
Abstract
Pongamia (Millettia pinnata) is a promising biofuel crop with multiple merits. Breeding of ideal Pongamia germplasm for industrial application demands substantial progress in molecular biology of this legume species, which has been largely hampered by the paucity of its genomic data. In this study, we constructed and characterized a comprehensive seed transcriptome by the high-throughput Illumina sequencing technology. We obtained over 83 million high-quality reads, which were processed and assembled into 53,586 unigenes with a mean length of 787 bp. Among these unigenes, 39,602 (73.90 %) and 24,078 (44.93 %) showed significant similarity to proteins in the NCBI non-redundant and the Swiss-Prot protein databases, respectively. Of the annotated unigenes, 30,619 (57.14 %) were classified into 56 Gene Ontology categories. Furthermore, 21,905 (40.88 %) unigenes were assigned to 128 pathways in the Kyoto Encyclopedia of Genes and Genomes pathway database. A set of 364 unigenes involved in five pathways closely related to oil biosynthesis and accumulation were screened out as candidates for future functional analyses. On the other hand, 5710 expressed sequence tag-simple sequence repeats (EST-SSRs) were identified in 4951 unigenes with a density of one SSR every 7.39-kb sequence. One hundred EST-SSRs were randomly selected to validate amplification and to assess polymorphism among 12 Pongamia individuals. Eighty-two primer pairs successfully amplified DNA fragments and 17 of them detected polymorphism, in which the polymorphism information content values ranged from 0.14 to 0.57. This transcriptome dataset will serve as a valuable basis for studies on functional genomics, molecular genetics and molecular breeding of Pongamia.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Pongamia (Millettia pinnata, formerly known as Pongamia pinnata) is a fast-growing, medium-sized tree that belongs to family Fabaceae, subfamily Papilionoideae. This species is widely distributed from the Indian subcontinent and southeast Asia, to Polynesia and northern Australia (Scott et al. 2008; Murphy et al. 2012). Traditionally, the Pongamia tree has been mainly utilized for medicines, timber, fodder, manure and landscaping (Chopade et al. 2008; Sangwan et al. 2010). In the past decade, it has received increasing attention as a sustainable biofuel crop with multiple advantages. First, the Pongamia trees are capable of producing high yield of seeds with high contents of non-edible oil, which can be readily extracted and converted into biodiesel by transesterification (Karmee and Chadha 2005; Naik et al. 2008). Second, the Pongamia seed oil possesses a high proportion of monounsaturated oleic acid and a relatively low amounts of saturated and polyunsaturated fatty acids (Bala et al. 2011; Pavithra et al. 2012), which may endow the biodiesel products with lower cloud and pour points. Third, being a tree legume, the Pongamia can undergo biological nitrogen fixation and thus reduce the application of nitrogen fertilizers, which may in turn mitigate the deleterious environmental impacts of greenhouse gas emissions or water pollution (Jensen et al. 2012; Biswas and Gresshoff 2014; Gresshoff et al. 2015). Fourth, with the ability to tolerate a wide range of abiotic stresses and to improve the soil nutrient status (Sangwan et al. 2010; Murphy et al. 2012), the Pongamia trees can grow on and ameliorate the marginal or degraded lands that are not viable for food production.
A prerequisite for the successful establishment of commercial Pongamia industry is the abundant supply of seed oils derived from large-scale plantations. Considering its outcrossing reproductive nature and the resulting high variability among progeny plants, the commercial plantations for Pongamia will first base on breeding of germplasm with favorable traits followed by vegetative propagation of genetically uniform trees (Kesari and Rangan 2010). In Pongamia, there might be two major types of breeding programs, i.e., the selection for elite germplasms from natural populations and the genetic manipulation for improved germplasms, both of which will benefit from a better understanding of the genetic background for this species.
The early efforts on selection of Pongamia germplasm primarily relied on the assessment of phenotypic traits, such as plant height, size and weight of pods and seeds, as well as oil content and composition (Kaushik et al. 2007; Kesari et al. 2008; Mukta et al. 2009; Sunil et al. 2009; Rao et al. 2011; Sahoo et al. 2011). While some of these phenotypic traits have been demonstrated to be with high broad-sense heritability in Pongamia and thus reliable for germplasm selection (Kaushik et al. 2007; Sunil et al. 2009; Rao et al. 2011), they are easily affected by environmental factors and are only assayable when the trees are mature. Molecular markers, on the other hand, can provide more informative and reproducible data for germplasm selection at a juvenile stage, and they can also aid in evaluating genetic diversity of extant populations and planning conservation actions for the long-term variability of a plant species. Several types of molecular markers have already been applied in Pongamia, including inter simple sequence repeat (ISSR) (Kesari et al. 2010; Sahoo et al. 2010; Sujatha et al. 2010; Jiang et al. 2012), random amplified polymorphic DNA (RAPD) (Kesari et al. 2010; Kesari and Rangan 2011) and amplified fragment length polymorphism (AFLP) (Kesari et al. 2010; Thudi et al. 2010; Sharma et al. 2011; Pavithra et al. 2014). However, owing to the difficulty in marker development using conventional methods, these studies only employed a limited number of marker loci, most of which were dominantly inherited and not feasible for distinguishing between homozygotes and heterozygotes. More numbers of genetic loci, especially those from codominant markers, are needed for the marker-assisted selection (MAS) and population genetics study of this outcrossing species.
