under four conditions (NS, CS, DS, and MS)] was isolated using the RNeasy Plant Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol and digested with DNAse I using on-column Qiagen DNAse set to remove putative genomic DNA, as per manufacturer’s instructions.
Intactness of the RNA extracted was verified by 1% agarose–TBE gel electrophoresis containing ethidium bromide, by evaluating the integrity of the 28S and 18S ribosomal RNA bands and absence of smears. All RNA samples were individually analyzed for the possible presence of DNA contamination by standard PCR reactions (35 cycles) using primers designed for ubiquitin (UBQ) gene (Table 2), in the absence of cDNA synthesis. Total RNA concentration and purity were further verified through NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies; Thermo Scientific) measurements to guarantee the same amount of starting material in subsequent cDNA synthesis. RNA concentrations ranged between 65.5 and 246.5 ng/μl, with OD 260:280 ratios always above 1.98. Six hundred and fifty micrograms of DNA-free total RNA was used to synthesize first-strand cDNAs using oligo-(dT)18 primers and the SuperScript II first-strand synthesis system, according to Invitrogen’s recommendations. Before qRT-PCR assays, each RT reaction was verified through a control standard RT-PCR, as before, to check for the amplification of the UBQ gene. All the RT-PCR reactions produced a single UBQ-specific band with the predicted molecular size (ca. 200 bp) on 1.5% agarose gels.
Selection of Reference Gene Sequences and Primer Design
A set of ten candidate genes were selected, comprising several conventional, commonly used reference genes in plants and others based on previous reports in Coffea (Cruz et al. 2009): actin, tubulin, ubiquitin, glyceraldehyde 3-phosphate dehydrogenase, eukaryotic initiation factor 4α (Elf4A), elongation factor 1α (EF1A), clathrin adaptor complexes subunit (AP47), adenine phosphoribosyltransferase (Apt), cyclophilin, and 40S ribosomal protein (S24). These selected genes belong to different functional classes to minimize the chance of co-regulation (Table 1).
Primers for the genes selected based on previous works in Coffea (Cruz et al. 2009) were used as published. Sequences for the other genes were obtained via searches against Coffea ESTs (expressed sequence tag) libraries on NCBI databases (Vidal et al. 2010; Mondego et al. 2011) (accession numbers are given in Table 2). All primers were designed using Primer3 software (Rozen and Skaletsky 2000). The length of the primers was set to be between 20 and 23 bp, with a GC content ranging from 45% to 60% and a melting temperature (T m value) between 62°C and 65°C. Amplicon length ranges were set to be between 80 and 150 bp. The probability of formation of hairpin structures and primer dimerization was subsequently checked using the Oligo Calculator (ver. 3.26) algorithm (Kibbe 2007). Primer sequences are given in Table 2.
Quantitative Real-Time PCR Conditions
qRT-PCR reactions were performed in 96-well plates using SYBR Green Master Mix (Fermentas). Reactions were prepared in volumes of 25 μl containing 150 ng of cDNA and 3 μM each primer, in 1× SYBR Green Master Mix (Fermentas, Vilnius, Lithuania). Reactions were run on the iQ™5 Real-Time Detection System (BioRad) using the following parameters: hot start activation of the TaqDNA polymerase at 95°C for 10 min, followed by 40 cycles of (denaturation 95°C for 15 s, annealing at 60°C for 30 s, elongation at 72°C for 30 s, and plate read). To verify the specificity of each amplification and the absence of primer dimers, dissociation curves were obtained for each amplicon at the end of the PCR run, by continuous fluorescence measurement from 55°C to 95°C, with sequential steps of 0.5°C for 15 s. Single peaks were obtained for every primer pair (representative traces are showed in Online Resource Fig. S1). Each reaction was prepared in at least triplicates and the mean Ct was used for data analyses. The full sample set was always included in each technical replicate to exclude any artifacts consequential of between-run variations. No signals were detected in non-template controls run in parallel for each primer set.
Calculation of PCR Efficiencies
Five-fold serial dilutions (1:1–1:625), spanning five orders of magnitude from pooled cDNAs that included equal-molar quantities of all samples independently for each genotype, were quantified in triplicates to generate standard curves for each primer pair. Based on the slopes of the standard curves, the PCR efficiency of each gene, for each genotype, was determined from the respective logarithm of the cDNA dilution, plotted against the mean threshold cycle (Ct) values. The reaction efficiency was calculated using the equation: E(%) = (10−(1/slope) − 1) × 100, where E is the efficiency, in percentage, and slope is the gradient of the best-fit line in the linear regression.
Gene Expression Stability and Rank Analysis
Global variability of gene expression for each gene was investigated via standard statistical parameters using Box Plot template for Excel software (Vertex42, LLC). The suitability of candidate reference genes was then evaluated by four different statistical approaches in order to determine the stability of the potential reference genes expression. Variation of gene expression was assessed by means of coefficient of variation (CV) calculations (CV = σ/μ), and three visual Basic Application (VBA) applets for Microsoft Excel: geNorm v. 3.5 (Vandesompele et al. 2002), BestKeeper v. 1.0 (Pfaffl et al. 2004), and NormFinder v. 0.953 (Andersen et al. 2004) as described in their respective manuals. In all cases, data was exported to Excel, and converted to appropriate input files, according to the requirements of each applet used. Ct data were either directly used for stability calculation (when using BestKeeper) or were first transformed into relative quantities via the ΔCt method (Livak and Schmittgen 2001), using the sample with the lowest Ct as calibrator and incorporating the calculated amplification efficiencies for each primer/genotype (in geNorm and NormFinder analyses), according to the formula: Q = E Δ(Ct), where Q are quantities and E is the efficiency calculated as stated above.
Merged Results: Consensus Ranking of Ordered Genes
Consensus rank of the ordered candidate reference genes was established by means of a non-weighted unsupervised rank aggregation method: RankAggreg v. 0.4-1 (Pihur et al. 2009) for R (v. 2.9.2; http://www.R-project.org). The input file was a matrix of rank-ordered genes according to the four stability measurements calculated for each dataset. The merged rank list was obtained from calculated Spearman footrule distances and the Cross-Entropy Monte Carlo algorithm to compare the relationship of the relative ordering of genes by different methods of analysis.
