
Abstract
Background and aims
It has frequently been shown that plants interact with soils to shape rhizosphere microbiomes. However, previous work has not distinguished between effects of soil properties per se, and effects attributable to the resident microbial communities of those soils. We aimed to test whether differences in the structure of bulk soil microbial communities, within a given soil type, would carry over to impact the structure of the rhizosphere microbial community.
Methods
We used repeated chemical amendments to develop divergent bulk soil microbial community starting points from which rhizosphere development proceeded. Additionally, we contrasted rhizosphere microbiomes associated with two different cultivars of corn (Zea mays).
Results
A wide range of bacterial and archaeal taxa responded to chemical resource amendments, which reduced bulk soil microbiome diversities. Corn genotypes P9714XR and 35F40 had largely similar impacts on rhizosphere microbiome development, although significant differences were evident in select treatments. Notably, in cases where resource amendments altered bulk soil microbial community composition, legacy effects persisted into the rhizosphere.
Conclusions
Our results suggest that rhizosphere microbial communities may develop into different states depending on site history and prior selective events. This work advances our understanding of soil microbiome dynamics and responsiveness to change in the form of simple resource amendments and the development of the rhizosphere.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Soil microbial communities are increasingly understood to interact extensively with plants and to have substantial influence on plant health and productivity. For instance, members of the soil microbiome may influence plant health, productivity and community dynamics through mechanisms ranging from production and modification of plant hormones (Sergeeva et al. 2007), to the imposition and suppression of disease (Klein et al. 2013), to impacts on nutrient availability (Vassilev et al. 2006) and enhanced tolerance toward abiotic stresses (Marquez et al. 2007).
Plants exert considerable influence over the rhizosphere microbiome, largely through the provision of exudates and other labile chemical resources [reviewed in (Bakker et al. 2012)]. Growing plant roots change the chemical identity, quantity and diversity of resources available to soil microbial communities (Zolla et al. 2013), through processes of both addition and removal of diverse chemical compounds from soil (Jones et al. 2004). In turn, this altered resource availability changes the selective pressures experienced by soil microbes. The outcome of these complex processes is the development of a rhizosphere microbial community that differs markedly from the source communities in bulk soil (Minz et al. 2013). In one example, plants engineered to produce novel carbon compounds were shown to quickly select for bacteria capable of metabolizing those compounds, even though such capabilities were undetectable among bacteria isolated before exposure to the plant (Oger et al. 2004). This is an indication of the strength and rapidity with which selection can act on soil microbial communities during the development of the rhizosphere. Several large-scale studies of host genotype effects on rhizosphere, rhizoplane and root endosphere microbiomes have recently been published, for Arabidopsis (Bulgarelli et al. 2012; Lundberg et al. 2012), maize (Peiffer et al. 2013), and rice (Edwards et al. 2015).
It is well known that the impact of host plants on soil microbial communities can vary with soil type (Girvan et al. 2003; Ulrich and Becker 2006). This suggests that microbiome dynamics observed in one soil type may not be generalizable to other soil types, and that it is important to consider microbiome dynamics across a range of soil types. There are a variety of mechanisms that could contribute to differences in microbiome assembly or dynamics across soil types. For instance, root exudation is impacted by soil characteristics such as nutrient availability or deficiency (Lu et al. 1999; Shen et al. 2001), and is sensitive to the microbes that colonize the root surface (De-la-Pena et al. 2008; Meharg and Killham 1995). Thus rhizosphere microbes may play a part in shaping their own selective environment by modulating plant root exudation. The adsorption of biologically active compounds on charged clay particles (Brady 1999) or interactive effects of pH with root exudation may also contribute to different microbiome dynamics in the rhizosphere of a common host across soils. Regardless of the mechanism, prior demonstrations of rhizosphere microbiome variation across soil types, even in the presence of a consistent host plant genotype, require that rhizosphere effects be considered across a range of soil types.
Because edaphic characteristics themselves also influence soil microbial communities (Carson et al. 2009; Girvan et al. 2003), many studies of plant genotype effects on the rhizosphere microbiome have confounded the impacts of soil type with the limitations of available rhizosphere colonizers. For instance, Schreiter et al. (2014) contrasted various soil types for influence on rhizosphere microbial community structure, demonstrating that both edaphic characteristics and bulk soil microbial community structures differed among the soil types they considered. It would be valuable to distinguish between these different mechanisms leading to variation among rhizosphere microbiomes within and across soil types. The rhizosphere microbial community that develops in response to selection by plant-driven forces must necessarily reflect the identity of the microbes that are present in the bulk soil. Furthermore, even if the same taxa are present between sites, but differ in relative abundance, rhizosphere microbial community development may proceed in different directions because initial root colonizers have better success than latecomers who attempt to establish on an already colonized root surface (Rainey 1999), and the taxa that are most abundant locally are probabilistically the most likely to be the first colonizers. Thus, the composition and structure of the initial microbial community colonizing a given plant root is likely to impact subsequent rhizosphere community structure and dynamics.