As for the genetic improvement of Pongamia germplasm, it is essential to identify candidate genes that participate in certain biological processes relevant to the quantity and quality of seed oils, and this serves as a foundation to create improved germplasm through genetic transformation. To date, dozens of Pongamia genes or genomic regions have been sequenced for molecular phylogenetic analyses, such as the phytochrome A gene (PHYA), the maturase R gene (matR), the ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit gene (rbcL) and the internal transcribed spacer (ITS) (Lavin et al. 1998; Hu et al. 2002; Shi et al. 2005; Arpiwi et al. 2013). In contrast, only less than ten genes have been isolated and characterized for functional study in Pongamia, including a chalcone isomerase (MpCHI) gene conferring the transgenic yeast strains with enhanced tolerance to salt stress (Wang et al. 2013), a stearoyl–acyl-carrier-protein desaturase (MpSAD) gene involved in the regulation of seed development (Ramesh et al. 2014) and four circadian clock genes (MpELF4, MpLCL1, MpPRR7 and MpTOC1) participating in flowering control (Winarto et al. 2015). Such a lack of gene resources will substantially hinder future application of genetic manipulation in Pongamia.
The high-throughput next-generation sequencing (NGS) technologies have provided an efficient way to generate an abundance of transcriptome data for both marker identification and gene discovery. In recent years, the NGS-based transcriptome analyses have already been applied in more than a dozen legume species (Garg et al. 2011; Zhang et al. 2012; Garg and Jain 2013; Liu et al. 2013; Hiz et al. 2014; Chen et al. 2015a, b; Souframanien and Reddy 2015). In a previous study, we employed the Illumina sequencing platform to produce a large-scale collection of Pongamia transcripts from root and leaf tissues for studying their transcriptome changes under salt treatments (Huang et al. 2012). In this study, a comprehensive seed transcriptome was also constructed for Pongamia by Illumina sequencing. We characterized the whole transcriptome with a special interest in screening functional genes associated with oil accumulation and mining potential SSR motifs. The former could serve as candidates for further functional research which would aid in exploring the molecular basis of seed oil accumulation and facilitate the genetic improvement of important traits, while the latter might extend the repertoire of molecular markers which would assist in genetic diversity analysis and germplasm selection.
Materials and methods
Plant materials and RNA preparation
Three 10-year-old Pongamia trees growing at the Garden Expo Park in Shenzhen, China, were used for seed sampling. Flowers at the lower position of these three trees were labeled with tags recording the first flowering date for each of them. Pods were harvested at a series of nine developmental stages from 12 weeks after flowering (WAF) till 36 WAF with an interval of 3 weeks. Meanwhile, the seeds were manually separated from pods, washed with distilled water, immediately frozen in liquid nitrogen and then stored at −80 °C before RNA extraction. At each stage, two seeds from each of the three trees were mixed together and subjected to RNA isolation using a modified CTAB method. The resulting RNA was further purified with the Qiagen RNeasy Plant Mini Kit (Qiagen, Germany). The concentration and quality of each RNA sample were determined by an Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Equal amounts of total RNA from each stage were pooled together for the following sequencing procedures.
Library construction and Illumina sequencing
A total of 20 μg of pooled RNA sample was used for cDNA library construction. Poly(A) mRNA was enriched from total RNA by Sera-mag Magnetic Oligo (dT) Beads (Thermo Fisher Scientific, USA), and then, the mRNA was digested into short fragments with fragmentation buffer (Ambion, USA). Taking these cleaved RNA fragments as templates, the first-strand cDNA was generated using random hexamer primers. Next, the second-strand cDNA was synthesized using the SuperScript Double-Stranded cDNA Synthesis Kit (Invitrogen, USA). The double-stranded cDNA fragments were purified with the QiaQuick PCR Extraction Kit (Qiagen, Germany) and resolved with EB buffer for end repair and dA tailing. The resulting short fragments were then ligated with sequencing adaptors and resolved on agarose gel. The suitable fragments were selected as templates for PCR amplification. Finally, the cDNA library was sequenced on an Illumina Hiseq™ 2000 platform (Illumina, USA) at the Beijing Genomics Institute (BGI, Shenzhen, China). Each sequencing pass could yield two 90-bp independent reads from either end of a cDNA fragment. All raw sequencing data have been deposited in NCBI Sequence Read Archive (SRA) under the accession number SRX862816.
De novo assembly of Pongamia seed transcriptome
After sequencing, the raw reads were initially cleaned by trimming adaptor sequences and removing reads with more than 10 % Q < 20 bases or reads with more than 5 % ambiguous sequences ‘N.’ Then, the clean reads were assembled by the short-read assembly program in the Trinity software (Grabherr et al. 2011). Firstly, the clean reads were split into smaller pieces, the k-mers, and were conjoined into contigs using the de Bruijn graph. After a trial of different k-mer sizes, 25-mer were chosen for this study to achieve a balance between the average sequence length, maximum sequence length and total number of transcripts (Table S1). Subsequently, the pair-end reads were used to identify the contigs from the same transcript and detect the distances between these contigs. Accordingly, the contigs were connected to get assembled sequences that could not be extended on either end. These assembled sequences were further processed by sequence splicing, redundancy removal and clustering with the TGICL software (Pertea et al. 2003) to create non-redundant transcripts defined as unigenes. This clustering step was based on terminal region matching for at least 40-bp overlap and 90 % identity. Moreover, to detect unigenes that might belong to the different parts of the same gene or represent the isoforms, a BLAST hit-based clustering was performed following the method described in Gahlan et al. (2012). The assembled sequence data have been deposited in NCBI with the BioProject ID PRJNA323432.
Sequence annotation and functional classification
All unigenes were annotated by BLAST against the publicly available databases including the NCBI (http://www.ncbi.nlm.nih.gov/) non-redundant protein (Nr) databases, the Swiss-Prot protein database (http://www.expasy.org/sprot/), the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (http://www.genome.jp/kegg/) and the Cluster of Orthologous Groups (COG) database (http://www.ncbi.nlm.nih.gov/COG/) with an E-value cutoff of 1.0e−5. The best alignments were extracted and used to predict the sequence orientations and the coding regions of the assembled transcripts. The incongruent results from different databases were settled under a priority order of Nr, Swiss-Prot, KEGG and COG. For the rest unigenes that were unaligned to the above databases, ESTScan was used to predict their sequence orientations (Iseli et al. 1999). Based on their Nr annotations, the unigenes were assigned Gene Ontology (GO) annotations using the Blast2GO program (Conesa et al. 2005), followed by GO functional classification to plot the distribution of gene functions using the WEGO software (Ye et al. 2006).