Determination of the Minimum Number of Reference Genes
Based on the rank order obtained after RankAggreg analyses, pairwise variations (V values) were calculated for each dataset to establish the minimum number of reference genes needed for an accurate normalization of data. In summary, V n / n + 1 is calculated between each set of two sequential NF (starting with the relative expression values of the two most stable genes, as ordered by RankAggreg) for all samples in each dataset. An array is calculated consisting of the log2-transformed NF ratios of every sequential combination of two NF in each sample. Finally, SD of the array data for each NF combination is calculated (V n / n + 1) and plotted to illustrate changes in expression stability of NF in comparison to the number of genes employed.
Quantification of the mRNA Transcription of the ApoA2 Gene
To verify how the normalization of the expression data for a gene of interest is affected by the use of different reference genes, the same 12 plant cDNA samples used for the stability analyses of reference genes were also analyzed by qRT-PCR for the expression of ApoA2, which encodes a reaction center protein of photosystem (PS) I. qRT-PCR reaction mixes and amplifications were done as described for reference gene expression and the sequences of the primer pair used are given in Table 2. ApoA2 relative gene expression values were normalized according to the criteria described in the “Results” sub-section and are given as mean value ± SE (standard error). SE of normalized expression levels were calculated according to the formula: SE = GInorm × ((SDNF/NF)2 + (SDGI/GI)2)0.5/m 0.5, where GInorm is the normalized relative expression of the gene of interest, SDNF is the standard deviation of the normalization factor, NF is the normalization factor, SDGI is the standard deviation of the quantities of the gene of interest, GI is the quantity calculated for the gene of interest, and m is the number of replicates, as described in Vandesompele et al. (2002).
Results
Plant Responses to Abiotic Stress Imposition
All genotypes were affected by drought as observed in gas exchange measurements, and all stressed plants showed severed wilted leaves when compared to well-watered plants. The decrease on water availability led to lower leaf water potential on the water-stressed plants (Table 3), and although the water content of plants had not been significantly affected by cold stress (Table 3), it impacted leaf shedding in a genotype-dependent way (data not shown). Moreover, a reduction in both photosynthetic capacity and photosynthetic rate was observed (Fig. 1a, b). These results confirm that plants used to sample RNA material were under actual stress.

Changes in physiological parameters with stress imposition form sample characterization. Changes in the values of (a) photosynthetic capacity (A max) and (b) photosynthetic rate. Values are given for no-stress plants (NS), cold stress (CS), drought stress (DS), and multiple stress (MS). Each value represents the mean of four measurements. Bars show the standard errors
Amplification Specificity and Efficiency
To examine the expression stability of the potential reference gene selected, transcript levels of the ten candidates (Table 1) were measured by qRT-PCR using gene-specific primer pairs (Table 2). Gene-specific amplification of each of the ten candidate genes was confirmed by the presence of a single peak in the dissociation curve analyses (Online Resource Fig. S1). Amplification efficiencies of every candidate gene were calculated individually for each genotype tested to investigate if primer efficiency varies accordingly to the genome background. All calibration curves exhibited significant linear relationships (coefficients of determination ranged from 0.983 to 0.999) between the fractional cycle number and the log of the initial copy number (Table 2). Each set of primers showed adequate efficiency percentages to be used in qRT-PCR experiments, with an overall efficiency that varied from 78% for EF-1A to 107% for GAPDH, both in ‘Icatu’ samples (Table 2). Some differences were observed according to the species (Table 2), advising the use of genotype-specific efficiency values for gene expression quantifications.
Expression Profiling of Candidate Reference Genes
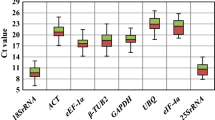
The range and distribution of the Ct values allowed a visualization of the variation of the levels of gene expression among the sample collection investigated. Analyses of the raw expression levels across all samples revealed that the transcription levels of the ten genes are not dramatically influenced by the treatment or genotype (Fig. 2) as gene expression analyses of the ten putative reference genes exhibited a narrow mean Ct value range across the entire experimental samples (Figs. 2 and 3). The variation ranges, medians, and quartiles of the Ct values are shown in Fig. 3 as a box plot. GAPDH was the most highly expressed gene of the set, with a Ct median of 20.2, while Apt displayed the lowest transcript accumulation with a median Ct of 26.3 (Fig. 3). Not only the stability but also the abundance of a reference gene affects the normalized results (Hu et al. 2009). Suitable reference genes should be equivalent in abundance to that of the target gene transcript, whose expression should not be very low (Ct > 30) or very high (Ct < 15) (Andersen et al. 2004). The ten candidate genes investigated here showed Ct levels between ca. 20 and 30 PCR cycles, which are comparable with usual target mRNA transcripts, allowing accurate subtraction of the baseline values in subsequent qRT-PCR data analyses.

Expression levels of the candidate reference genes across all experimental samples. Values are qRT-PCR quantification cycle numbers (Ct values). The lower the Ct value, the higher the expression of the gene. Each Ct is the average of three replicates

Box plot representation of expression profiling range of each candidate reference gene. The ten genes were examined in 12 samples. The median Ct values are shown as lines, 25th to 75th percentile as boxes, and the range of Ct values as whiskers. Coefficient of variance (CV) of each gene among all samples is given in percentage
Analysis of the gene transcription variation between the samples investigated also offers a first picture of its expression stability. S24, Cycl, and elf-4A showed the narrowest Ct ranges among the different samples and therefore the least deviation from the Ct median (with CVs of 2.03%, 6.22%, and 2.94%, respectively). In contrast, Ap47, Apt, and EF-1A were the most variable ones (with CVs of 5.46%, 5.59%, and 5.02%, respectively). The high CV observed for Cycl is explained by the presence of two outliers in the data.
Gene Expression Stability Data Analysis
The expression stability of the selected reference genes was addressed by four different algorithms: GeNorm, BestKeeper, NormFinder packages, and the calculated CV. The results were analyzed by dividing the data into five different experimental datasets: all samples in the assay, according to individual stresses (cold or drought stress), to multiple stress (cold and drought imposed simultaneously), and to genotype.
