
Abstract
Targeted gene regulation on a genome-wide scale is a powerful strategy for interrogating, perturbing, and engineering cellular systems. Recent advances with the RNA-mediated Cas9 endonuclease derived from clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated proteins (Cas) systems have dramatically transformed our ability to specifically modify intact genomes of diverse cells and organisms. The CRISPR–Cas system has been adapted as an efficient, facile, and robust gene-targeting technology with the potential for high-throughput and multiplexed genome engineering. Exciting breakthroughs in understanding the mechanisms of the CRISPR–Cas system and its enormous potential for applications across basic science, agricultural and biotechnology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the past decade, precise and efficient genome targeting technologies have emerged that enable systematic reverse engineering of causal genetic variations by allowing selective perturbation of individual genetic elements. RNA interference (RNAi), Zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) have provided valuable technologies for targeted gene regulation in a diverse range of cell types and model organisms (Joung and Sander 2012; Zhang et al. 2010, 2013b; Li et al. 2012; Gaj et al. 2013; Mussolino and Cathomen 2013; Streubel et al. 2012; Chang et al. 2013; Hockemeyer et al. 2011; Shan et al. 2013a; Hannon 2002). RNAi can be used in a relatively rapid, inexpensive, powerful, reliable, and high-throughput method for genome-wide loss-off unction screening (Berns et al. 2004; Boutros et al. 2004), however, RNAi is also imperfect because it only temporary inhibition of gene function and can exhibit unpredictable off-target effects on other mRNAs (Echeverri et al. 2006; Kaelin 2012). In addition, custom DNA-binding proteins, ZFNs and TALENs are hybrid proteins created by fusing ZF or TALE DNA-binding domain to the nonspecific cleavage domain of FokI endonuclease (Nekrasov et al. 2013; Gaj et al. 2013; Bogdanove and Voytas 2011). The FokI endonuclease nonspecific cleavage domain must dimerize to cleave the DNA target (Klug 2010; Moscou and Bogdanove 2009). ZFNs and TALENs can be programmed to cleave genomes in specific locations, however, these technologies demand elaborate design and assembly of individual DNA-binding proteins for each DNA target sequence. These chimeric nucleases have been successful in genome modifications by generating DNA double-strand breaks (DSBs) that stimulate the standard cellular DNA repair mechanisms, including error-prone non-homologous end joining (NHEJ) and homology-directed repair (HDR) (Weinthal et al. 2010; Gaj et al. 2013; Charpentier and Doudna 2013). NHEJ-mediated repair typically leads to the indels and introduction of small deletions/insertions at the site of the break, resulting in knockout of gene function via frameshift mutations (Gaj et al. 2013; Chang et al. 2013). HDR, however, requires a homologous DNA segment as a template to correct or replace existing genes (Weinthal et al. 2010; Charpentier and Doudna 2013; Gaj et al. 2013). Although the low efficiency of HDR in a variety of cell types and organisms, it can be used to generate precise, defined modifications at the target site.
Recently, another markedly simple, versatile, efficient and breakthrough genome engineering technology for genome editing, the clustered regularly interspaced short palindromic repeats-CRISPR-associated protein (CRISPR–Cas) system, was developed.
Mechanisms of the CRISPR–Cas defense system
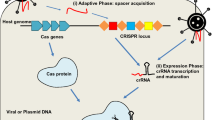
In Ishino et al. (1987), a research team observed an unusual repetitive segment of neighbouring bacterial gene. Before 2005, many researchers assumed that these odd sequences were junk, however, three various groups reported that these segments often matched the sequences of phages or plasmids, and indicating a possible role for CRISPR in immunity against transmissible genetic elements (Bolotin et al. 2005; Mojica et al. 2005; Pourcel et al. 2005). CRISPR–Cas systems constitute a widespread class of immunity systems that protect bacteria and archaea from invading viruses and plasmids via RNA-guided DNA cleavage in three steps (Wiedenheft et al. 2012; Gaj et al. 2013; Marx 2007) (Fig. 1). During the acquisition phase, recognition and subsequent integration of viral or plasmid DNA-derived spacers between two adjacent repeat units within the CRISPR loci (Barrangou et al. 2007; Garneau et al. 2010; Yosef et al. 2012; Deveau et al. 2008; Swarts et al. 2012; Datsenko et al. 2012; Cady et al. 2012; Lopez-Sanchez et al. 2012; Deltcheva et al. 2011) (Fig. 1). During the expression phase, the CRISPR loci are transcribed as a precursor CRISPR RNA (pre-crRNA) containing the full set of CRISPR repeats and embedded invader-derived sequences from the leader region (Deltcheva et al. 2011). Next, specific endoribonucleases cleave the pre-crRNAs into short guide CRISPR RNAs (crRNAs) consisting of unique single repeat-spacer element (Deltcheva et al. 2011; Brouns et al. 2008; Carte et al. 2008; Haurwitz et al. 2010; Hatoum-Aslan et al. 2011; Garside et al. 2012; Gesner et al. 2011; Sashital et al. 2011; Charpentier and Doudna 2013) (Fig. 1). During the interference phase, the mature crRNA is incorporated into a large multiprotein complex, called CRISPR-associated complex for antiviral defense (CASCADE), can recognize and base-pair specifically with regions of incoming cognate-invading nucleic acids that have perfect complementarity, triggering degradation or silencing of the foreign sequences (Garneau et al. 2010; Wiedenheft et al. 2012; Deveau et al. 2010; Horvath and Barrangou 2010; Koonin and Makarova 2009; Marraffini and Sontheimer 2008, 2010a, b; Sorek et al. 2008; van der Oost et al. 2009; Waters and Storz 2009; Hale et al. 2009; Beloglazova et al. 2011; Jore et al. 2011; Mulepati and Bailey 2011; Wiedenheft et al. 2011a, b; Makarova et al. 2011) (Fig. 1).