Gene validation by T–A cloning and sequencing
Nine unigenes with potential roles in fatty acid and glycerolipid metabolism were chosen for validation by Sanger sequencing to check the quality of sequence assembly and annotation from the Illumina sequencing. All these nine assembled unigenes were predicted to contain the complete ORF. Specific PCR primers were designed with sequences from the 5′ and 3′ untranslated regions. PCR was performed in a total volume of 25 μL containing 2.0 mM Mg2+, 0.15 mM dNTPs, 0.4 mM of each primer, 0.6 U Taq DNA polymerase and 15 ng cDNA under the reaction conditions as follows: an initial denaturation step at 95 °C for 1 min, 35 cycles at 94 °C for 45 s, 56 °C for 45 s, and 72 °C for 80 s, a final extension step at 72 °C for 10 min and hold at 4 °C. The PCR products were separated and ligated into the pMD18-T vector (Takara Bio Inc, China) and then transformed into E. coli DH5α. Positive clones were sequenced with ABI 3730 (Applied Biosystems, USA).
EST-SSR detection and primer design
The perl script program MISA (Thiel et al. 2003) was employed to detect and locate SSRs in the assembled transcripts. The searching criteria were set as follows: at least six contiguous repeats for di-, five repeats for tri- and tetra- and four repeats for penta- and hexa-nucleotide motifs. Considering the difficulties of distinguishing genuine mononucleotide repeats from polyadenylation products or single nucleotide stretch errors generated by sequencing, this type of repeats was excluded from the searching process. The maximal number of bases interrupting two SSRs in a compound microsatellite was 100. Based on MISA results, primer pairs flanking each SSR locus were designed using Primer3 (Rozen and Skaletsky 2000) with the core criteria of the predicted product size ranging from 80 to 200 bp, the GC percentage between 40 and 60 %, the optimum primer length of 22 bp and the melting temperature between 50 and 60 °C.
Survey of EST-SSR polymorphism
Twelve Pongamia individuals from four populations located along the seashore of South China were selected for polymorphism investigation with 100 randomly chosen EST-SSR loci. Genomic DNA was isolated from the leaves of each individual using the Wolact Plant Genomic DNA Purification Kit (Vicband Life Sciences, Hongkong, China) following the manufacturer’s instructions. PCR amplification was also conducted in a 25-μL reaction mixture under the reaction cycling profile as described above. The annealing temperature differed according to the T m value of each primer set. The PCR products were first electrophoresed on agarose gel. Then, those amplified products with expected fragment sizes were further separated on a Fragment Analyzer Automated CE System (Advanced Analytical Technologies, USA). The number of alleles (N a), observed heterozygosity (H o), expected heterozygosity (H e) and Shannon’s information index (I) at each locus were calculated using the GenAlEx software version 6.4 (Peakall and Smouse 2006). The polymorphic information content (PIC) values were obtained with the program PowerMarker version 3.25 (Liu and Muse 2005).
Results
Sequencing and de novo assembly of Pongamia seed transcriptome
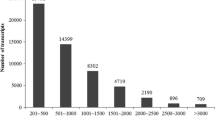
To achieve a comprehensive overview of the Pongamia seed transcriptome, a cDNA library constructed with pooled RNA samples from nine stages after flowering was subjected to pair-end sequencing on the Illumina platform. These nine stages have covered the cell division phase, the cell expansion phase and the desiccation phase of seed development. A total of 83,890,994 raw reads were yielded. After filtration of adaptor-containing and low-quality sequences, 80,212,402 clean reads consisting of 7,219,116,180 nucleotides were obtained. The average Q20 percentage and GC percentage of these clean reads were 99.20 and 44.77 %, respectively. These clean reads were first assembled into 108,731 contigs with an average length of 365 bp (Table S2). Then, the contigs were subjected to pair-end joining and gap filling steps which resulted in 60, 235 assembled sequences. These assembled sequences were further processed by sequence splicing, redundancy removal and similarity-based clustering to yield 53,586 unigenes. These unigenes ranged in length from 200 to 6401 bp, with an average length of 787 bp and a total length of about 42.2 Mb (Table S2). Among them, there were 27,226 (50.81 %) unigenes more than 500 bp long (Fig. 1). The sequencing depth for each unigene ranged from 0.38- to 517,623-fold, with an average of 130.7-fold (Table S3).

Length distribution of assembled unigenes
Functional annotation and classification of the unigenes
To assign putative functions to the unigenes in this transcriptome, we conducted a sequence similarity search against the public databases. A BLASTx of 53,586 unigenes with an E-value threshold of 1.0e−5 against the Nr protein database and the Swiss-Prot protein database retrieved 39,602 (73.90 %) and 24,078 (44.93 %) matches, respectively. Only 54.77 % of the unigenes shorter than 500 bp had significant BLAST hits in the Nr database. In contrast, the proportion of unigenes with significant hits increased sharply to 98.24 % for those with a size over 1000 bp (Fig. S1). The longer unigenes were more likely to have BLAST matches in the protein databases. The E-value distribution revealed that over 60 % of the top hits in the Nr database showed strong homology with the E-value <1.0e−45 (Fig. S2a). Meanwhile, the identity distribution indicated that nearly 60 % of the matched sequences had a similarity higher than 80 % (Fig. S2b). Based on their BLAST hits, another sequence clustering step was carried out to identify unigenes probably representing isoforms or different parts of the same gene. This step reduced the total number of transcripts from 53,586 to 43,122, which might be more close to the number of actual unique genes.