This is an important nuance because of the high spatial variability of soil microbial communities (Bakker et al. 2013; Nunan et al. 2002); across a single field, a population of host plants may experience a common soil type but divergent initial soil microbial community composition and structure. Understanding how this variation in initial microbial communities plays out during plant-driven selection in the rhizosphere will inform our understanding of plant-microbe interactions in both managed and natural systems, and will offer insights into the feasibility of ‘engineering’ the rhizosphere for our own ends (Ryan et al. 2009).
Our objectives in this work were to: i) introduce variation in microbial community structure within a given soil type; ii) to observe whether these induced differences in bulk soil microbial community structure persisted as observable differences in the rhizosphere; iii) compare rhizosphere microbial community structure for two different host plant genotypes; iv) to perform these contrasts across a number of different soil types. We used amendments of defined exogenous chemical resources to induce changes in the structure of microbial communities in four different soils. From these divergent community starting points, we allowed for rhizosphere development over several weeks by two different genotypes of Z. mays. We assessed changes in root-associated microbial (bacteria + archaea) community structure by sequencing 16S rRNA gene fragments from rhizosphere soil, allowing us to contrast the effects of Z. mays genotypes and to observe how diverse microbial starting points influence the development of the rhizosphere microbiome.
Materials and methods
Soil collection
Four soils with differing textures and history of management and plant cover were collected (Table 1). Each soil was sieved through a 2 mm mesh to remove plant roots and large clods before being further homogenized by hand. Soils were stored at 4 °C and were used for experiments within 4 months.
Resource amendments to shift soil microbial community structure
Soils were distributed into magenta box containers at a consistent depth (110 g for Soil 1, 95 g Soil 2, 75 g Soil 3, 100 g Soil 4) and wetted to 50 % of field capacity. Ten replicate microcosms were established for each treatment. Three different resource amendments were applied to each soil, at a rate of 1 mg carbon gram−1 dry soil. The amendments consisted of either glucose, a mix of seven sugars and sugar alcohols (inositol, galactose, glucose, maltose, mannitol, sorbitol, sucrose; each component contributing an equal amount of carbon), or soluble starch. The goal in choosing these amendments was to span a range of chemical diversity and complexity, while still allowing for rapid metabolism such that a minimum quantity of the exogenous chemicals would remain after the introduction of a plant. Amendments were added four times, at weekly intervals, and were incorporated by gently mixing with a spatula. One week after the final amendment, soil was collected from a random subset of samples in each treatment for chemical analysis and DNA extraction. Soil carbon content (% C), nitrogen content (% N) and pH were assessed at the Soil, Water and Plant Testing Laboratory at Colorado State University, using standard procedures.
Rhizosphere microbial community structure
Ten days after the final resource amendment, one corn seed was planted per container (five replicate microcosms each of P9714XR or 35F40, seed provided by Dupont Pioneer). These cultivars are derived from different hybrid families and have different rates of developmental progression (relative maturity: 97 d for P9714XR, 105 d for 35F40). Corn seeds were surface-disinfested to avoid the influence of seed-borne microbes on subsequent rhizosphere community composition, by shaking for 2 min in 70 % ethanol, 25 min in 0.5 % NaOCl with 0.01 % Tween 20, and rinsing four times with sterile distilled water. After planting, boxes were arranged in a randomized block design on a light table. Plants were watered as needed, and were fertilized twice with 25 mL of 1/5 strength Hoagland’s solution (Phytotechnology Laboratories). After 4 weeks of growth, rhizosphere soil was collected for microbial community structure analyses. Plants were upturned over a paper towel and bulk soil was worked free from the roots with a spatula. Soil remaining attached to the root system was considered to be rhizosphere soil.
Sequencing and sequence processing
Soil DNA extraction was performed with the PowerSoil DNA Extraction Kit (MoBio; 96 well plate format), according to the manufacturer’s instructions. Samples were arranged in random order among wells in the DNA extraction plates. The concentration of extracted DNA was measured with a Nanodrop spectrophotometer and samples were diluted with molecular biology grade water to 10 ng μL−1. Amplification of 16S rRNA gene fragments was performed with primers targeting positions 515–532 (5′- GTG YCA GCM GCC GCG GTA A -3′) and 1052–1071 (5′- GAR CTG RCG RCR RCC ATG CA -3′). These primers offer good coverage for known taxa of both bacteria and archaea (Wang and Qian 2009). Each PCR was performed in a 25 μL volume, consisting of 10 μL HotMasterMix (5 Prime, Inc.), 1 μL primer mix (5 μM each), 1 μL DNA template, and 13 μL molecular biology grade water. The thermocycler program consisted of 3 min at 94 °C, 35 cycles of (45 s at 94 °C, 1 min at 50 °C, 1.5 min at 72 °C), and a final extension step of 10 min at 72 °C. Three separate PCRs were performed for each sample, and successful amplification of each reaction was verified by agarose gel electrophoresis with ethidium bromide staining. Replicate PCRs were pooled and amplicons were purified with the UltraClean PCR clean-up kit (MoBio), according to the manufacturer’s instructions. Amplicons were prepared for sequencing with the Nextera XT DNA sample preparation kit and associated index kit (Illumina), according to the manufacturer’s instructions. Samples were dual indexed, and were sequenced across two runs of a MiSeq sequencing machine (Illumina). Sequencing was performed at the Genome Center at Yale University, using a MiSeq version 2 300 cycle kit. The raw sequence data have been deposited into the NCBI Sequence Read Archive as accession SRP018039.