Diversity of CRISPR-mediated adaptive immune systems. CRISPR–Cas systems act in three stages: acquisition, expression and interference. Specific protospacers (with an adjacent PAM) of double-stranded DNA from a invading virus or plasmid are acquired at the leader end of a CRISPR array on host DNA. Each CRISPR locus consists of a series of direct repeats separated by unique spacer sequences acquired from protospacers (Marraffini and Sontheimer 2008, 2010b). After the initial recognition step, Cas1 and Cas2 usually located in the vicinity of the CRISPR array, most probably incorporate the protospacers into the CRISPR locus to form spacers. Pre-crRNA is transcribed from the leader region by RNA polymerase and further processed into short mature crRNAs. The interference process is different in the Type I, II, and III systems. In type I and III, the CASCADE complex binds pre-crRNA, which is cleaved by a CRISPR-specific endoribonuclease, resulting in crRNAs with a typical 8-nt upstream of each spacer sequence (Gesner et al. 2011; Haurwitz et al. 2010; Carte et al. 2010). In type III, Cas6 is responsible for the processing step, but the crRNAs seem to be transferred to a specific Cas complex (Csm in subtype III-A and Cmr in subtype III-B) (Carte et al. 2008). In Type II, a tracrRNA with the repeat region of the pre-crRNA, followed by cleavage within the repeats by the host RNase III in the presence of Cas9 (Deltcheva et al. 2011). The final step results in cleavage of invading nucleic acid and proceeds compelling differences in all systems. In Type I, crRNA with CASCADE complex along with the Cas3 subunit can target that contain complementary target DNA and is probably responsible for cleavage of invading DNA (Sontheimer and Marraffini 2010; Jore et al. 2011; Wiedenheft et al. 2011b; Sinkunas et al. 2011). The two subtypes of CRISPR–Cas type III systems target either DNA (subtype III-A Marraffini and Sontheimer 2008) or RNA (subtype III-B Hale et al. 2009) and a PAM does not appear to be required for the activity of Type III. In Type II, Cas9 loaded with crRNA can probably target invading DNA for cleavage (open orange triangle) in a process that requires the PAM (Haurwitz et al. 2010). Modified from (Makarova et al. 2011)
Architecture and characters of CRISPR systems
CRISPR loci typically consist of several noncontiguous, highly conserved direct repeats separated by stretches of variable sequences called spacers which mostly correspond to sequences of captured viral and plasmid sequences and are often adjacent to groups of conserved protein-encoding genes, named cas genes (Horvath and Barrangou 2010). Based on recent bioinformatic analyses, cas genes encode a large and heterogeneous family of proteins that carry identifiable functional domains typical of nucleases, helicases, polymerases, and polynucleotide-binding proteins, which led to the initial speculation that they may be part of a novel DNA repair system. CRISPR–Cas system can be divided into two partially independent subsystems: the highly conserved ‘information processing’ subsystem involved in the adaptation phase and requires the universally present core proteins, Cas1 and Cas2, and the ‘executive’ subsystem, involved in crRNA processing and interference with invading foreign nucleic acid, and is quite diverse (Bhaya et al. 2011; Makarova et al. 2011; Horvath and Barrangou 2010; van der Oost et al. 2009). Repeat-associated mysterious proteins (RAMPs or Cmr) that constitute a large superfamily of Cas proteins, contain at least one RNA recognition motif (RRM; it is also called the ferredoxin-fold domain), which is somewhat functionally analogous to CASCADE, and have been shown to be involved in the processing of pre-crRNA transcripts (Makarova et al. 2011; Hale et al. 2009; Horvath and Barrangou 2010).