In terms of species distribution, the top hits for those annotated unigenes came from 208 species (Table S4). The majority of unigenes were annotated by protein accessions from plants (39,515 unigenes; 99.78 %), while the remaining less than one percentage of unigenes received annotations from algae (62 unigenes; 0.16 %) and fungi (25 unigenes; 0.06 %). All the top three species with BLAST hits, i.e., Glycine max, Medicago truncatula and Lotus corniculatus, belonged to the Fabaceae family. There were also 70 unigenes with matched sequences from Pongamia protein accessions, which had already been characterized by a handful of previous studies. Taken together, more than 90 % of the annotated unigenes (36,601 out of 39,602) had BLAST hits from legume species. These results reflected that the sequences of the Pongamia transcripts were properly assembled and annotated in the current study.
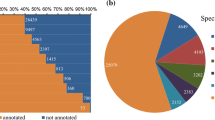
To categorize the functions of the Pongamia unigenes, GO assignments were performed following their Nr annotations. Out of the 53,586 assembled unigenes, 30,619 (57.14 %) were assigned at least one GO term within 56 functional groups (Fig. 2). Among these unigenes, 23,811 were assigned with terms from the Biological Process category, 23,489 were assigned with terms from the Molecular Function category, 24,052 were assigned with terms from the Cellular Component category, and 16,196 unigenes had an assignment in all three categories. The remaining unigenes with Nr annotations failed to obtain a GO term, largely due to their uninformative descriptions (e.g., unknown or hypothetical proteins). In the Biological Process category, the two most abundantly represented groups were cellular process (19,276 unigenes) and metabolic process (18,895 unigenes). The unigenes involved in response to stimulus (9207 unigenes) and biological regulation (7950 unigenes) were also well represented. Within the Molecular Function category, most of the unigenes were functionally correlated with binding (15,464 unigenes) and catalytic activity (15,431 unigenes). As for the Cellular Component category, most of the unigenes produced gene products located in cell (22,576 unigenes) and organelle (18,267 unigenes).

Gene Ontology classifications of assembled unigenes
For further functional prediction and classification, all the assembled unigenes were subjected to a search against the COG database. In total, 13,147 of 53,586 (24.53 %) unigenes were assigned into 25 COG categories (Fig. S3). The largest number of unigenes were classified into the group of ‘General function prediction only’ (4390 unigenes), followed by ‘Transcription’ (2639 unigenes), ‘Replication, recombination and repair’ (2366 unigenes), ‘Posttranslational modification, protein turnover, chaperones’ (1960 unigenes), ‘Signal transduction mechanisms’ (1833 unigenes) and ‘Translation, ribosomal structure and biogenesis’ (1562 unigenes). The predominance of unigenes in these functional categories is typical in plants. Besides, a set of 1088 unigenes with putative involvement in ‘Cell cycle control, cell division, chromosome partitioning’ were also of particular interest for further research on seed development. On the other hand, only a few unigenes were classified into ‘Nuclear structure’ and ‘Extracellular structures’ (21 and 7 unigenes, respectively).
Metabolic pathway assignment by KEGG analysis
To gain insights into the biological pathways activated in Pongamia seed, we mapped all the assembled unigenes to the reference pathways in the KEGG database. As a whole, 21,905 out of 53,586 (40.88 %) unigenes were assigned to 128 pathways. The number of unigenes in each pathway ranged from 1 to 4731 (Table S5). The categories of metabolic pathways (4731 unigenes), biosynthesis of secondary metabolites (2179 unigenes), plant hormone signal transduction (1219 unigenes) and plant–pathogen interaction (1169 unigenes) were well represented in this transcriptome. It was worth noting that glycerophospholipid metabolism (691 unigenes) and ether lipid metabolism (538 unigenes) were also among the ten most abundantly represented pathways, which indicated that lipid metabolism was particularly active in Pongamia seed tissues.
There were approximately 2100 unigenes participating in 14 cellular activities of lipid metabolism (Fig. S4). Specifically, a set of 364 unigenes were found to be homologous to previously identified genes involved in five KEGG pathways that were most closely related to oil accumulation. In most cases, more than one unigene was annotated as encoding the same enzyme. Such unigenes might represent different regions of the same transcript or different members of a gene family. Hence, there were 58 unigenes encoding 13 enzymes in fatty acid biosynthesis, 36 unigenes encoding 7 enzymes in fatty acid elongation, 57 unigenes encoding 11 enzymes in biosynthesis of unsaturated fatty acids, 128 unigenes encoding 8 enzymes in fatty acid degradation and 145 unigenes encoding 17 enzymes in glycerolipid metabolism. Among them, 60 unigenes encoding nine enzymes were shared by two different pathways. For example, there were 11 unigenes coding for 3-oxoacyl-[acyl-carrier-protein] reductase (fabG) that was involved in both fatty acid biosynthesis and biosynthesis of unsaturated fatty acids. We combined the pathways of fatty acid biosynthesis, fatty acid elongation and biosynthesis of unsaturated fatty acids into one category and listed all the enzymes under three categories (Table 1).
For these 364 unigenes, 329 (90.38 %) were annotated with protein accessions from legume species. Furthermore, 107 out of these 364 assembled unigenes were predicted to contain the complete ORF. From them, we selected nine unigenes to check the quality of sequence assembly by cloning and sequencing the full-length cDNA with the Sanger method. The sequencing results showed that all these nine assembled unigenes had covered the complete ORF of the corresponding genes (Table S6).