Sequence processing was performed with Mothur v. 1.28.0 (Schloss et al. 2009). Forward and reverse reads were merged into contigs. Contigs were culled if they were shorter than 100 nucleotides in length, contained any ambiguous characters or homopolymeric runs longer than 15 nucleotides, or failed quality screening (qwindowsize = 50, qwindowaverage = 35). Contigs were classified against the combined SILVA bacterial and archaeal databases (Pruesse et al. 2007), using the naive Bayesian classifier (Wang et al. 2007) embedded in Mothur, with a confidence threshold of 75 %. Contigs that could not be confidently assigned to the phylum level were dropped from the analysis. Samples with fewer than 10,000 contigs remaining at this point were dropped from the analysis and random subsampling was performed to the depth of the smallest remaining sample (11,140 contigs). Contigs were clustered into family-level phylotypes based on the taxonomic classification of each contig.
Patterns of similarity in microbial community structure were visualized via principal coordinates analysis (PCoA), using function cmdscale in the stats package for R (R Core Development Team 2011), with pairwise Bray-Curtis distances as the input data. Differences in community structure among treatments were tested using analysis of molecular variance (AMOVA), as implemented in Mothur. For phylotype diversity estimates, the method of Leinster and Cobbold (Leinster and Cobbold 2012) was used. This approach expresses diversities as effective numbers, across a range of sensitivities toward low abundance community members. We used a similarity-informed version of the index, assigning similarities of 1 for phylotypes in the same family (the diagonal on the similarity matrix), 0.9 for shared order, 0.8 for shared class, 0.7 for shared phylum, 0.6 for shared domain, and 0.5 for phylotypes from different domains (i.e., bacteria vs. archaea). Although assigning similarity to pairs of phylotypes is imprecise given our dearth of knowledge about most microbial taxa, even crude estimates of similarity among taxa represent an improvement over the more common, naïve approach to estimating diversity which assumes that all pairs of taxa are completely dissimilar from each other (Leinster and Cobbold 2012).
MANOVA was used to test the significance of soil and of resource amendment on the relative abundance of phyla. ANOVA with Dunnett’s test identified significant changes within each resource amendment class. In summarizing the response of each phylum to resource amendments, the glucose and sugar mix amendment treatments were combined into ‘sugar’ (n = 8 for sugar, n = 4 for starch). The number of significant increases or decreases was divided by the total number of tested cases.
Root exudate collection and analysis
To test whether Z. mays cultivars P9714XR and 35F40 differed in root exudation, exudates were collected in an axenic hydroponic system and analyzed by gas chromatography coupled with mass spectrometry (see Electronic supplementary material for details).
Results
Impacts of resource amendments on soil pH, C and N
Measured edaphic properties were rarely significantly influenced by the resource amendments. Relative to pre-amendment soils, pH increased slightly for glucose and starch amendments in Soil 2 (Supplementary Figure S1; p < 0.01, ANOVA with Dunnett’s test). Soil carbon content increased only for the starch amendment in Soil 1, while soil nitrogen content was not significantly influenced by any of the resource amendments in any of the soils (Supplementary Figure S1).
Impacts of resource amendments on the bulk soil microbiome
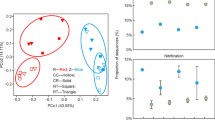
Our goal was to shift the structure of the bulk soil microbiomes through resource amendments. This approach was effective in most cases; in the sandy loam (Soil 1), communities with a history of exposure to either glucose, a mix of sugars, or starch were all distinct from each other (AMOVA, p < 0.05). In the clay soils (Soil 2 and Soil 4), communities with a history of exposure to either glucose or a mix of sugars were distinct from those that had been amended with starch (AMOVA, p < 0.05). Communities in the silt loam (Soil 3) were the most resistant to change, and did not differ significantly regardless of resource amendment history (AMOVA, p > 0.05). Visual depictions of similarity among samples from each soil are shown via principal coordinate analysis plots (Fig. 1).