geNorm
The geNorm algorithm calculates the gene expression stability measure (M value) for each gene based on its average pairwise expression ratio relative to each of the other genes in the analysis. A gene displaying a high M value has a high variance in its expression. All candidate reference genes, except TUB in the genotype series, displayed acceptable stability values lower than 1.5 (Vandesompele et al. 2002). Consequently, TUB was excluded from this specific dataset. A typical automatic geNorm analyses does a stepwise exclusion of the least stable gene and recalculates M values until only two genes are left, these being the most stably expressed (those results are provided in Online Resource Fig. S2 as an indication, but were not considered in the methodology adopted). This approach has as advantage the possibility of a fine-tuned control of variations in the amount of template cDNA because fluctuations linked to technical variability affect equally both paired genes. However, the possibility of occurrence of co-regulated genes may bias the results since it would be expected to show a lower level of pairwise variation than independently regulated genes (Vandesompele et al. 2002). Due to the risk of co-regulation related to the imposition of stress conditions, in this study we used geNorm only to calculate M values when the entire set of candidate reference genes was considered (except TUB in the genotype set, as stated). Moreover, the results obtained with geNorm are largely dependent of the sample set and the exclusion of a single sample leads geNorm to alter the classification of a gene from unstable to a top-ranked gene (Silberberg et al. 2009), suggesting a lack of robustness of this algorithm compared with other methods (Silberberg et al. 2009). In fact, the geNorm software assigns the M values assuming that two optimal reference genes exist within all experimental samples, regardless of differences that may exist within an experiment. Our results show that geNorm indicated GAPDH and UBQ10 as reliable reference genes for experiments in Coffea. These two genes were the top-ranked ones in all series, except for genotype, where UQB10 still ranked third (Table 4). The less stable genes varied with the experiment, although TUB was consistently among the lower stable genes (Table 4).
BestKeeper
The stability of gene expression for individual reference genes is evaluated by BestKeeper based on standard deviation (SD), coefficient of correlation (r), and percentage covariance of data (Pfaffl et al. 2004). Variations and pairwise correlations in the expression levels are calculated between all possible candidates and well-correlated genes are combined into an index (BestKeeper Index) representing the average of the most stable candidates (Pfaffl et al. 2004). A high Pearson’s coefficient of correlation between a reference gene and the index indicates that these gene pairs had very similar overall expression patterns, revealing high stability. In general, BestKeeper indicated S24, elf-4A, EF1-A, and TUB as unreliable reference genes in every experiment (Table 5). ACT, Ap47, GAPDH, and UBQ10 had strong correlation with the BestKeeper Index (r > 0.9), and always ranked among the top five genes (Table 5). Additional descriptive statistics of the BestKeeper analyses are provided in Online Resource Table S1. The variation in expression of some candidate reference genes was greater than two-fold (corresponding to SD >1.0) for Ap47 and Apt in every experimental setup; Cycl in all stress assays and genotype (but not when the whole set of samples was analyzes in the same dataset); ACT in drought stress, cold stress, and genotype; and GAPDH in the genotype series.
NormFinder
NormFinder estimates both the overall expression variation of the candidate reference genes and the variation between sample sub-groups of the sample set and ranks the best gene or pair of genes based on its minimal combined inter- and intra-group expression variation. The intra-group variation is calculated across all samples, whether the inter-group is calculated between groups of samples. In this sense, two different sub-groups were established: genotype or type of stress. The results of the NormFinder analysis applied to our data sets are summarized in Table 6. Genes displaying the lowest stability values are top-ranked. Similarly to BestKeeper, GAPDH, ACT, and UBQ10 (except UBQ10 in the drought stress series) were always included in the four top-ranked genes. Elf-4A consistently ranked poorly in all treatments. Apt and TUB were also among the least stable genes in all series, except in drought stress and multiple stress for Apt and TUB, respectively. Analyzing the dataset grouped by stress or genotype (Table 6) or without designation of stress treatments (data not shown) did not affect the ranking of the genes, although the stability values were modified. When genotypes were used to group the samples, lower stability values were obtained in all genes (Table 6). The NormFinder software also provides the most appropriate combination of two genes and its corresponding stability value (Online Resource Table S2).
Coefficient of Variance
Stability of a particular candidate gene may be inferred by the percentage of variance of the relative quantities calculated from Ct values and has been proposed to be an alternative to NormFinder when the sample set cannot be properly subdivided (Exposito-Rodriguez et al. 2008). As in NormFinder, this approach does not rely on pairwise analyses of stability relative to the entire set of tested reference genes. In this approach, the higher the variation, the less stably the gene is expressed in the experimental setup (Dheda et al. 2004). S24, elf-4A, and UBQ10 showed the least variation in all datasets, ranking always in the top five least variable genes, while ACT, Ap47, and Apt were the most variable ones (Table 7). This is in accordance with the results shown in Fig. 3. In fact, when CV were calculated from raw Ct values, S24 (2.03%), elf-4A (2.94%), and GAPDH (4.26%) also showed the lowest percentage of CV, whereas Ap47 (5.46%), Apt (5.59%), and ACT (5.96) were among the genes with higher CV percentage.
Consensus Ranking of Gene Stability Based on the Four Algorithms
In this work, a certain but not a total agreement was obtained using the four different statistical models (Table 8 for the “whole assay” data). In fact, differences between the different models and assumptions in which the algorithms are based can account for a notable effect on the consequential gene stability ranking.
The methodology suggested in this work was adapted from the approach followed by Mallona et al. (2010): four lists were generated according to rank positions obtained based on the stability calculated by the four statistical approaches and used to produce a consensus aggregate order that defined the optimal ranking of reference genes, according to their stability, in each experimental dataset (Fig. 4). The data yielded by all the strategies merged and used in parallel indicated GAPDH, Cycl, UBQ10, and ACT to be the genes showing the most invariant expression among the candidates evaluated (Fig. 4a). In general, these genes showed also good stability in all of the other setups, after GenAggreg ranking (Fig. 4b–e). In the stress conditions series, differences in the ranking of the candidate reference genes were more pronounced in the drought stress series than in other stress series, particularly due to the comparatively less stability values of UBQ10 and top ranking of the Apt gene in this dataset. On the other hand, the approach that produced most dissimilar ranking was CV (see Table 8 for results when all samples were considered), which can be explained by the influence of outliers in some gene/sample combinations. As shown in Fig. 3, ACT and Cycl, which are often top-ranked with the other algorithms, contain outliers in the data, and therefore are in the ranking generated by CV. Interestingly, S24 and elf-4A are clearly recommended as the two best genes by the CV approach, being among the less stable genes when assessed by geNorm (Table 4), BestKeeper (Table 5), and NormFinder (Table 6).