Based on this classification that integrates phylogeny, gene conservation, locus organization, and content, CRISPR–Cas system have recently been classified into three distinct, type I, type II, and type III (Bhaya et al. 2011; Wiedenheft et al. 2012; Makarova et al. 2011) (Fig. 1). The classification reflects an evolution of the defense system into subtype-specific molecular mechanisms for expression and maturation of crRNAs and interference with invaders (Makarova et al. 2011). The type I and III systems share some biochemically and structurally features: multiple specialized Cas proteins that form CASCADE-like complexes with demonstrated RNAse activity are present in several copies in both type I and III system. Cas endonucleases processes pre-crRNA into mature crRNAs, and each crRNA assembles into a large Cas effector complexes use these processed crRNAs to recognize and cleave cognate-invading nucleic acids (Haurwitz et al. 2010; Jinek et al. 2012; Makarova et al. 2006, 2011; Wiedenheft et al. 2012) (Fig. 1). In contrast, type II systems have evolved distinct pre-crRNA processing and interference mechanisms in which a trans-activating crRNA (tracrRNA) binds to the repeat sequences of pre-crRNA forming a dual-RNA. Doubles tranded (ds) RNA-specific ribonuclease RNase III cleaves an RNA duplex formed by the CRISPR repeat and a trans-activating CRISPR RNA (tracrRNA) (Jinek et al. 2012; Deltcheva et al. 2011; Bhaya et al. 2011; Chylinski et al. 2013; Wiedenheft et al. 2012; Makarova et al. 2011) (Fig. 1). In addition, the three types of CRISPR–Cas system show a distinctly non-uniform distribution, with the type I system have been found in both bacteria and archaea, whereas the type III system appear more commonly in archaea. In particular, the type II system are exclusively widespread in bacteria so far (Makarova et al. 2011; Terns and Terns 2011; Bhaya et al. 2011).
Cas9 as an RNA-guided nuclease for genome editing
The best-studied Type II systems are the simplest of the three CRISPR–Cas types, with only four cas genes, one of which is always Cas9 (formerly Csn1) (Chang et al. 2013; Jinek et al. 2013). Cas9 is a single protein, a crRNA-guided double-stranded DNA endonuclease with two nuclease domains, an HNH (McrA-like) nuclease domain that cleaves the complementary DNA strand and a RuvC-like nuclease domain that cleaves the noncomplementary DNA strand (Jinek et al. 2012, 2014; Bikard et al. 2013; Chylinski et al. 2013; Fonfara et al. 2013; Jiang et al. 2013a) (Fig. 2). To form a functional DNA-targeting complex, target recognition and cleavage by the Cas9 protein requires a chimeric single-guide RNA (sgRNA) consisting of a fusion of crRNA (Each crRNA unit then contains a 20-nt guide sequence and a partial direct repeat) and tracrRNA and a short conserved sequence motif downstream of the crRNA-binding region, called CRISPR motifs or protospacer adjacent motif (PAM) (Jinek et al. 2012; Garneau et al. 2010; Jiang et al. 2013a; Feng et al. 2013; Fu et al. 2013; Hsu et al. 2013; Carroll 2012) (Fig. 2). In the CRISPR–Cas system derived from the bacterium Streptococcus pyogenes, the target DNA must immediately precede a 5′-NGG PAM (Jinek et al. 2012), whereas, it has been shown that many type II systems have differing PAM requirements, which may constrain their ease of targeting (Mali et al. 2013b; Cong et al. 2013; Garneau et al. 2010; Gasiunas et al. 2012; Sapranauskas et al. 2011; Zhang et al. 2013a). RNA-guided Cas9 activity creates site-specific DSBs, which are then repaired by either NHEJ or HDR, the sequence at the repair site can be modified or new genetic information inserted (Cong et al. 2013; Mali et al. 2013c; Cho et al. 2013) (Fig. 2). More intriguingly, the Cas9 protein and the sgRNA are the only a minimal set of two molecules necessary for induction of targeted invading DNA cleavage.

Targeted genome editing with RNA-guided Cas9. In a type II CRISPR–Cas system, Cas9 generates a blunt-ended double-stranded break 3 bp upstream of PAM through a process mediated by two catalytic domains in the protein, an HNH domain and a RuvC-like domain each of which cleaves one strand within the target DNA (Mali et al. 2013a; Jinek et al. 2012). Cas9 nucleases carry out strand-specific cleavage (Jinek et al. 2012; Ran et al. 2013b). Nuclease-induced DSBs can be repaired by NHEJ-mediated disruption of the genome and HDR-mediated modification of the genome (Mali et al. 2013a, b, c; Cong et al. 2013)