Principal coordinates analysis (Bray-Curtis distance index) plots as a visualization of differences in microbial community structure between samples. All samples were included in a single principle coordinates analysis, but samples are plotted separately by soil in order to improve clarity. The first principal coordinate axis explained 39.3 % and the second axis explained 21.9 % of the observed variation
Resource amendments shifted the relative abundances of bacterial and archaeal phyla in soil (Fig. 2). Across the initial and resource-amended soils, the dominant phylum ranged from Actinobacteria (most samples), to Proteobacteria (Soil 2 initial or with starch amendment, Soil 4 initial), to Firmicutes (Soil 1 with glucose or sugar mix amendments). The relative abundances of bacterial and archaeal phyla were impacted by both soil type and the identity of resource amendments (p < 0.01, MANOVA). Taxa significantly varying in relative abundance with soil type included Proteobacteria, Actinobacteria, Bacteroidetes, Firmicutes, Crenarchaeota, Acidobacteria, Planctomycetes and Gemmatimonadetes (p < 0.01, 2-way ANOVA).

The relative abundance of bacterial and archaeal phyla at the onset of the experiment (‘Initial’), following resource amendments, and after rhizosphere development. Phyla having a mean relative abundance of at least 1 ‰ are shown individually; less abundant phyla are grouped into “Other.” Data from the two Z. mays genotypes were combined for this visual summary because rhizosphere bacterial community structures were very similar. Mean values across replicates are shown (3 ≤ n ≤ 10)
All common phyla (those represented by at least 1 ‰ of sequence reads) exhibited a significant change in relative abundance compared to the corresponding pre-amendment soil in response to resource amendments in at least one case (Table 2). Actinobacteria, Chloroflexi and Firmicutes increased significantly in response to sugar amendments in some cases, while no bacterial or archaeal phylum showed a significant increase in relative abundance in response to starch amendment (Table 2). Where phyla responded to resource amendments, decreases in relative abundance were more common than increases in relative abundance. This may be a result of our measuring relative, rather than absolute, abundances. For instance, if a small subset of the community responded to resource amendmentswith a large increase in absolute abundance, the relative abundances of all other taxa would automatically decline.
The addition of simple chemical resource amendments to soil tended to reduce bacterial phylotype diversity; amendment with glucose, a mix of sugars, or with starch uniformly reduced phylotype diversity compared to the pre-amendment level across all four soil types (Fig. 3). There was a particularly large drop in bulk soil microbial diversity in the sandy loam-glucose treatment (Fig. 3; Soil 1). In this treatment, just five phylotypes comprised over 90 % of the sequence reads, and all five of these phylotypes belonged to the class Bacilli. This dominance of Bacilli was so extreme that other taxa may have been extirpated in this treatment. A similar, although less extreme enrichment of Bacilli (phylum Firmicutes) occurred in the sugar mix treatment in this soil (Fig. 2). Also notable was the minor change in bulk soil microbial diversity upon starch addition to Soil 2 (clay), compared to the much larger drop in diversity when this soil was amended with sugars (Fig. 3). In the silt loam soil, diversities were nearly identical across amendment treatments (Fig. 3; Soil 3), which is consistent with the indication via analysis of molecular variance (see above) that microbiome structure did not differ among amendment treatments in this soil.

Phylotype diversities of bulk soil microbiomes either prior to, or following repeated amendment with simple chemical resources. Low abundance phylotypes contribute less to the diversity statistic as the sensitivity parameter, q, increases. Mean values are shown for each treatment (3 ≤ n ≤ 4)
Soil microbial community structure in the rhizosphere
Similar dynamics were observed across most communities as the rhizosphere developed from bulk soil communities. For instance, Actinobacteria and Firmicutes typically declined in relative abundance, while Proteobacteria, Bacteroidetes, Verrucomicrobia and Acidobacteria increased in relative abundance (Fig. 2; shown are averages across corn cultivars).
There were few statistically significant differences between the two Z. mays cultivars in impact on rhizosphere microbiomes. Following the resource amendments, the only pairwise contrast that showed a significant difference in rhizosphere microbiome structure between the two cultivars was in the silt loam soil (Soil 3) with glucose amendment (AMOVA, p = 0.034); in all other treatments, rhizosphere microbiome structure was not significantly different between the two cultivars (AMOVA, p > 0.05; visual summary via principal coordinates plot in Fig. 4).