Rank aggregation of the candidate genes lists. Representations of rank aggregation using the Monte Carlo algorithm with the Spearman footrule distances on rank lists generated from the four algorithms in series a total assay, b genotype, c drought stress, d cold stress, and e multiple stress. Genes are ordered in the plot based on the solved ranked position, according to each stability measurement (gray). In each plot, the mean rank position of each candidate reference gene (black) and the model computed by the Monte Carlo algorithm (red) are shown. The RankAggreg (v. 0.4-1) and gTools (v. 2.6.2) routine packages for R (v. 2.9.2) were used in the analyses
Estimation of the Number of Reference Genes for Normalization
The optimal number of reference genes that should be used for accurate normalization is determined by calculating V values as a pairwise variation (V n /V n + 1) between two consecutively ranked normalization factors (NF) after the stepwise addition of the subsequent more stable reference gene (NF n and NF n + 1), as per Vandesompele et al. (2002), and is included in the geNorm package. However, in the present work, we calculated V values manually from NFs computed based on the consensus ranked list of genes generated by RankAggreg (see above) and not by the geNorm’s automatic analysis. Our findings indicate that a combination of three or four reference genes is suitable for every dataset series investigated in this work (Fig. 5). Using only two reference genes, the calculated V values were generally high (V > 0.2) across all datasets (Fig. 5). When all genotypes and all stress-treated samples were considered together in the same sample set, the use of three reference genes is enough for accurate normalization, as shown by the V 3/4 <0.15 (i.e., less than 15% variation in normalization factors). Furthermore, the addition of a fourth gene increases the variability, so, in this case, the use of the geometric mean of the three most stably expressed genes should be used to normalize gene expression in this dataset.

Determination of the optimal number of reference genes for normalization by pairwise variation analysis, based on the RankAggreg ordered list for each experimental dataset. Average pairwise variations V n / n + 1 are calculated between NF n and NF n + 1. Bars indicate the change in normalization factors after the inclusion of an additional gene. Arrows correspond to the number of genes selected
Conversely, as showed in Fig. 5, a significant decrease in the pairwise variation was observed in all other datasets with the inclusion of a fourth gene, as unveiled by the differences in the V values of V 3/4 and V 4/5, indicating that, in these cases, the normalization factor used to correct gene expression should be calculated as the geometric mean of four reference genes. In the cold- and multiple-stress assays, the calculated V 4/5 values are higher than 0.15. However, the pairwise variation threshold value of 0.15 is not a strict cut-off and the observed trend of changing V values is recognized to be equally informative (Hong et al. 2008; Hu et al. 2009; Reid et al. 2006). Therefore, even in these datasets, the decrease in variability obtained by adding one (multiple stress) or two (cold stress) additional genes does not outweigh the drawbacks of time consumption and additional costs accompanying its inclusion in the analysis.
Validation of the Selected Reference Genes
To evaluate the robustness of the selected reference genes using the strategy proposed, the GAPDH and the Apt genes were selected as representing stable and unstable genes, respectively, to check their expression ratios when normalized with the geometric mean of the three most stable genes, as determined by RankAggreg and V 3/4 values in the “total assay” experiment. The expression pattern of GAPDH shows a stable expression with a relative expression ratio around 1 (red line) across all samples (Fig. 6a). In contrast, when the less stable gene, Apt, was normalized against the same NF, a large variability was observed (Fig. 6b), illustrating the low expression stability of this gene in the sample set.

Relative quantification of selected genes for validation of gene stability measures and normalization strategies. Gene expression of a stable gene (a) (GAPDH) and an unstable gene (b) (Apt), both normalized by the geometric mean of the expression levels of the three reference genes determined to be the best genes for data normalization in the whole experimental set (GAPDH, Cycl, and UBQ10). Relative expression around the arrow line (no-fold change) means stable gene expression. The target ApoA2 gene expression in the 12 samples was normalized by (c) the geometric mean of the three reference genes determined to be suitable for data normalization in the whole experimental set, (d) the four reference genes proper for each individual condition, and (e) the inappropriate gene Apt. Error bars show the standard error of three replicas
The expression profile of a target gene was also used as an example to examine the effect of using different normalization genes on the estimation of its relative expression. Photosynthesis efficiency is drastically reduced due to abiotic stresses. The particular stresses studied in this work (drought and cold) are well known to induce stomatal closure, causing a reduction in the photosyntheticrate (Flexas and Medrano 2002; Zhu 2002), as well as extensive impact on both PS activity, particularly in PSI, upon chilling exposure (Batista-Santos et al. 2011). Therefore, a member of the PSI P700 apoprotein A2 (ApoA2) gene family, which encodes a reaction center protein of PSI, was chosen to be used as an example concerning the effect of different normalization strategies in the results. The transcription of ApoA2 was quantified by qRT-PCR in the same 12 cDNA samples used for selection of reference genes, following the same protocol. The PCR efficiencies for ApoA2 were 93.6% for ‘Icatu’ samples, 104% for ‘Apoatã’, and 98% for ‘Obatã’, with coefficients of determination (r 2) of 0.999, 0.996, and 0.998, respectively. The amplicon size was 100 bp. Melting curves showed a single peak, indicating absence of non-specific amplifications (data not shown).
The ApoA2 gene expression across the entire set of samples used in this work was normalized using three different strategies: (1) the geometric mean of the relative quantities of the three most stably expressed reference genes selected by RankAggreg in the whole-sample set (GAPDH, Cycl, and UBQ10), (2) the four reference genes selected in each individual treatment (genotype—GAPDH, UBQ10, Ap47, and EF-1A; cold stress—UBQ10, GAPDH, ACT, and EF-1A; drought stress—GAPDH, ACT, EF-1A, and Apt; multiple stress—UBQ10, GAPDH, ACT, and elf-4A), and (3) the least stably expressed gene, Apt.
As expected, ApoA2 expression was detected in all samples, with lower transcription rates in samples from cold stress when compared with non-stressed plants (Fig. 6c, d) in all genotypes. This is in accordance with the measured photosynthetic rates, both net (P n) and maximal values (A max) in these samples (Fig. 1) and previous results of coffee acclimation under chilling (Batista-Santos et al. 2011). A reduction in both P n and A max, and in ApoA2 gene expression was similarly observed in drought and multiple stresses, with the exception of ApoA2 gene expression that increases in Apoatã during water deficit conditions. Stomatal aperture in Apoatã (C. canephora) is suggested to be less responsive to decreasing water availability than C. arabica (DaMatta and Ramalho 2006), which can be related to the fact that C. canephora grows in regions with lower drought episodes, hence, exhibit a lower requirement for efficient stomatal control of evaporation in this species. Therefore, the up-regulation of ApoA2in Apoatã may be explained as part of the plant responses to assure a lower impact on PSI integrity and performance, when compared to C. Arabica, when it experiences hydric stress situations. The magnitude of the variations was different according to the specific genotype, suggesting different impacts and reflecting different acclimation abilities among genotypes, thus providing additional support to the validation results. In a second approach, each sample was normalized with the NF build with the geometric mean of the four reference genes determined to be more stable for each treatment, as ranked by RankAggreg and per differences in the pairwise variation values (V values) (see previous sub-sections) (Fig. 6d). The same pattern was generated by the two approaches used for normalization, only with slight differences in the estimated relative transcription abundance (Fig. 6c, d).