Identification and characterization of EST-SSR markers
In order to develop SSR markers in Pongamia, we screened all the 53,586 unigenes in this transcriptome to mine EST-derived SSR motifs. In total, 5710 putative EST-SSRs were detected in 4951 unigenes, including 4335 and 616 unigenes with one and more than one SSR, respectively. 352 EST-SSRs were present in compound formation. On average, one EST-SSR could be found every 7.39 kb in the unigenes. The major repeat types of the identified EST-SSRs were dinucleotide (2715; 47.55 %) and trinucleotide (2400; 42.03 %). Within these EST-SSRs, 177 motif sequence types were identified, of which di-, tri-, tetra-, penta- and hexa-nucleotide repeats had 4, 10, 16, 47 and 100 types, respectively. The most abundant motif of dinucleotide repeat was AG/CT (1913; 33.50 %), followed by AC/GT (495; 8.67 %) and AT/AT (302; 5.29 %). As for the trinucleotide repeats, AAG/CTT (715; 12.52 %) was the most common motif, followed by AAC/GTT (366; 6.41 %) and ATC/ATG (349; 6.11 %). The remaining 171 types of motifs accounted for 27.50 % in total (Fig. 3). EST-SSRs with six tandem repeats (1640; 28.72 %) were the most frequently observed, followed by those with five tandem repeats (1408; 24.66 %), seven tandem repeats (985; 17.25 %) and eight tandem repeats (484; 8.48 %) (Table S7). Approximately 85 % of the identified EST-SSRs were 12–20 bp in length, while there were only 19 EST-SSRs longer than 24 bp.

Frequency distribution of SSRs based on motif types
Validation and polymorphism of EST-SSR markers
Among the 5710 putative EST-SSRs, 1071 had sufficient flanking sequence to allow for PCR primer design, which mostly contained dinucleotide (326; 30.44 %) and trinucleotide (621; 57.98 %) motifs. From these candidates, 100 primer pairs were randomly chosen and synthesized for a preliminary polymorphism assay across 12 Pongamia individuals from natural populations in South China (Table S8). Eighty-two of the 100 primer pairs successfully amplified fragments, while 18 did not yield amplified products under different reaction conditions. Among the 82 working primer pairs, 13 generated multiple bands and 16 resulted in weak amplifications or bands with unexpected size. The remaining 53 primer pairs could produce distinct bands with expected size. Only 17 microsatellite loci, covering di-, tri-, tetra-, penta- and hexa-nucleotide repeat motifs, showed polymorphic allelic patterns among 12 individuals (Table 2). For these 17 polymorphic loci, the number of different alleles ranged from 2 to 4 with an average of 2.47 alleles per locus, the H o values varied from 0 to 1.00 with an average of 0.40, and the H e values varied from 0.16 to 0.67 with an average of 0.38. Besides, the Shannon’s information index for these loci ranged from 0.29 to 1.14 with an average of 0.61, while the PIC values ranged from 0.14 to 0.57 with an average of 0.31. Out of these 17 loci, 15 were located in unigenes with BLAST hits in the Nr database, including seven loci in the 5′UTR, four loci in the CDS region and four loci in the 3′UTR. Three of these polymorphic loci (MPM4, MPM10 and MPM73) were located in unigenes encoding enzymes in relation to fatty acid and glycerolipid metabolism.
Discussion
Pongamia is an adaptable legume crop whose seed oil has been recognized as a desirable source for sustainable biodiesel production (Karmee and Chadha 2005; Scott et al. 2008). Despite the interest in this crop, little is known on the molecular biology of Pongamia. For such a woody tree species with around 1300 Mb per haploid genome (Choudhury et al. 2014), it is still difficult to perform the whole genome sequencing. Alternatively, the NGS-based transcriptome sequencing provides a quick and cost-effective approach for generating a wealth of molecular data. In this study, we made use of the pooled RNA samples from various developmental stages to exploit the transcriptomic sequence resource of Pongamia seed.
Transcriptome sequencing and de novo assembly resulted in 53,586 Pongamia unigenes with a mean length of 787 bp. Compared with the results from other legume species generated by the Illumina sequencing platform, the mean length of unigenes for Pongamia was longer than those for chickpea (523 bp) (Garg et al. 2011), peanut (619 bp) (Zhang et al. 2012), common bean (777 bp) (Wu et al. 2014) and black gram (443 bp) (Souframanien and Reddy 2015), but shorter than those for alfalfa (803 bp) (Liu et al. 2013), adzuki bean (1213 bp) (Chen et al. 2015a) and mung bean (874 bp) (Chen et al. 2015b). Moreover, the average sequencing depth of all unigenes was up to 130.7-fold. Both the mean length and the sequencing depth of unigenes in this transcriptome showed significant enhancements relative to those in the transcriptome constructed from leaf and root tissues as reported in our previous study (Huang et al. 2012).
For gene annotation, the sequence similarity search was performed against the public databases including Nr, Swiss-Prot, GO, COG and KEGG. In total, 42,932 unigenes (80.11 %) had at least one hit in these databases. The percentage of annotated unigenes in this transcriptome was also much higher than that in the transcriptome of leaf and root tissues, largely owing to the general increase in sequence length and the explosive growth of sequence information in the above databases. With regard to Nr annotation, it was not surprising that more than 90 % of the annotated unigenes matched protein accessions from legume species, especially from the three legumes with the earliest complete genome sequences, i.e., G. max, M. truncatula and L. corniculatus (Sato et al. 2008; Schmutz et al. 2010; Young et al. 2011). More specifically, the number of unigenes with hits from soybean (31,249) was far more than that from M. truncatula (3715) or L. corniculatus (914). This observation could most possibly be explained by a closer phylogenetic relationship between Pongamia and soybean, both of which belonged to the Millettioids within the subfamily Papilionoideae, in comparison with M. truncatula and L. corniculatus that belonged to the Hologalegina (Wojciechowski et al. 2004).
KEGG analysis revealed that 21,905 unigenes were involved in 128 pathways. Similar to other plant transcriptomes, metabolic pathways, biosynthesis of secondary metabolites, plant hormone signal transduction and plant–pathogen interaction were well represented in this transcriptome. Notably, there were nearly 2100 unigenes (9.6 %) belonging to 14 cellular activities under the category of lipid metabolism. This percentage was much higher than that in the transcriptome of leaf and root tissues (6.8 %) (Huang et al. 2012), suggesting that lipid metabolism was more active in the oil-bearing seed than in other tissues of Pongamia. Similarly, the unigenes involved in lipid metabolism were abundant in the seed transcriptome of peanut (Yin et al. 2013). Nevertheless, lipid metabolism was not well represented by unigenes in the seed transcriptome of black gram, another legume species not rich in seed oil (Souframanien and Reddy 2015).