Principal coordinates analysis (Bray-Curtis distance index) plot as a visualization of differences in rhizosphere microbial community structure between two Z. mays cultivars. For clarity, the mean scores across replicates for the first and second principal coordinate axes are plotted, and points corresponding to the two cultivars in the same soil-amendment treatment are connected. Soil and prior resource amendments are indicated in the legend. * indicates treatments for which rhizosphere microbiome structure differed significantly between the two host plant genotypes (p < 0.05, AMOVA)
The diversity of rhizosphere microbiomes differed between the two Z. mays cultivars in certain cases. Microbial diversity in the rhizosphere of cultivar 35F40 was higher than for cultivar P9714XR in the silt loam soil following the sugar mix amendment (T-test, p < 0.05; Fig. 5). Following starch amendment, the rhizosphere of 35F40 was more diverse than for P9714XR in one clay soil (Soil 2), but the opposite pattern was observed in the other clay soil (Soil 4; T-test, p < 0.05; Fig. 5). These contrasting results highlight the interactive effects of soil factors and host plant genotype in shaping rhizosphere microbiomes.

Phylotype diversities of rhizosphere microbiomes following repeated amendment with simple chemical resources. Low abundance phylotypes contribute less to the diversity statistic as the sensitivity parameter, q, increases. Mean values are shown for each treatment (3 ≤ n ≤ 5). * indicates significant differences in diversity between the two Z. mays cultivars (T-test, p < 0.05 at any given value of q)
For each soil, rhizosphere microbiomes developing after starch amendments were distinct from those developing after glucose or sugar mix amendments (AMOVA, p < 0.05 for all pairwise contrasts except for P9714XR rhizosphere following sugar mix vs. starch amendments, and 35F40 rhizosphere following glucose vs. starch in the silt loam soil; see also Fig. 4). Rhizosphere microbiome structures following glucose vs. sugar mix amendments were generally not distinguishable within a given soil (AMOVA, p > 0.05).
Across all soil-amendment treatments, rhizosphere microbiomes developing after glucose and sugar mix amendment in the sandy loam soil (Soil 1) were clearly distinct from all other treatments (Figs. 2 and 4). This suggests a legacy effect of resource amendments that continued to influence soil microbial community structure in the rhizosphere. We have highlighted above how Bacilli (phylum Firmicutes) were strongly selected within the bulk soil microbiome in these treatments, perhaps to the exclusion of other taxa. After rhizosphere microbiome development following glucose and sugar mix amendment in the sandy loam soil, otherwise common phyla such as Chloroflexi, Crenarchaeota and Planctomycetes remained nearly absent (Fig. 2). Notably, the absence of these taxa was not accommodated by a uniform increase in relative abundance across the remaining taxa. For instance, rhizosphere communities in these treatments had much higher relative abundance of Bacteroidetes, and lower abundance of Actinobacteria than was observed across other treatments (Fig. 2). Thus, rhizosphere microbiomes developed into different states as a function of the microbial taxa available for colonization.
Because we stopped chemical amendments when seeds were planted, some portion of the observed changes between bulk soil and rhizosphere microbiomes may have been due to the cessation of new inputs of glucose, sugar mix, or starch. However, the observed changes did not represent a return to the initial community structure that existed prior to resource amendments (particularly in Soils 1 & 4; see Fig. 2), suggesting that changes were not due primarily to the removal of exogenous temporary resource amendments and accompanying selective forces.
Root exudate analysis
Profiling of root exudation in an axenic system did not reveal substantial differences between Z. mays cultivars P9714XR and 35F40. There was no clear clustering of samples by cultivar when exudate profiles were plotted with principal components analysis (data not shown). It is possible that exudation profiles may have differed to a greater extent if the plants had been grown in a solid substrate or in interaction with soil organisms.
Discussion
Impacts of carbon source amendments on bulk soil microbial community structure
Our results shed light on the responsiveness of soil microbiomes to changes in resource availability, and the consistency of resulting microbiome dynamics across soils. We demonstrate that microbiome dynamics may differ widely across soil types. For instance, microbial communities in the sandy loam soil were much more sensitive and responsive to changing resource availability than were the microbial communities of the clay or silt loam soils. The low carbon content of the sandy loam soil likely offered the most scarce resource base to its bulk soil microbiome. In contrast, endogenous resources in Soils 2, 3 and 4 may have buffered microbial communities against change driven by new resource inputs.
Differences among amendment treatments can be attributed to the chemical nature of the amendments. However, not all differences from the initial community state may be due to the amendments themselves; some portion of the observed effects on bulk soil microbiomes may have been due to microcosm incubation conditions or to the prolonged maintenance of soil moisture at levels suitable for microbial physiological activity. Nevertheless, this does not impinge upon our primary objective, which was to accentuate differences in microbiome structure within given soil types prior to introducing a plant to initialize rhizosphere development.