When ApoA2 normalization was performed with Apt, a gene ranking poorly according to all individual algorithms, a significant over- or under-estimation of transcription of the gene of interest was observed in some samples, particularly in non-stressed ‘Icatu’ plants and in drought stressed ‘Obatã’ (Fig. 6e). Besides differences in the gene expression patterns, note that the relative expression, reflecting transcript abundance, also showed significant differences, being severely affected by poor selection of the correct reference gene set.
Discussion
Gene expression analysis represents a powerful tool to assist our understanding of metabolic pathways underlying cellular and developmental processes in plants, including coffee (e.g., de Oliveira et al. 2010; Barreto et al. 2011). Housekeeping genes have been used extensively to normalize gene expression data, but it is currently accepted that the stability of potential reference genes must be systematically determined prior to their use (Guenin et al. 2009; Gutierrez et al. 2008). Normalization errors caused by the use of reference genes without prior validation of their stability have been considered the most common error in qRT-PCR experiments and normalization based on a single reference gene can also lead to erroneous expression differences of more than 3- and 6-fold in 25% and 10% of the experiments, respectively (Vandesompele et al. 2009).
In this work, we evaluated a set of potential reference genes to be used in gene expression studies in Coffea experiments that includes genotypes from different Coffea species submitted to single or multiple (drought and cold) abiotic stresses.
Although some candidate reference genes had shown acceptable stability when assessed with each of the four algorithms employed in the analyses, the choice of the specific set and the number of genes to use in gene expression studies is not straightforward if one does not want to consider just a single algorithm, but to retrieve the information supplied by different methods. In fact, the differences observed in the stability-based ranking are likely to reflect the diverse models and assumptions inherent to each model employed in the statistical analysis (Exposito-Rodriguez et al. 2008; Reid et al. 2006; Schmidt and Delaney 2010).
geNorm, BestKeeper, and NormFinder have been the most popular software used for evaluation of candidate reference genes, with a huge preponderance of the former (Vandesompele et al. 2009). geNorm and BestKeeper statistics rely on the principle that the expression ratio of two ideal reference genes is constant in all samples, independently of the experimental conditions, whereas the rational of NormFinder is that the variation in the average of multiple genes is smaller than the variation in individual genes, and that contributions from reference genes with bias for different groups minimize this variation (Vandesompele et al. 2009). NormFinder and CV are based on the analysis of the level of variation in each gene tested rather than pairwise analysis of gene stability relative to a set of potential reference genes; therefore, the stability of each single reference gene is examined independently instead of being relative to the other genes, which allows avoidance of artificial selection of co-regulated genes. This aspect can have large significance considering our narrow knowledge regarding gene co-regulation. Although the genes analyzed in this study are involved in diverse cellular functions, co-regulation should never be excluded in the context of multiple stresses since, in these cases, the limiting conditions may have a broad effect on the expression of multiple classes of genes or may affect a set of assumedly unrelated genes with a similar pattern. In fact, based on gene regulation, it is quite common that several metabolic pathways respond similarly not only under environmental stresses in plants but also in somatic embryogenesis (Lin and Lai 2010) or human aging (Zampieri et al. 2010). In plants, such co-regulation would promote simultaneously a better protection and/or repair of damaged structures/molecules, as well as qualitative changes (e.g., of membrane lipids), leading to a maintenance of efficient functioning of cell metabolism. Among those quite different but concurrent pathways are the components of the antioxidant system, photosynthetic pathway, and those promoting lipid turnover, namely at chloroplast level in Coffea spp. (Fortunato et al. 2010). Hence, despite the higher popularity of geNorm (which is strongly affected if co-regulation occurs), other strategies, like NormFinder, can be more appropriate in some experiments. Notably, in our data, the stability ranking generated by BestKeeper was very close to NormFinder, which relies on different statistical principles, with GAPDH, ACT, and UBQ10 showing higher stability in all datasets, contrasting with regular poor stability for TUB, elf4A, and S24.
On the other hand, BestKeeper examine primarily Ct values, whereas geNorm and Normfinder evaluate relative quantities, with consequences in that PCR efficiency dissimilarities are more likely to affect the stability measurements. Our results reveal that differences concerning amplification efficiencies exist between genotypes belonging to different species, which strengthens the importance of this methodological feature. These differences are probably due to sequence polymorphisms that exist between species, which in Coffea results from the fact that C. arabica is an allotetraploid (Lashermes et al. 1999) and can produce significant effects on the qRT-PCR results (Taris et al. 2008). Even a few or single sequence polymorphism in priming sites are known to affect primer binding and amplification efficiency (Stevenson et al. 2005; Taris et al. 2008). Our results strengthen the need to examine the sequences to be amplified, to accurately determine primer efficiency independently for each species, and to use the species-specific efficiency percentage when estimating gene expression data in assays involving multiple species. Therefore, to compare transcript accumulation in plants belonging to different species from the same genus, we carefully assessed the stability of candidate reference genes based on input data corrected for the amplification efficiency calculated individually in each genotype. Differences in gene expression between the same tissue in different genotypes were previously reported in several species such as Petunia (Mallona et al. 2010), soybean (Hu et al. 2009; Jian et al. 2008), or coffee (Cruz et al. 2009; Singh et al. 2011). However, in these works, the genotypes analyzed belong to the same species; hence, the presence of a number of polymorphisms affecting amplification efficiencies would not be a strong issue.