What makes the CRISPR–Cas9 system even more attractive is the ease, high efficiency, and versatility of the technology. Martin Jinek, designed a single RNA molecule of dual-tracrRNA:crRNA (sgRNA), successfully mixed it with specific Cas9 and showed that the synthetic complexes could target and cleave any dsDNA sequence of interest (Jinek et al. 2012). The type II CRISPR system from bacteria has been rapidly applied to achieve efficient robust RNA-guided genome editing in different species (Horvath and Barrangou 2010; Jiang et al. 2013a; Jinek et al. 2012; Makarova et al. 2011; Marraffini and Sontheimer 2010a; Sorek et al. 2008; Wiedenheft et al. 2012). Significantly, recent studies demonstrate that CRISPR–Cas system can function in human cells. Several researchers engineered a synthetic sgRNA consisting of a fusion of crRNA and tracrRNA can direct ‘humanized’ Cas9 endonuclease in various human cell lines, including induced pluripotent stem cells, they observed the expected alterations to the target DNA (Cong et al. 2013; Jinek et al. 2013; Mali et al. 2013c; Fu et al. 2013; Cho et al. 2013). Cas9 endonucleases that have also been shown to act as a nickases, enabling an additional level of control over the mechanism of DNA repair (Cong et al. 2013; Mali et al. 2013a). Up to now, in addition to human cells, CRISPR–Cas system has been successfully applied to achieve efficient genome editing in many eukaryotic organisms including Saccharomyces cerevisiae (DiCarlo et al. 2013), Caenorhabditis elegans (Dickinson et al. 2013; Friedland et al. 2013), Drosophila (Yu et al. 2013), zebrafish (Chang et al. 2013; Hwang et al. 2013; Jao et al. 2013), mouse (Shen et al. 2013; Li et al. 2013a; Wang et al. 2013a; Wu et al. 2013; Yang et al. 2013), rat (Li et al. 2013a, 2013c), and, at the same time, the feasibility and efficacy of CRISPR–Cas system has also been successfully demonstrated in the plants Arabidopsis thaliana (Feng et al. 2013; Li et al. 2013b; Jiang et al. 2013b), Nicotiana benthamiana (Li et al. 2013b; Nekrasov et al. 2013), and cultivated food crop rice (Oryza sativa) (Feng et al. 2013; Miao et al. 2013; Shan et al. 2013b; Jiang et al. 2013b), wheat (Triticum aestivum) (Shan et al. 2013b) and sorghum (Jiang et al. 2013b) (Fig. 3). Indeed, these findings hint that RNA-guided Cas9 might be useful for engineering other multicellular organisms, including animals and plants. Recently, both research groups demonstrated that further functionality of RNA-guided CRISPR–Cas9 system in both human and mouse cells and that multiplex editing of target genes is feasible upon introduction of multiple sgRNAs at the same time (Cong et al. 2013; Mali et al. 2013c; Pennisi 2013). Use Cas9 system, Li and colleagues have successfully targeted five target genes in Arabidopsis or N. benthamiana, and achieve efficient targeted mutagenesis in all cases (Li et al. 2013b). Subsequently, Gao’s and Zhu’s team have highly efficient targeted mutagenesis in multiple genes in rice (Shan et al. 2013b; Feng et al. 2013). Importantly, stable expression of the Cas9 system in transgenic animals and plants led to mutations in target genes. Impressively, the system was modified to create a more efficient and well-suited, enabling multiple endogenous genes editing by programming Cas9 to edit several sites in a genome simultaneously by simply using multiple guide RNAs (Shan et al. 2013b; Li et al. 2013a, b; Wang et al. 2013a). These pioneering experiments provide dramatic evidence that the technique could be used to engineer these model plant systems and crucial crop species.

Potential application of CRISPR–Cas9 systems. In addition to immunity systems that protect bacteria and archaea from invading viruses and plasmids. The diverse potential applications of Cas9 range from targeted genome editing to targeted genome regulation and possibly to one capable of introducing custom changes in the complex epigenome
In addition to genome editing, CRISPR interference (CRISPRi) can efficiently and selectively repress or activate transcription of targeted genes using a modified Cas9 protein lacking endonucleolytic activity (Qi et al. 2013; Gilbert et al. 2013; Larson et al. 2013) (Fig. 3). Thus, CRISPRi has the potential to be utilized as an efficient and flexible platform for engineering transcriptional regulatory networks control without altering the target DNA sequence. Furthermore, Cas9nuclease-null has been used to target proteins with specific functions to edit the epigenome (Rusk 2014). Further regulation will be able to occur through histone modification (acetylation and methylation) and, hence, change chromatin states and DNA methylation (Fig. 3).