Several studies have now reported on the responsiveness of particular microbial taxa to artificial resource inputs. Our results support previous observations that amendment with simple resources decreases microbial diversity (Zhou and Wu 2012). Fierer et al. (2007) reported Acidobacteria declining and Bacteroidetes and β-Proteobacteria increasing in relative abundance in response to amendments of sugar compounds. We show comparable responses for Acidobacteria, which declined significantly in relative abundance in 50 % of treatments receiving sugar amendments. However, Bacteroidetes responded oppositely in our study compared to the previous report, also declining significantly in relative abundance in 50 % of treatments receiving sugar amendments. The phylum Proteobacteria, or certain classes within this phylum, has been proposed as a copiotrophic lineage (Fierer et al. 2007; Pascault et al. 2010), but we did not find any significant increases in the relative abundance of this phylum in soils that had been amended with sugars. Interestingly, Firmicutes have been previously reported as non-responsive to sugar amendments (Fierer et al. 2007), yet in our experiment they were the most consistently responsive of all phyla, increasing significantly in relative abundance in 62.5 % of treatments receiving sugar amendments. Together, these contrasts suggest that the responses of particular taxa to resource amendments may not be consistent across soil types or in the context of distinct soil microbiomes. Alternately, members of these broadly defined taxa may simply include too wide a range of ecological variation for such simple classification schemes to hold merit.
Impacts of host plant genotype on rhizosphere microbial community structure
The historical legacy created by resource amendments sometimes persisted into the development of the rhizosphere microbiome. This was most evident where conditioning by resource amendments led to severe declines in the abundance of particular taxa, as in the apparent extirpation of Crenarchaeota and Planctomycetes following glucose amendment to the sandy loam soil. These results provide empirical support for the logical notion that the composition of the starting microbial community available to a plant can fundamentally constrain the outcomes of selection in the rhizosphere. Previous work has investigated changes in the development of rhizosphere bacterial communities when Z. mays roots are inoculated with particular bacterial strains (Baudoin et al. 2009; Herschkovitz et al. 2005), but the present work substantially extends our understanding of rhizosphere microbial community development as a function of the composition and structure of microbial communities available to plant roots.
We found only subtle differences among rhizosphere microbiomes associated with two different genotypes of Z. mays. For instance, rhizosphere microbial community structure differed significantly between these genotypes in only one out of 12 treatments (4 soils × 3 resource amendments), while rhizosphere microbiome diversity differed between genotypes in three treatments.
A number of recent studies have also used Z. mays to explore the impacts of host plant genotype on rhizosphere microbial communities. Aira et al. (2010) used phospholipid fatty acid analysis to detect differences in bacterial communities associated with corn genotypes derived from the same hybrid cross. The particular genotypes contrasted in this case were chosen for known genetic differences related to the storage of sugars vs. starch, which have a strong probability of impacting associated microbial communities. Thus this study represented a ‘best-case’ scenario for detecting microbial community differences associated with fine-scale differences in host genotype. Other cases in which Z. mays genotypes have been documented to differ in impact on soil microorganisms have come from contrasting genotypes with dramatic genetic differences, such as inbred vs. hybrid varieties (Picard and Bosco 2005; Picard and Bosco 2006) or inbred lines from widely different major genetic groups within the species (Bouffaud et al. 2012). Studies contrasting Z. mays lines with more subtle genetic variation have sometimes failed to detect differences in impact on associated soil bacterial communities (Chiarini et al. 1998; Dohrmann et al. 2013; Fang et al. 2005; Philippot et al. 2006), although not always (Castaldini et al. 2005). Recent work in Z. mays under field conditions has indicated that the magnitude of heritable effects on soil microbial communities is modest, and is heavily influenced by interactions with the environment (Peiffer et al. 2013). Rhizosphere microbial communities associated with Z. mays also experience on-going change as the host plant develops (Li et al. 2014). Thus our finding of similarity in impact of two Z. mays cultivars from different hybrid families on rhizosphere microbiome structure is consistent with prior research.
More pronounced host genotype-specific effects may require longer growth periods or repeated cultivation in order for cumulative effects to become evident. Because our experimental approach relied on environmental DNA, it is possible that we detected inactive community members. This may have dampened our ability to detect fine differences in bulk soil microbial community structure due to resource amendments, or in rhizosphere microbiome structure among host genotypes. It has been shown that differences are evident when root-associated microbiomes are profiled via RNA-based methods, compared to DNA-based approaches (Ofek et al. 2014). Simultaneous consideration of soil fungal community dynamics may also have provided additional insight into the observed bacterial and archaeal community changes.
In conclusion, this work constitutes a step forward in our understanding of soil microbiome assembly and functioning, and sheds light on the responsiveness of soil microbial communities across a range of soil types to resource amendments. We demonstrate differences in microbiome response to resource amendments across soil types, and show that legacy effects of prior selection on microbiomes may continue to influence rhizosphere microbial community structure. We find that two Z. mays genotypes differing in genetic background and maturity class foster rhizosphere microbiomes that could rarely be distinguished from each other.