From the discussed above, to date there is no consensus on which algorithm should be used to evaluate and choose reference genes. Each of the different approaches has it own advantages and pitfalls. Therefore, to pursue an unbiased estimation of the stability of the selected ten candidate reference genes, a multiple analytical approach was followed. The strategy relies on evaluating the dispersion and calculating the expression stability using four methods in parallel, and then ranking the list of ordered stable genes to obtain a consensus stability-based ranking (Mallona et al. 2010). Here, we additionally propose that the number of genes needed to calculate the normalization factors for accurate and reliable data normalization is determined based on this merged ranking of stability from all algorithms considered together (Fig. 7). Typically, selection of reference genes is based solely in the results from one software and, to our knowledge, inference on the number of genes to be used is only made based on geNorm stability values. Our proposed flowchart (Fig. 7) has the advantage of not discarding putatively useful information provided by different algorithms. Merged data from the stability ranked obtained using four strategies, based on a non-weighted unsupervised rank aggregation method, places GAPDH, Cycl, and UBQ10 as the overall three most suitable reference genes when considering the all-the-sample pools, providing the most robust platform for transcript normalization across multiple stresses and multi-species. However, different sub-sets of samples had their own specific best reference genes, as follows: genotype—GAPDH, UBQ10, Ap47, and EF-1A; cold stress—UBQ10, GAPDH, ACT, and EF-1A; drought stress—GAPDH, ACT, EF1A, and Apt; and multiple stress—UBQ10, GAPDH, ACT, and elf-4A.

Schematic diagram flow chart illustrating data analysis. Efficiency values were calculated independently for each species and used to calculate relative quantities. Raw Ct values or relative quantities were used to calculate stability measurements and stability-based ranked lists according to four methods. The four lists were used as input in RankAggreg to obtain a consensus list of ranked genes. Based on the consensus list, the number of gene needed for normalization was determined by pairwise variation analysis
In drought-stressed leaves, although the combined expression of UBQ10 and GAPDH has been previously suggested to be suitableto use in C. arabica, the stability of both individual genes was poorly ranked both by geNorm and NormFinder (Cruz et al. 2009). In that work, S24 was the reference gene with best stability values. Our results ranked S24 poorly under stress conditions, maybe due to the use of more than one species in the analyses. Also in C. arabica under various stages of development and biotic stress, GAPDH showed good stability but polyubiquitin was the least secure gene for normalization (Barsalobres-Cavallari et al. 2009). A combination of GAPDH, Cycl (two genes determined to be stable in our study), and triose-phosphate isomerase (not evaluated in this work), but not UBQ10, proved to be suitable to compare gene expression in shoot tips, using two cultivars (Singh et al. 2011). On the other hand, Marraccini et al. (2011) normalized drought-related qRT-PCR data in Coffea with a single reference gene, choosing either GAPDH or UBQ10, depending on the Coffea species background, consolidating the relevance of the results reported here. In seed development, UBQ-10 was similarly used as a single gene for normalization (Salmona et al. 2007; Joët et al. 2010). These genes show also the widest distribution among Coffea spp. cDNA libraries (Mondego et al. 2011).
Genes related to the ubiquitin degradation process proved to be among the most stable ones in Arabidopsis (Czechowski et al. 2005) or during tomato development (Exposito-Rodriguez et al. 2008). On the contrary, UBQ10 proved to be an inappropriate reference gene in development-related gene expression in rice (Jain et al. 2006), soybean (Jian et al. 2008), peach (Tong et al. 2009), or Fraxinus spp. (Rivera-Vega et al. 2011). Similarly, genes encoding GAPDH have been shown to be stably expressed under a range experimental conditions and species such as grapevine (Reid et al. 2006) or coffee (Barsalobres-Cavallari et al. 2009) but not in peach (Tong et al. 2009) or rice (Jain et al. 2006). Other authors do not favor the use of actin as reference gene in plants (Gutierrez et al. 2008; Nicot et al. 2005). However, in woody species, Populus (Brunner et al. 2004) and Vitis (Reid et al. 2006), tubulin and actin proved to be stably expressed. In our dataset and in tomato development (Exposito-Rodriguez et al. 2008), tubulin was among the least stable genes. Notably, in Vitis, even using the same tissue from the same genotype, and the same experiment conditions in two different years, differences the ranking of candidate reference genes based on their stability were observed, using geNorm (Reid et al. 2006). These few examples illustrate the lack of reliability of reference genes across species/tissues and a high specificity for a particular experimental situation. That strongly advises to carefully examine the expressional stability of candidate reference genes in every individual experimental setup and sample collection to be analyzed.
Coffea breeding aims to obtain genotypes more adapted and productive in its traditional growing areas. It is also important to breed for genotypes that will endure the predictable strong impact that will result from restrictions on suitable cultivation areas due to the variations in environmental conditions, particularly in the context of climate changes. Taking advantage of the contrasting acclimation abilities observed within the Coffea genus, namely to cold (Fortunato et al. 2010; Partelli et al. 2009; Ramalho et al. 2003), such goal could be explored by means of breeding for inter-species hybrids. In addition to opening the possibility to improve the selection criteria of reference genes in qRT-PCR analysis, our work allows studying the biological relevance of differences in the expression of target genes among genotypes. Considering financial costs and restricted sample availability in many experimental designs, it is not always possible to systematically test large sets of candidate genes for selection. Supported on our results, we recommend the inclusion of these top-ranked candidates in the minimal set of reference candidates to be evaluated in future Coffea transcriptomic studies aimed to investigate Coffea responses to other experimental variables.