Based on KEGG assignments, we further singled out 364 unigenes within five pathways associated with fatty acid and glycerolipid metabolism. These unigenes have covered all essential enzymes responsible for oil biosynthesis and accumulation according to the lipid gene catalog of Arabidopsis (Beisson et al. 2003). The oil biosynthesis was proposed to be limited by the supply of fatty acids (Bao and Ohlrogge 1999), and the most committed step in fatty acid biosynthesis was catalyzed by acetyl-CoA carboxylase (ACC). Reducing ACC activity could to some extent lower the fatty acid content in transgenic seeds (Thelen and Ohlrogge 2002). In this study, 28 unigenes were found to code for all the four ACC subunits, among which accC was matched by the largest number of unigenes. Comparatively, the transcripts encoding accD were absent in the seed transcriptome of peanut (Yin et al. 2013), while the transcripts encoding accB were most abundant in the seed transcriptome of Jatropha (King et al. 2011). Oleoyl desaturase (FAD2) and linoleate desaturase (FAD3) were two key enzymes controlling the conversion of oleic acid to polyunsaturated fatty acids. Combinations of mutant FAD2 and FAD3 genes could produce high oleic acid and low linolenic acid soybean oil (Pham et al. 2012). In this transcriptome, seven and five unigenes were found to code for FAD2 and FAD3, respectively. Diacylglycerol acyltransferase (DGAT) played a vital role in the formation of triacylglycerol (TAG). Previous studies showed that ectopic expression of DGAT could enhance seed oil content in Arabidopsis (Jako et al. 2001) and soybean (Lardizabal et al. 2008). Although the lack of DGAT transcripts was observed in the developing seeds of castor bean (Lu et al. 2007) and Jatropha (King et al. 2011), unigenes coding for DGAT were well represented in Pongamia seeds. Taken together, the above examples suggested that the seed transcriptome of Pongamia would provide substantial candidate genes for further functional studies aiming at improving oil content and composition in plants.
Molecular markers have been widely and effectively applied in plants for genetic diversity and association analysis. Prior to this study, the genotyping of Pongamia germplasm has been restricted to ISSR, RAPD and AFLP markers (Kesari et al. 2010; Sahoo et al. 2010; Sujatha et al. 2010; Thudi et al. 2010; Kesari and Rangan 2011; Sharma et al. 2011; Jiang et al. 2012; Pavithra et al. 2014). No codominant SSR markers have been developed for this outcrossing species. In the present study, 5710 putative EST-SSRs were identified from the transcriptome dataset. The distribution density of one SSR per 7.39 kb was higher than those reported for alfalfa (1/12.06 kb) (Liu et al. 2013) and black gram (1/11.90 kb) (Souframanien and Reddy 2015), but was lower than those in chickpea (1/5.80 kb) (Garg et al. 2011) and common bean (1/4.70 kb) (Wu et al. 2014). Though the transcriptomes of these legume species were all generated by Illumina sequencing, the differences in SSR abundance might still be caused by inconsistency in genome structure or composition, dataset size, search method and criteria. Likewise, the dominant repeat type and motif type of EST-SSRs also varied among these legume transcriptomes.
To assess the quality of the newly developed EST-SSR markers, 100 primer pairs were randomly chosen for amplification. Eighty-two of them successfully yielded amplicons from the target genomic DNA. Such high rate of successful amplification (82 %) again proved that the majority of unigenes were accurately assembled. The failure of the remaining primer pairs to generate amplicons might mainly be attributed to the long introns preventing genomic amplification or the location of primers across splice sites. For those EST-SSR markers with amplified products, only 17 exhibited polymorphism among 12 Pongamia germplasms. The polymorphic ratio of these markers was relatively low, yet it was still comparable to the ratios for some EST-SSR markers and genomic-SSR markers in legumes (Liu et al. 2013; Chen et al. 2015a; Wang et al. 2015). Besides, the polymorphic ratio of SSR markers might also enhance with the number of germplasms tested. The PIC values for these 17 EST-SSR markers were generally higher than those for AFLP markers (Thudi et al. 2010; Pavithra et al. 2014), but lower than those for RAPD and ISSR markers applied in Pongamia (Kesari et al. 2010; Kesari and Rangan 2011). Most of the 17 polymorphic EST-SSRs were located in 5′UTR or 3′UTR. Interestingly, three polymorphic EST-SSRs were located in those among the set of 364 unigenes related to oil accumulation. MPM4 was located in unigene encoding dihydroxyacetone kinase (DAK) for glycerolipid metabolism, while MPM10 and MPM73 were in unigenes encoding 3-oxoacyl-ACP synthase (fabH) and 3-oxoacyl-ACP reductase (fabG) for fatty acid biosynthesis, respectively. Previously, two studies have demonstrated that SSRs in FAD2 or FAD5 gene could contribute to variations in oleic acid or stearic acid content in soybean (Spencer et al. 2003; Bachlava et al. 2008). In this sense, the EST-SSR markers validated in this transcriptome would provide useful targets for seed oil quantitative trait loci (QTL) mapping and association analysis.
In summary, we employed the Illumina sequencing platform to generate a seed transcriptome of Pongamia with a large number of unigenes. A high proportion of unigenes were annotated by similarity search against the commonly used public databases. The annotated unigenes offered enough coverage to allow for the discovery of almost all genes involved in the pathways related to oil biosynthesis and accumulation. In addition, we identified an abundance of SSR markers from the transcriptome dataset, which were tightly linked with functional genes and held tremendous potential for future genetic research and molecular breeding of Pongamia germplasm.