Limitation and expansion of the Cas9 system
Although CRISPR–Cas system show great promise and flexibility for genetic engineering, sequence requirements within the PAM sequence may constrain some applications. In addition to the targeting range, another key question concerning the specificity of CRISPR–Cas RNA-guided endonucleases is whether off-target cleavage is required to evaluate. The issue of specificity is paramount for all the targetable nucleases. Currently, off-target cleavage by ZFNs and TALENs has been reduced by modifying the cleavage domain to require the formation of heterodimers (Carroll 2013). Present early-phase versions of the Cas9 system may also suffer to some degree from the same problem. In the CRISPR–Cas system, earlier studies have demonstrated that, although each base within the 20 nt guide sequence contributes to overall specificity, some base mismatches between the guide RNA and target DNA are tolerated depending on the quantity, position, and base identity of mismatches leading to potential off-target DSBs formation (Cong et al. 2013; Fu et al. 2013; Hsu et al. 2013; Jiang et al. 2013a). It has been reported that there is a high frequency of off-target effect of CRISPR–Cas-induced mutagenesis in human cells (Pattanayak et al. 2013; Fu et al. 2013) and a lower off-target effect in mice and zebrafish (Yang et al. 2013; Hruscha et al. 2013). Besides several studies using genome-wide sequencing found no detectable off-target genome modifications in Arabidopsis and N. benthamiana (Feng et al. 2014; Nekrasov et al. 2013). Nevertheless, more comprehensive studies are required to thoroughly address the off-target issue for the CRISPR–Cas system in other plant species or for other target genes. For routine application of Cas9, it is important to consider ways to reduce the frequency of unexpected mutations from off-target genome modification and to be able to detect the presence of off-target cleavage (Hsu et al. 2013; Fu et al. 2013; Jiang et al. 2013a). Although imperfect Cas9 specificity is a major reason for concern, there are several methods of potentially improving this. The challenges will be to analyse and address possible off-target effects and improve the efficiency and specificity of the system. Potential attractive strategy minimizes off-target mutagenesis include exploiting different Cas9 homologs identified through bioinformatics and directed evolution of these nucleases toward higher specificity. Alternatively, the range of targetable sites could be expanded through the use of homologs with different cognate PAM sequences. Additionally, a previously report shown that a Cas9 nickase mutant (Cas9n) cut only one DNA strand, and facilitated HDR at on-target sites can potentially increase the specificity of target recognition (Cong et al. 2013). More recently, a double-nicking strategy of combining Cas9n with paired guide RNAs by comparison, maintains high on-target efficiencies while drastically reducing off-target modifications to background levels (Ran et al. 2013a). In particular, a more thorough sequencing analysis for a large number of sgRNAs will also provide more information about the potential off-target cleavage of the CRISPR–Cas system and lead to a better prediction of potential off-target sites.
Comparison with other genome editing technologies
ZFNs, TALENs, and RNA-guided DNA endonucleases are transformative tools that have broad implications for synthetic biology, the direct and multiplexed perturbation of gene networks, and targeted in vitro and in vivo gene therapy (Gaj et al. 2013). Whereas, CRISPR–Cas9 system offers several potential advantages over ZFNs and TALENs. The complex designs of ZNFs or TALENs for each target gene and the efficiency of targeting may vary substantially, no multiplexed gene-targeting has been reported to date. However, compared with ZFNs and TALENs, CRISPR–Cas9 system not only offer a simpler means of attaining specificity and demonstrate equal or greater cleavage efficacy, but also provide a gene editing tool that can more easily be targeted to one or more genomic loci. Furthermore, ZNFs and TALENs locate target sequences using proteins that are often difficult and costly to produce. Given that the CRISPR–Cas9 system’s sgRNAs are now much easier to make than proteins exploited in ZNFs and TALENs genome engineering technologies. CRISPR systems have stormed onto the scene, promising to even out-compete ZNFs and TALENs. Ultimately, CRISPR may take a place beside ZNFs and TALENs, with the choice of editing tool depending on the particular application.
Future directions
The discovery and application of bacterial systems, have revolutionized molecular biology in the past. But for now, despite the intricacies of significant progress has been made in the last few years, many central aspects remain obscure. An important question is how safe, effective and specific are CRISPR–Cas9 system is not well understood. In addition, what is the optimal RNA scaffold for powerful application of CRISPR–Cas9 in multiple eukaryotic systems is still unclear. Furthermore, how effective is Cas9 systems as a basis for generating versatile and heritable modifications specifically at target genes between animals and plants also await elucidation. Intriguingly, more research will raise new questions and highlight the areas with the greatest potential for future research.
Given the dizzying rate at which CRISPR-targeting publications are appearing, researchers are clearly eager to capitalize on these advantages. Ideally, most research teams are to build a library of CRISPRs that can be harnessed to target any sequences in an organism’s entire genome, including promoters, enhancers, introns, and intergenic regions, which are inaccessible by means of RNAi (Shalem et al. 2013). In particular, null off-target mutagenesis using the CRISPR–Cas9 system could overcome one of the major limitations of RNAi, which would allow access to an entirely new repertoire of regulation of gene function (Wang et al. 2013b). Just a few days ago, two studies were published using CRISPRs for genome-scale loss-of-function screens in human cells. Moreover, relative to other methods of plant genome engineering and editing, the CRISPR–Cas9 system should be applicable to a wide range of higher plants. Notably, the CRISPR–Cas9 system facilitates HDR, in the future the technique will be successfully applied to precisely insert into a specific location of other cereals with more complicated genomes, which requires future investigation. Further, this creates a valuable new tool holds significant promise for plant biologists and breeders. Looking forward, the versatility and ease of use afforded by RNA-guided Cas9 enzymes coupled with its singular ability to bring together RNA and DNA in a fully programmable fashion will form the basis of a versatile tool for rewriting genomic sequence information that has the potential to explore and reshape any genome and constitute a new and promising paradigm to understand.