References
Aira M, Gómez-Brandón M, Lazcano C, Bååth E, Domínguez J (2010) Plant genotype strongly modifies the structure and growth of maize rhizosphere microbial communities. Soil Biol Biochem 42:2276–2281
Bakker MG, Manter DK, Sheflin AM, Weir TL, Vivanco JM (2012) Harnessing the rhizosphere microbiome through plant breeding and agricultural management. Plant Soil 360:1–13
Bakker MG, Bradeen JM, Kinkel LL (2013) Effects of plant host species and plant community richness on streptomycete community structure. FEMS Microbiol Ecol 83:596–606
Baudoin E, Nazaret S, Mougel C, Ranjard L, Moënne-Loccoz Y (2009) Impact of inoculation with the phytostimulatory PGPR Azospirillum lipoferum CRT1 on the genetic structure of the rhizobacterial community of field-grown maize. Soil Biol Biochem 41:409–413
Bouffaud M-L, Kyselková M, Gouesnard B, Grundmann G, Muller D, Moënne-Loccoz Y (2012) Is diversification history of maize influencing selection of soil bacteria by roots? Mol Ecol 21:195–206
Brady N (1999) The nature and properties of soils, 12th edn. Prentice Hall, Upper Saddle River
Bulgarelli D, Rott M, Schlaeppi K, van Themaat EVL, Ahmadinejad N, Assenza F, Rauf P, Huettel B, Reinhardt R, Schmelzer E, Peplies J, Gloeckner FO, Amann R, Eickhorst T, Schulze-Lefert P (2012) Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488:91–95
Carson JK, Campbell L, Rooney D, Clipson N, Gleeson DB (2009) Minerals in soil select distinct bacterial communities in their microhabitats. FEMS Microbiol Ecol 67:381–388
Castaldini M et al (2005) Impact of Bt corn on rhizospheric and soil eubacterial communities and on beneficial mycorrhizal symbiosis in experimental microcosms. Appl Environ Microbiol 71:6719–6729
Chiarini L, Bevivino A, Dalmastri C, Nacamulli C, Tabacchioni S (1998) Influence of plant development, cultivar and soil type on microbial colonization of maize roots. Appl Soil Ecol 8:11–18
R Core Development Team (2011) R: a language and environment for statistical computing R foundation for statistical computing: Vienna, Austria
De-la-Pena C, Lei Z, Watson BS, Sumner LW, Vivanco JM (2008) Root-microbe communication through protein secretion. J Biol Chem 283:25247–25255
Dohrmann AB, Kuting M, Junemann S, Jaenicke S, Schluter A, Tebbe CC (2013) Importance of rare taxa for bacterial diversity in the rhizosphere of Bt- and conventional maize varieties. ISME J 7:37–49
Edwards J, Johnson C, Santos-Medellin C, Lurie E, Podishetty NK, Bhatnagar S, Eisen JA, Sundaresan V (2015) Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci U S A. doi:10.1073/pnas.1414592112
Fang M, Kremer RJ, Motavalli PP, Davis G (2005) Bacterial diversity in rhizospheres of nontransgenic and transgenic corn. Appl Environ Microbiol 71:4132–4136
Fierer N, Bradford MA, Jackson RB (2007) Toward an ecological classification of soil bacteria. Ecology 88:1354–1364
Girvan MS, Bullimore J, Pretty JN, Osborn AM, Ball AS (2003) Soil type is the primary determinant of the composition of the total and active bacterial communities in arable soils. Appl Environ Microbiol 69:1800–1809
Herschkovitz Y, Lerner A, Davidov Y, Rothballer M, Hartmann A, Okon Y, Jurkevitch E (2005) Inoculation with the plant-growth-promoting rhizobacterium Azospirillum brasilense causes little disturbance in the rhizosphere and rhizoplane of maize (Zea mays). Microb Ecol 50:277–288
Jones DL, Hodge A, Kuzyakov Y (2004) Plant and mycorrhizal regulation of rhizodeposition. New Phytol 163:459–480
Klein E, Ofek M, Katan J, Minz D, Gamliel A (2013) Soil suppressiveness to Fusarium disease: shifts in root microbiome associated with reduction of pathogen root colonization. Phytopathology 103:23–33
Leinster T, Cobbold CA (2012) Measuring diversity: the importance of species similarity. Ecology 93:477–489
Li XZ, Rui JP, Mao YJ, Yannarell A, Mackie R (2014) Dynamics of the bacterial community structure in the rhizosphere of a maize cultivar. Soil Biol Biochem 68:392–401
Lu Y, Wassmann R, Neue HU, Huang C (1999) Impact of phosphorus supply on root exudation, aerenchyma formation and methane emission of rice plants. Biogeochemistry 47:203–218
Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, del Rio TG, Edgar RC, Eickhorst T, Ley RE, Hugenholtz P, Tringe SG, Dangl JL (2012) Defining the core Arabidopsis thaliana root microbiome. Nature 488:86–90
Marquez LM, Redman RS, Rodriguez RJ, Roossinck MJ (2007) A virus in a fungus in a plant: three-way symbiosis required for thermal tolerance. Science 315:513–515
Meharg AA, Killham K (1995) Loss of exudates from the roots of perennial ryegrass inoculated with a range of microorganisms. Plant Soil 170:345–349