References
Arpiwi N, Yan G, Barbour E, Plummer J (2013) Genetic diversity, seed traits and salinity tolerance of Millettia pinnata (L.) Panigrahi, a biodiesel tree. Genet Resour Crop Evol 60(2):677–692
Bachlava E, Dewey R, Auclair J, Wang S, Burton J, Cardinal A (2008) Mapping genes encoding microsomal omega-6 desaturase enzymes and their cosegregation with QTL affecting oleate content in soybean. Crop Sci 48(2):640–650
Bala M, Nag TN, Kumar S, Vyas M, Kumar A, Bhogal NS (2011) Proximate composition and fatty acid profile of Pongamia pinnata, a potential biodiesel crop. J Am Oil Chem Soc 88(4):559–562
Bao X, Ohlrogge J (1999) Supply of fatty acid is one limiting factor in the accumulation of triacylglycerol in developing embryos. Plant Physiol 120(4):1057–1062
Beisson F, Koo A, Ruuska S et al (2003) Arabidopsis genes involved in acyl lipid metabolism. A 2003 census of the candidates, a study of the distribution of expressed sequence tags in organs, and a web-based database. Plant Physiol 132(2):681–697
Biswas B, Gresshoff P (2014) The role of symbiotic nitrogen fixation in sustainable production of biofuels. Int J Mol Sci 15(5):7380–7397
Chen H, Liu L, Wang L, Wang S, Somta P, Cheng X (2015a) Development and validation of EST-SSR markers from the transcriptome of adzuki bean (Vigna angularis). PLoS One 10(7):e0131939
Chen H, Wang L, Wang S, Liu C, Blair M, Cheng X (2015b) Transcriptome sequencing of mung bean (Vigna radiata) genes and the identification of EST-SSR markers. PLoS One 10(4):e0120273
Chopade V, Tankar A, Pande V, Tekade A, Gowekar N, Bhandari S, Khandake S (2008) Pongamia pinnata: phytochemical constituents, traditional uses and pharmacological properties—a review. Int J Green Pharm 2(2):72–75
Choudhury R, Basak S, Ramesh A, Rangan L (2014) Nuclear DNA content of Pongamia pinnata L. and genome size stability of in vitro-regenerated plantlets. Protoplasma 251(3):703–709
Conesa A, Gotz S, Garcia-Gomez J, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21(18):3674–3676
Gahlan P, Singh H, Shankar R, Sharma N, Kumari A, Chawla V, Ahuja P, Kumar S (2012) De novo sequencing and characterization of Picrorhiza kurrooa transcriptome at two temperatures showed major transcriptome adjustments. BMC Genom 13:126
Garg R, Jain M (2013) Transcriptome analyses in legumes: a resource for functional genomics. Plant Genome 6(3):1–9
Garg R, Patel R, Tyagi A, Jain M (2011) De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res 18(1):53–63
Grabherr M, Haas B, Yassour M et al (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29(7):644–652
Gresshoff P, Hayashi S, Biswas B et al (2015) The value of biodiversity in legume symbiotic nitrogen fixation and nodulation for biofuel and food production. J Plant Physiol 172:128–136
Hiz M, Canher B, Niron H, Turet M (2014) Transcriptome analysis of salt tolerant common bean (Phaseolus vulgaris L.) under saline conditions. PLoS One 9(3):e92598
Hu J, Lavin M, Wojciechowski M, Sanderson M (2002) Phylogenetic analysis of nuclear ribosomal ITS/5.8S sequences in the tribe Millettieae (Fabaceae): Poecilanthe–Cyclolobium, the core Millettieae, and the Callerya group. Syst Bot 27(4):722–733
Huang J, Lu X, Yan H, Chen S, Zhang W, Huang R, Zheng Y (2012) Transcriptome characterization and sequencing-based identification of salt-responsive genes in Millettia pinnata, a semi-mangrove plant. DNA Res 19(2):195–207
Iseli C, Jongeneel C, Bucher P (1999) ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol 1999:138–148
Jako C, Kumar A, Wei Y, Zou J, Barton D, Giblin E, Covello P, Taylor D (2001) Seed-specific over-expression of an Arabidopsis cDNA encoding a diacylglycerol acyltransferase enhances seed oil content and seed weight. Plant Physiol 126(2):861–874
Jensen E, Peoples M, Boddey R, Gresshoff P, Hauggaard-Nielsen H, Alves B, Morrison M (2012) Legumes for mitigation of climate change and the provision of feedstock for biofuels and biorefineries. A review. Agron Sustain Dev 32(2):329–364
Jiang Q, Yen S, Stiller J, Edwards D, Scott P, Gresshoff P (2012) Genetic, biochemical, and morphological diversity of the legume biofuel tree Pongamia pinnata. J Plant Genome Sci 1(3):54–67
Karmee S, Chadha A (2005) Preparation of biodiesel from crude oil of Pongamia pinnata. Bioresour Technol 96(13):1425–1429
Kaushik N, Kumar S, Kumar K, Beniwal R, Kaushik N, Roy S (2007) Genetic variability and association studies in pod and seed traits of Pongamia pinnata (L.) Pierre in Haryana, India. Genet Resour Crop Evol 54(8):1827–1832
Kesari V, Rangan L (2010) Development of Pongamia pinnata as an alternative biofuel crop—current status and scope of plantations in India. J Crop Sci Biotechnol 13(3):127–137
Kesari V, Rangan L (2011) Genetic diversity analysis by RAPD markers in candidate plus trees of Pongamia pinnata, a promising source of bioenergy. Biomass Bioenergy 35(7):3123–3128
Kesari V, Krishnamachari A, Rangan L (2008) Systematic characterisation and seed oil analysis in candidate plus trees of biodiesel plant, Pongamia pinnata. Ann Appl Biol 152(3):397–404
Kesari V, Sathyanarayana V, Parida A, Rangan L (2010) Molecular marker-based characterization in candidate plus trees of Pongamia pinnata, a potential biodiesel legume. AoB Plants 2010:plq017