References
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315(5819):1709–1712
Beloglazova N, Petit P, Flick R, Brown G, Savchenko A, Yakunin AF (2011) Structure and activity of the Cas3 HD nuclease MJ0384, an effector enzyme of the CRISPR interference. EMBO J 30(22):4616–4627
Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B (2004) A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428(6981):431–437
Bhaya D, Davison M, Barrangou R (2011) CRISPR–Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet 45:273–297. doi:10.1146/annurev-genet-110410-132430
Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA (2013) Programmable repression and activation of bacterial gene expression using an engineered CRISPR–Cas system. Nucleic Acids Res 41(15):7429–7437. doi:10.1093/nar/gkt520
Bogdanove AJ, Voytas DF (2011) TAL effectors: customizable proteins for DNA targeting. Science 333(6051):1843–1846
Bolotin A, Quinquis B, Sorokin A, Ehrlich SD (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151(8):2551–2561
Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B, Haas SA, Paro R, Perrimon N (2004) Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 303(5659):832–835
Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, Van Der Oost J (2008) Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321(5891):960–964
Cady KC, Bondy-Denomy J, Heussler GE, Davidson AR, O’Toole GA (2012) The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J Bacteriol 194(21):5728–5738
Carroll D (2012) A CRISPR approach to gene targeting. Mol Ther 20(9):1658–1660
Carroll D (2013) Staying on target with CRISPR–Cas. Nat Biotechnol 31(9):807–809
Carte J, Wang R, Li H, Terns RM, Terns MP (2008) Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev 22(24):3489–3496
Carte J, Pfister NT, Compton MM, Terns RM, Terns MP (2010) Binding and cleavage of CRISPR RNA by Cas6. RNA 16(11):2181–2188
Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X, Xiong JW, Xi JJ (2013) Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res 23(4):465–472. doi:10.1038/cr.2013.45
Charpentier E, Doudna JA (2013) Biotechnology: rewriting a genome. Nature 495(7439):50–51
Cho SW, Kim S, Kim JM, Kim J-S (2013) Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31:230–232
Chylinski K, Le Rhun A, Charpentier E (2013) The tracrRNA and Cas9 families of type II CRISPR–Cas immunity systems. RNA Biol 10(5):726–737. doi:10.4161/rna.24321
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823
Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, Semenova E (2012) Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun 3:945
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471(7340):602–607. doi:10.1038/nature09886
Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S (2008) Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190(4):1390–1400. doi:10.1128/JB.01412-07
Deveau H, Garneau JE, Moineau S (2010) CRISPR/Cas system and its role in phage-bacteria interactions. Annu Rev Microbiol 64:475–493
DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM (2013) Genome engineering in Saccharomyces cerevisiae using CRISPR–Cas systems. Nucleic Acids Res 41(7):4336–4343. doi:10.1093/nar/gkt135
Dickinson DJ, Ward JD, Reiner DJ, Goldstein B (2013) Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods 10(10):1028–1034
Echeverri CJ, Beachy PA, Baum B, Boutros M, Buchholz F, Chanda SK, Downward J, Ellenberg J, Fraser AG, Hacohen N (2006) Minimizing the risk of reporting false positives in large-scale RNAi screens. Nat Methods 3(10):777–779
Feng Z, Zhang B, Ding W, Liu X, Yang DL, Wei P, Cao F, Zhu S, Zhang F, Mao Y, Zhu JK (2013) Efficient genome editing in plants using a CRISPR/Cas system. Cell Res 23(10):1229–1232
Feng Z, Mao Y, Xu N, Zhang B, Wei P, Yang DL, Wang Z, Zhang Z, Zheng R, Yang L, Zeng L, Liu X, Zhu JK (2014) Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc Natl Acad Sci USA. doi:10.1073/pnas.1400822111
Fonfara I, Le Rhun A, Chylinski K, Makarova KS, Lecrivain AL, Bzdrenga J, Koonin EV, Charpentier E (2013) Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR–Cas systems. Nucleic Acids Res. doi:10.1093/nar/gkt1074
Friedland AE, Tzur YB, Esvelt KM, Colaiácovo MP, Church GM, Calarco JA (2013) Heritable genome editing in C. elegans via a CRISPR–Cas9 system. Nat Methods 10(8):741–743
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD (2013) High-frequency off-target mutagenesis induced by CRISPR–Cas nucleases in human cells. Nat Biotechnol 31(9):822–826. doi:10.1038/nbt.2623
Gaj T, Gersbach CA, Barbas CF 3rd (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31(7):397–405. doi:10.1016/j.tibtech.2013.04.004
Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan AH, Moineau S (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468(7320):67–71. doi:10.1038/nature09523
Garside EL, Schellenberg MJ, Gesner EM, Bonanno JB, Sauder JM, Burley SK, Almo SC, Mehta G, MacMillan AM (2012) Cas5d processes pre-crRNA and is a member of a larger family of CRISPR RNA endonucleases. RNA 18(11):2020–2028
Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci 109(39):E2579–E2586
Gesner EM, Schellenberg MJ, Garside EL, George MM, MacMillan AM (2011) Recognition and maturation of effector RNAs in a CRISPR interference pathway. Nat Struct Mol Biol 18(6):688–692
Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154(2):442–451. doi:10.1016/j.cell.2013.06.044
Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP (2009) RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139(5):945–956. doi:10.1016/j.cell.2009.07.040