Minz D, Ofek M, Hadar Y (2013) Plant rhizosphere microbial communities. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (eds) The prokaryotes. Springer, Berlin, pp 56–84
Nunan N, Wu K, Young IM, Crawford JW, Ritz K (2002) In situ spatial patterns of soil bacterial populations, mapped at multiple scales, in an arable soil. Microb Ecol 44:296–305
Ofek M, Voronov-Goldman M, Hadar Y, Minz D (2014) Host signature effect on plant root-associated microbiomes revealed through analyses of resident vs. active communities. Environ Microbiol 16:2157–2167
Oger P, Mansouri H, Nesme X, Dessaux Y (2004) Engineering root exudation of lotus toward the production of two novel carbon compounds leads to the selection of distinct microbial populations in the rhizosphere. Microb Ecol 47:96–103
Pascault N, Nicolardot B, Bastian F, Thiebeau P, Ranjard L, Maron P-A (2010) In situ dynamics and spatial heterogeneity of soil bacterial communities under different crop residue management. Microb Ecol 60:291–303
Peiffer JA et al (2013) Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci U S A 110:6548–6553
Philippot L, Kuffner M, Cheneby D, Depret G, Laguerre G, Martin-Laurent F (2006) Genetic structure and activity of the nitrate-reducers community in the rhizosphere of different cultivars of maize. Plant Soil 287:177–186
Picard C, Bosco M (2005) Maize heterosis affects the structure and dynamics of indigenous rhizospheric auxins-producing Pseudomonas populations. FEMS Microbiol Ecol 53:349–357
Picard C, Bosco M (2006) Heterozygosis drives maize hybrids to select elite 2,4-diacethylphloroglucinol-producing Pseudomonas strains among resident soil populations. FEMS Microbiol Ecol 58:193–204
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO (2007) SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acid Res 35:7188–7196
Rainey PB (1999) Adaptation of Pseudomonas fluorescens to the plant rhizosphere. Environ Microbiol 1:243–257
Ryan PR, Dessaux Y, Thomashow LS, Weller DM (2009) Rhizosphere engineering and management for sustainable agriculture. Plant Soil 321:363–383
Schloss PD et al (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Schreiter S, Ding G-C, Heuer H, Neumann G, Sandmann M, Grosch R, Kropf S, Smalla K (2014) Effect of the soil type on the microbiome in the rhizosphere of field-grown lettuce. Front Microbiol 5:144
Sergeeva E, Hirkala DLM, Nelson LM (2007) Production of indole-3-acetic acid, aromatic amino acid aminotransferase activities and plant growth promotion by Pantoea agglomerans rhizosphere isolates. Plant Soil 297:1–13
Shen H, Wang XC, Shi WM, Cao ZH, Yan XL (2001) Isolation and identification of specific root exudates in elephantgrass in response to mobilization of iron- and aluminum-phosphates. J Plant Nut 24:1117–1130
Ulrich A, Becker R (2006) Soil parent material is a key determinant of the bacterial community structure in arable soils. FEMS Microbiol Ecol 56:430–443
Vassilev N, Vassileva M, Nikolaeva I (2006) Simultaneous P-solubilizing and biocontrol activity of microorganisms: potentials and future trends. Appl Microbiol Biotechnol 71:137–144
Wang Y, Qian P-Y (2009) Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS ONE 4:e7401
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Zhou X, Wu F (2012) Effects of amendments of ferulic acid on soil microbial communities in the rhizosphere of cucumber (Cucumis sativus L.). Eur J Soil Biol 50:191–197
Zolla G, Bakker MG, Badri DV, Chaparro JM, Sheflin AM, Manter DK, Vivanco J (2013) Understanding root-microbiome interactions. In: de Bruijn FJ (ed) Molecular microbial ecology of the rhizosphere. Wiley, Hoboken, pp 743–754
Acknowledgments
Shawn Salley provided assistance in locating soils and performed the texture analysis. MGB was supported by a USDA NIFA Postdoctoral Fellowship grant (2011-67012-30938) for a portion of this work. Mention of trade names or commercial products is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. This article was the work of U.S. Government employees engaged in their official duties and is exempt from copyright.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Gera Hol.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 210 kb)
Rights and permissions
About this article

Cite this article
Bakker, M.G., Chaparro, J.M., Manter, D.K. et al. Impacts of bulk soil microbial community structure on rhizosphere microbiomes of Zea mays . Plant Soil 392, 115–126 (2015). https://doi.org/10.1007/s11104-015-2446-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-015-2446-0