References
Andersen CL, Jensen JL, Orntoft TF (2004) Normalization of real-time quantitative reverse transcription–PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64:5245–5250
Barber RD, Harmer DW, Coleman RA, Clark BJ (2005) GAPDH as a housekeeping gene: analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol Genomics 21:389–395
Barreto HG, Lazzari F, Ságio SA, Chalfun-Junior A, Paiva LV, Benedito VA (2011) In silico and quantitative analyses of the putative FLC-like homologue in coffee (Coffea arabica L.). Plant Mol Biol Rep. doi:10.1007/s11105-011-0310-9
Barsalobres-Cavallari C, Severino F, Maluf M, Maia I (2009) Identification of suitable internal control genes for expression studies in Coffea arabica under different experimental conditions. BMC Mol Biol 10:1
Batista-Santos P, Lidon FC, Fortunato A, Leitão AE, Lopes E, Partelli F, Ribeiro AI, Ramalho JC (2011) The impact of cold on photosynthesis in genotypes of Coffea spp.—photosystem sensitivity, photoprotective mechanisms and gene expression. J Plant Physiol 168:792–806
Brunner AM, Yakovlev IA, Strauss SH (2004) Validating internal controls for quantitative plant gene expression studies. BMC Plant Biol 4:14
Bustin SA, Benes V, Nolan T, Pfaffl MW (2005) Quantitative real-time RT–PCR—a perspective. J Mol Endocrinol 34:597–601
Chen K, Fessehaie A, Arora R (2011) Selection of reference genes for normalizing gene expression during seed priming and germination using qPCR in Zea mays and Spinacia oleracea. Plant Mol Biol Rep. doi:10.1007/s11105-011-0354-x
Cruz F, Kalaoun S, Nobile P, Colombo C, Almeida J, Barros L, Romano E, Grossi-de-Sá M, Vaslin M, Alves-Ferreira M (2009) Evaluation of coffee reference genes for relative expression studies by quantitative real-time RT–PCR. Mol Breed 23:607–616
Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible W-R (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 139:5–17
DaMatta FM, Ramalho JDC (2006) Impacts of drought and temperature stress on coffee physiology and production: a review. Braz J Plant Physiol 18:55–81
de Oliveira RR, Chalfun-Junior A, Paiva LV, Andrade AC (2010) In silico and quantitative analyses of MADS-Box genes in Coffea arabica. Plant Mol Biol Rep 28:460–472
Dheda K, Huggett J, Bustin S, Johnson M, Rook G, Zumla A (2004) Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques 37:112–114
Exposito-Rodriguez M, Borges A, Borges-Perez A, Perez J (2008) Selection of internal control genes for quantitative real-time RT–PCR studies during tomato development process. BMC Plant Biol 8:131
Flexas J, Medrano H (2002) Drought-inhibition of photosynthesis in C3 plants: stomatal and non-stomatal limitations revisited. Ann Bot 89:183–189
Fortunato AS, Lidon FC, Batista-Santos P, Eduardo-Leitão A, Pais IP, Ribeiro AI, Ramalho JC (2010) Biochemical and molecular characterization of the antioxidative system of Coffea sp. under cold conditions in genotypes with contrasting tolerance. J Plant Physiol 167:333–342
Guenin S, Mauriat M, Pelloux J, Van Wuytswinkel O, Bellini C, Gutierrez L (2009) Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. J Exp Bot 60:487–493
Gutierrez L, Mauriat M, Guénin S, Pelloux J, Lefebvre J-F, Louvet R, Rusterucci C, Moritz T, Guerineau F, Bellini C, Wuytswinkel OV (2008) The lack of a systematic validation of reference genes: a serious pitfall undervalued in reverse transcription–polymerase chain reaction (RT–PCR) analysis in plants. Plant Biotechnol J 6:609–618
Hochstrasser M (2000) Evolution and function of ubiquitin-like protein-conjugation systems. Nat Cell Biol 2:E153
Hong S-Y, Seo P, Yang M-S, Xiang F, Park C-M (2008) Exploring valid reference genes for gene expression studies in Brachypodium distachyon by real-time PCR. BMC Plant Biol 8:112
Hu R, Fan C, Li H, Zhang Q, Fu Y-F (2009) Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT–PCR. BMC Mol Biol 10:93
Jain M, Nijhawan A, Tyagi AK, Khurana JP (2006) Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochem Biophys Res Commun 345:646–651
Jian B, Liu B, Bi Y, Hou W, Wu C, Han T (2008) Validation of internal control for gene expression study in soybean by quantitative real-time PCR. BMC Mol Biol 9:59
Joët T, Salmona J, Laffargue A, Descroix F, Dussert S (2010) Use of the growing environment as a source of variation to identify the quantitative trait transcripts and modules of co-expressed genes that determine chlorogenic acid accumulation. Plant Cell Environ 33:1220–1233
Katsuyoshi S, Atsushi H, Paul S, Nobuo K, De-Maw C (1996) Evidence that glyceraldehyde-3-phosphate dehydrogenase is involved in age-induced apoptosis in mature cerebellar neurons in culture. J Neurochem 66:928–935
Kibbe WA (2007) OligoCalc: an online oligonucleotide properties calculator. Nucl Acids Res 35:W43–W46
Lashermes P, Combes M-C, Robert J, Trouslot P, D'Hont A, Anthony F, Charrier A (1999) Molecular characterisation and origin of the Coffea arabica L. genome. Mol Gen Genet 261:259–266
Li Q-F, Sun SSM, Yuan D-Y, Yu H-X, Gu M-H, Liu Q-Q (2010) Validation of candidate reference genes for the accurate normalization of real-time quantitative RT–PCR data in rice during seed development. Plant Mol Biol Rep 28:49–57
Lima ALS, DaMatta FM, Pinheiro HA, Totola MR, Loureiro ME (2002) Photochemical responses and oxidative stress in two clones of Coffea canephora under water deficit conditions. Environ Exp Bot 47:239–247
Lin YL, Lai ZX (2010) Reference gene selection for qPCR analysis during somatic embryogenesis in longan tree. Plant Sci 178:359–365
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-[Delta][Delta]CT method. Methods 25:402
Mallona I, Lischewski S, Weiss J, Hause B, Egea-Cortines M (2010) Validation of reference genes for quantitative real-time PCR during leaf and flower development in Petunia hybrida. BMC Plant Biol 10:4
Marraccini P, Freire LP, Alves GSC, Vieira NG, Vinecky F, Elbelt S, Ramos HJO, Montagnon C, Vieira LGE, Leroy T, Pot D, Silva VA, Rodrigues GC, Andrade AC (2011) RBCS1 expression in coffee: Coffea orthologs, Coffea arabica homeologs, and expression variability between genotypes and under drought stress. BMC Plant Biol 11:85
Mittler R (2006) Abiotic stress, the field environment and stress combination. Trends Plant Sci 11:15–19
Mondego JM, Vidal RO, Carazzolle MF, Tokuda EK, Parizzi LP, Costa GG, Pereira LF, Andrade AC, Colombo CA, Vieira LGE, Pereira GAG (2011) An EST-based analysis identifies new genes and reveals distinctive gene expression features of Coffea arabica and Coffea canephora. BMC Plant Biology 11:30
Nicot N, Hausman JF, Hoffmann L, Evers D (2005) Housekeeping gene selection for real-time RT–PCR normalization in potato during biotic and abiotic stress. J Exp Bot 56:2907–2914