Hannon GJ (2002) RNA interference. Nature 418(6894):244–251
Hatoum-Aslan A, Maniv I, Marraffini LA (2011) Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc Natl Acad Sci 108(52):21218–21222
Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA (2010) Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329(5997):1355–1358. doi:10.1126/science.1192272
Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, Cost GJ, Zhang L, Santiago Y, Miller JC (2011) Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol 29(8):731–734
Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327(5962):167–170
Hruscha A, Krawitz P, Rechenberg A, Heinrich V, Hecht J, Haass C, Schmid B (2013) Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 140(24):4982–4987
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31(9):827–832. doi:10.1038/nbt.2647
Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JJ, Joung JK (2013) Efficient genome editing in zebrafish using a CRISPR–Cas system. Nat Biotechnol 31(3):227–229
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169(12):5429–5433
Jao L-E, Wente SR, Chen W (2013) Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci 110(34):13904–13909
Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA (2013a) RNA-guided editing of bacterial genomes using CRISPR–Cas systems. Nat Biotechnol 31(3):233–239. doi:10.1038/nbt.2508
Jiang W, Zhou H, Bi H, Fromm M, Yang B, Weeks DP (2013b) Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res 41(20):e188. doi:10.1093/nar/gkt780
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821
Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J (2013) RNA-programmed genome editing in human cells. Elife 2:e00471
Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E, Doudna JA (2014) Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science (New York, NY)
Jore MM, Lundgren M, van Duijn E, Bultema JB, Westra ER, Waghmare SP, Wiedenheft B, Pul Ü, Wurm R, Wagner R (2011) Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat Struct Mol Biol 18(5):529–536
Joung JK, Sander JD (2012) TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14(1):49–55
Kaelin WG (2012) Use and abuse of RNAi to study mammalian gene function. Science 337(6093):421–422
Klug A (2010) The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem 79:213–231
Koonin EV, Makarova KS (2009) CRISPR–Cas: an adaptive immunity system in prokaryotes. F1000 biology reports 1:95
Larson MH, Gilbert LA, Wang X, Lim WA, Weissman JS, Qi LS (2013) CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protoc 8(11):2180–2196. doi:10.1038/nprot.2013.132
Li T, Liu B, Spalding MH, Weeks DP, Yang B (2012) High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat Biotechnol 30(5):390–392
Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X (2013a) Heritable gene targeting in the mouse and rat using a CRISPR–Cas system. Nat Biotechnol 31(8):681–683
Li JF, Norville JE, Aach J, McCormack M, Zhang D, Bush J, Church GM, Sheen J (2013b) Multiplex and homologous recombination–mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol 31:688–691. doi:10.1038/nbt.265010.1038/nbt.2623
Li W, Teng F, Li T, Zhou Q (2013c) Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR–Cas systems. Nat Biotechnol 31(8):684–686
Lopez-Sanchez MJ, Sauvage E, Da Cunha V, Clermont D, Ratsima Hariniaina E, Gonzalez-Zorn B, Poyart C, Rosinski-Chupin I, Glaser P (2012) The highly dynamic CRISPR1 system of Streptococcus agalactiae controls the diversity of its mobilome. Mol Microbiol 85(6):1057–1071
Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV (2006) A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1(1):7
Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF (2011) Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol 9(6):467–477. doi:10.1038/nrmicro2577
Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM (2013a) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31(9):833–838. doi:10.1038/nbt.2675
Mali P, Esvelt KM, Church GM (2013b) Cas9 as a versatile tool for engineering biology. Nat Methods 10(10):957–963. doi:10.1038/nmeth.2649
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013c) RNA-guided human genome engineering via Cas9. Science 339(6121):823–826
Marraffini LA, Sontheimer EJ (2008) CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322(5909):1843–1845
Marraffini LA, Sontheimer EJ (2010a) CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet 11(3):181–190. doi:10.1038/nrg2749
Marraffini LA, Sontheimer EJ (2010b) Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463(7280):568–571
Marx J (2007) New bacterial defense against phage invaders identified. Science 315(5819):1650–1651
Miao J, Guo D, Zhang J, Huang Q, Qin G, Zhang X, Wan J, Gu H, Qu LJ (2013) Targeted mutagenesis in rice using CRISPR–Cas system. Cell Res 23(10):1233–1236. doi:10.1038/cr.2013.123
Mojica FJ, García-Martínez J, Soria E (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60(2):174–182
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Science 326(5959):1501
Mulepati S, Bailey S (2011) Structural and biochemical analysis of nuclease domain of clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein 3 (Cas3). J Biol Chem 286(36):31896–31903
Mussolino C, Cathomen T (2013) RNA guides genome engineering. Nat Biotechnol 31(3):208–209
Nekrasov V, Staskawicz B, Weigel D, Jones JD, Kamoun S (2013) Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat Biotechnol 31(8):691–693
Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR (2013) High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31(9):839–843. doi:10.1038/nbt.2673
Pennisi E (2013) The CRISPR craze. Science 341:833–836
Pourcel C, Salvignol G, Vergnaud G (2005) CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151(3):653–663
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152(5):1173–1183. doi:10.1016/j.cell.2013.02.022
Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F (2013a) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154(6):1380–1389. doi:10.1016/j.cell.2013.08.021
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013b) Genome engineering using the CRISPR–Cas9 system. Nat Protoc 8(11):2281–2308. doi:10.1038/nprot.2013.143
Rusk N (2014) CRISPRs and epigenome editing. Nat Methods 11(1):28
Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V (2011) The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res 39(21):9275–9282
Sashital DG, Jinek M, Doudna JA (2011) An RNA-induced conformational change required for CRISPR RNA cleavage by the endoribonuclease Cse3. Nat Struct Mol Biol 18(6):680–687
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F (2013) Genome-scale CRISPR–Cas9 knockout screening in human cells. Science 303:84–87
Shan Q, Wang Y, Chen K, Liang Z, Li J, Zhang Y, Zhang K, Liu J, Voytas DF, Zheng X (2013a) Rapid and efficient gene modification in rice and Brachypodium using TALENs. Molecular plant 6(4):1365–1368
Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu J-L (2013b) Targeted genome modification of crop plants using a CRISPR–Cas system. Nat Biotechnol 31(8):686–688
Shen B, Zhang J, Wu H, Wang J, Ma K, Li Z, Zhang X, Zhang P, Huang X (2013) Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res 23:720–723
Sinkunas T, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V (2011) Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J 30(7):1335–1342
Sontheimer EJ, Marraffini LA (2010) Microbiology: slicer for DNA. Nature 468(7320):45–46
Sorek R, Kunin V, Hugenholtz P (2008) CRISPR—a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat Rev Microbiol 6(3):181–186
Streubel J, Blücher C, Landgraf A, Boch J (2012) TAL effector RVD specificities and efficiencies. Nat Biotechnol 30(7):593–595
Swarts DC, Mosterd C, van Passel MW, Brouns SJ (2012) CRISPR interference directs strand specific spacer acquisition. PLoS One 7(4):e35888
Terns MP, Terns RM (2011) CRISPR-based adaptive immune systems. Curr Opin Microbiol 14(3):321–327
van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJ (2009) CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci 34(8):401–407
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R (2013a) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153(4):910–918. doi:10.1016/j.cell.2013.04.025
Wang T, Wei JJ, Sabatini DM, Lander ES (2013b) Genetic screens in human cells using the CRISPR/Cas9 system. Science 303:80–84
Waters LS, Storz G (2009) Regulatory RNAs in bacteria. Cell 136(4):615–628
Weinthal D, Tovkach A, Zeevi V, Tzfira T (2010) Genome editing in plant cells by zinc finger nucleases. Trends Plant Sci 15(6):308–321
Wiedenheft B, Lander GC, Zhou K, Jore MM, Brouns SJ, van der Oost J, Doudna JA, Nogales E (2011a) Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature 477(7365):486–489
Wiedenheft B, van Duijn E, Bultema JB, Waghmare SP, Zhou K, Barendregt A, Westphal W, Heck AJ, Boekema EJ, Dickman MJ (2011b) RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc Natl Acad Sci 108(25):10092–10097
Wiedenheft B, Sternberg SH, Doudna JA (2012) RNA-guided genetic silencing systems in bacteria and archaea. Nature 482(7385):331–338. doi:10.1038/nature10886
Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S, Yan Z, Li D, Li J (2013) Correction of a genetic disease in mouse via use of CRISPR–Cas9. Cell Stem Cell 13(6):659–662. doi:10.1016/j.stem.2013.10.016
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R (2013) One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154(6):1370–1379. doi:10.1016/j.cell.2013.08.022
Yosef I, Goren MG, Qimron U (2012) Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res 40(12):5569–5576
Yu Z, Ren M, Wang Z, Zhang B, Rong YS, Jiao R, Gao G (2013) Highly efficient genome modifications mediated by CRISPR/Cas9 in Drosophila. Genetics 195(1):289–291
Zhang F, Maeder ML, Unger-Wallace E, Hoshaw JP, Reyon D, Christian M, Li X, Pierick CJ, Dobbs D, Peterson T (2010) High frequency targeted mutagenesis in Arabidopsis thaliana using zinc finger nucleases. Proc Natl Acad Sci 107(26):12028–12033
Zhang Y, Heidrich N, Ampattu BJ, Gunderson CW, Seifert HS, Schoen C, Vogel J, Sontheimer EJ (2013a) Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell 50(4):488–503
Zhang Y, Zhang F, Li X, Baller JA, Qi Y, Starker CG, Bogdanove AJ, Voytas DF (2013b) Transcription activator-like effector nucleases enable efficient plant genome engineering. Plant Physiol 161(1):20–27
Acknowledgments
We thank anonymous reviewers and the journal editor for their critical comments on the manuscript. We apologise to colleagues for not being able to cite all relevant and earlier papers because of space limitations and the focus of the article on recent research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, L., Fan, XD. CRISPR–Cas system: a powerful tool for genome engineering. Plant Mol Biol 85, 209–218 (2014). https://doi.org/10.1007/s11103-014-0188-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-014-0188-7