Partelli FL, Vieira HD, Viana AP, Batista-Santos P, Leitão AE, Ramalho JC (2009) Cold impact on photosynthetic related parameters in coffee genotypes. Pesq Agrop Bras 44:1404–1415
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett 26:509–515
Phillips MA, D’Auria JC, Luck K, Gershenzon J (2009) Evaluation of candidate reference genes for real-time quantitative PCR of plant samples using purified cDNA as template. Plant Mol Biol Rep 27:407–416
Pihur V, Datta S, Datta S (2009) RankAggreg, an R package for weighted rank aggregation. BMC Bioinforma 10:62
Pinheiro HA, DaMatta FM, Chaves ARM, Fontes EPB, Loureiro ME (2004) Drought tolerance in relation to protection against oxidative stress in clones of Coffeacanephora subjected to long-term drought. Plant Sci 167:1307–1314
Ramalho JC, Campos PS, Teixeira M, Nunes MA (1998) Nitrogen dependent changes in antioxidant system and in fatty acid composition of chloroplast membranes from Coffea arabica L. plants submitted to high irradiance. Plant Sci 135:115–124
Ramalho JC, Campos PS, Quartin VL, Silva MJ, Nunes MA (1999) High irradiance impairments on photosynthetic electron transport, ribulose-1,5-bisphosphate carboxilase/oxygenase and N assimilation as a function of N availability in Coffea arabica L. plants. J Plant Physiol 154:319–326
Ramalho JC, Pons TL, Groeneveld HW, Azinheira HG, Nunes MA (2000) Photosynthetic acclimation to high light conditions in mature leaves of Coffea arabica L.: role of xanthophylls, quenching mechanisms and nitrogen nutrition. Funct Plant Biol 27:43–51
Ramalho JC, Quartin VL, Leitão E, Campos PS, Carelli MLC, Fahl JI, Nunes MA (2003) Cold acclimation ability and photosynthesis among species of the tropical Coffea genus. Plant Biol 5:631–641
Reid K, Olsson N, Schlosser J, Peng F, Lund S (2006) An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT–PCR during berry development. BMC Plant Biol 6:27
Rivera-Vega L, Mamidala P, Koch JL, Mason ME, Mittapalli O (2011) Evaluation of reference genes for expression studies in ash (Fraxinus spp.). Plant Mol Biol Rep. doi:10.1007/s11105-011-0340-3
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana, Totowa, pp 365–386
Salmona J, Dussert S, Descroix F, de Kochko A, Bertrand B, Joët T (2007) Deciphering transcriptional networks that govern Coffea arabica seed development using combined cDNA array and real-time RT–PCR approaches. Plant Mol Biol 66:105–124
Schmidt G, Delaney S (2010) Stable internal reference genes for normalization of real-time RT–PCR in tobacco (Nicotiana tabacum) during development and abiotic stress. Mol Gen Genom 283:233–241
Schölander PF, Bradstreet ED, Hemmingsen EA, Hammel HT (1965) Sap pressure in vascular plants: negative hydrostatic pressure can be measured in plants. Science 148:339–346
Silberberg G, Baruch K, Navon R (2009) Detection of stable reference genes for real-time PCR analysis in schizophrenia and bipolar disorder. Anal Biochem 391:91
Singh R, Irikura B, Nagai C, Albert HH, Kumagai M, Paull RE, Moore PH, Wang M-L (2011) Characterization of prolyl oligopeptidase genes differentially expressed between two cultivars of Coffea arabica L. Tropical Plant Biol. doi:10.1007/s12042-011-9082-5
Stevenson J, Hymas W, Hillyard D (2005) Effect of sequence polymorphisms on performance of two real-time PCR assays for detection of herpes simplex virus. J Clin Microbiol 43:2391–2398
Taris N, Lang R, Camara M (2008) Sequence polymorphism can produce serious artefacts in real-time PCR assays: hard lessons from Pacific oysters. BMC Genomics 9:234
Tong Z, Gao Z, Wang F, Zhou J, Zhang Z (2009) Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol Biol 10:71
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT–PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:34.31–34.11
Vandesompele J, Kubista M, Pfaffl M (2009) Reference gene validation software for improved normalization. In: Logan J, Edwards K, Saunders N (eds) Real-time PCR: current technology and applications. Caister Academic, Norfolk, pp 47–63
Vidal RO, Mondego JMC, Pot D, Ambrósio AB, Andrade AC, Pereira LFP, Colombo CA, Vieira LGE, Carazzolle MF, Pereira GAG (2010) A high-throughput data mining of single nucleotide polymorphisms in Coffea species Expressed Sequence Tags suggests differential homeologous gene expression in the allotetraploid Coffea arabica. Plant Physiol 154:1053–1066
Zampieri M, Ciccarone F, Guastafierro T, Bacalini MG, Calabrese R, Moreno-Villanueva M, Reale A, Chevanne M, Bürkle A, Caiafa P (2010) Validation of suitable internal control genes for expression studies in aging. Mech Ageing Dev 131:89–95
Zhu J-K (2002) Salt and drought stress signal transduction in plants. Ann Rev Plant Biol 53:247–273
Acknowledgments
Financial support was provided by “Fundaçãopara a Ciência e a Tecnologia” (FCT) (Project PTDC/AGR-AAM/64078/2006) and by the European Fund FEDER. A.S. Fortunato acknowledges a postdoctoral grant from FCT (SFRH/BPD/47563/2008). The authors acknowledge Isabel Palos and Sandra Anjos for excellent technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Luis F. Goulao and Ana S. Fortunato contributed equally to this work.
Online Resource Material
Below is the link to the electronic supplementary material.
Online Resource Fig. S1
Confirmation of primer specificity of studied genes in qRT-PCR amplifications. Melting curves generated for all genes, showing the presence of a single peak and lack of non-specific fragments or primer dimers. Identical traces were obtained for the other two genotypes (not shown) (PPT 166 kb)
Online Resource Fig. S2
Gene expression stability and ranking of the ten reference genes as predicted, automatically, by geNorm. Plots are based on mean expression stability (M values) calculated in (a) the total assay, (b) the genotype series, (c) in the drought stress assay, (d) in the cold stress, and (e) in the multiple stress assay. V values were obtained using geNorm as the pairwise variation (V n / n + 1) between NF n and NF n + 1, based on the ranked genes, according to the stepwise exclusion of the least stable genes and M value recalculation, as implemented in the algorithm (PPT 392 kb)
Online Resource Table S1
BestKeeper software statistics for the ten reference genes based on Ct values in the experimental series studied (XLS 53 kb)
Online Resource Table S2
Stability values for the best combination of two reference genes, based on the NormFinder output (XLS 34 kb)
Rights and permissions
About this article
Cite this article
Goulao, L.F., Fortunato, A.S. & C. Ramalho, J. Selection of Reference Genes for Normalizing Quantitative Real-Time PCR Gene Expression Data with Multiple Variables in Coffea spp.. Plant Mol Biol Rep 30, 741–759 (2012). https://doi.org/10.1007/s11105-011-0382-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-011-0382-6