King A, Li Y, Graham I (2011) Profiling the developing Jatropha curcas L. seed transcriptome by pyrosequencing. Bioenergy Res 4(3):211–221
Lardizabal K, Effertz R, Levering C, Mai J, Pedroso M, Jury T, Aasen E, Gruys K, Bennett K (2008) Expression of Umbelopsis ramanniana DGAT2A in seed increases oil in soybean. Plant Physiol 148(1):89–96
Lavin M, Eshbaugh E, Hu J, Mathews S, Sharrock R (1998) Monophyletic subgroups of the tribe Millettieae (Leguminosae) as revealed by phytochrome nucleotide sequence data. Am J Bot 85(3):412–433
Liu K, Muse S (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21(9):2128–2129
Liu Z, Chen T, Ma L, Zhao Z, Zhao P, Nan Z, Wang Y (2013) Global transcriptome sequencing using the Illumina platform and the development of EST-SSR markers in autotetraploid alfalfa. PLoS One 8(12):e83549
Lu C, Wallis JG, Browse J (2007) An analysis of expressed sequence tags of developing castor endosperm using a full-length cDNA library. BMC Plant Biol 7:42
Mukta N, Murthy IYLN, Sripal P (2009) Variability assessment in Pongamia pinnata (L.) Pierre germplasm for biodiesel traits. Ind Crop Prod 29(2–3):536–540
Murphy H, O’Connell D, Seaton G et al (2012) A common view of the opportunities, challenges, and research actions for Pongamia in Australia. Bioenergy Res 5(3):778–800
Naik M, Meher L, Naik S, Das L (2008) Production of biodiesel from high free fatty acid Karanja (Pongamia pinnata) oil. Biomass Bioenergy 32(4):354–357
Pavithra H, Gowda B, Kumar K, Prasanna K, Shivanna M (2012) Oil, fatty acid profile and Karanjin content in developing Pongamia pinnata (L.) Pierre seeds. J Am Oil Chem Soc 89(12):2237–2244
Pavithra H, Shivanna M, Chandrika K, Prasanna K, Gowda B (2014) Genetic analysis of Pongamia pinnata (L.) Pierre populations using AFLP markers. Tree Genet Genomes 10(1):173–188
Peakall R, Smouse P (2006) Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6(1):288–295
Pertea G, Huang X, Liang F et al (2003) TIGR gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19(5):651–652
Pham A-T, Shannon G, Bilyeu K (2012) Combinations of mutant FAD2 and FAD3 genes to produce high oleic acid and low linolenic acid soybean oil. Theor Appl Genet 125(3):503–515
Ramesh A, Kesari V, Rangan L (2014) Characterization of a stearoyl–acyl carrier protein desaturase gene from potential biofuel plant, Pongamia pinnata L. Gene 542(2):113–121
Rao G, Shanker A, Srinivas I, Korwar G, Venkateswarlu B (2011) Diversity and variability in seed characters and growth of Pongamia pinnata (L.) Pierre accessions. Trees 25(4):725–734
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Sahoo D, Aparajita S, Rout G (2010) Inter and intra-population variability of Pongamia pinnata: a bioenergy legume tree. Plant Syst Evol 285(1–2):121–125
Sahoo D, Rout G, Das S, Aparajita S, Mahapatra A (2011) Genotypic variability and correlation studies in pod and seed characteristics of Pongamia pinnata (L.) Pierre in Orissa, India. Int J Forest Res 2011:1–6
Sato S, Nakamura Y, Kaneko T et al (2008) Genome structure of the legume, Lotus japonicus. DNA Res 15(4):227–239
Schmutz J, Cannon S, Schlueter J et al (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Scott P, Pregelj L, Chen N, Hadler J, Djordjevic M, Gresshoff P (2008) Pongamia pinnata: an untapped resource for the biofuels industry of the future. Bioenergy Res 1(1):2–11
Sharma S, Negi M, Sinha P, Kumar K, Tripathi S (2011) Assessment of genetic diversity of biodiesel species Pongamia pinnata accessions using AFLP and three endonuclease-AFLP. Plant Mol Biol Rep 29(1):12–18
Shi S, Huang Y, Zeng K, Tan F, He H, Huang J, Fu Y (2005) Molecular phylogenetic analysis of mangroves: independent evolutionary origins of vivipary and salt secretion. Mol Phylogenet Evol 34(1):159–166
Souframanien J, Reddy K (2015) De novo assembly, characterization of immature seed transcriptome and development of genic-SSR markers in black gram [Vigna mungo (L.) Hepper]. PLoS One 10(6):e0128748
Spencer M, Pantalone V, Meyer E, Landau-Ellis D, Hyten D (2003) Mapping the Fas locus controlling stearic acid content in soybean. Theor Appl Genet 106(4):615–619
Sujatha K, Rajwade A, Gupta V, Hazra S (2010) Assessment of Pongamia pinnata (L.)—a biodiesel producing tree species using ISSR markers. Curr Sci 99(10):1327–1329
Sunil N, Kumar V, Sivaraj N, Lavanya C, Prasad R, Rao B, Varaprasad K (2009) Variability and divergence in Pongamia pinnata (L.) Pierre germplasm—a candidate tree for biodiesel. GCB Bioenergy 1(6):382–391
Thelen J, Ohlrogge J (2002) Metabolic engineering of fatty acid biosynthesis in plants. Metab Eng 4(1):12–21
Thiel T, Michalek W, Varshney R, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106(3):411–422
Thudi M, Manthena R, Wani S, Tatikonda L, Hoisington D, Varshney R (2010) Analysis of genetic diversity in Pongamia [Pongamia pinnata (L.) Pierrre] using AFLP markers. J Plant Biochem Biotechnol 19(2):209–216
Wang H, Hu T, Huang J, Lu X, Huang B, Zheng Y (2013) The expression of Millettia pinnata chalcone isomerase in Saccharomyces cerevisiae salt—sensitive mutants enhances salt-tolerance. Int J Mol Sci 14(5):8775–8786
Wang L, Elbaidouri M, Abernathy B, Chen H, Wang S, Lee S, Jackson S, Cheng X (2015) Distribution and analysis of SSR in mung bean (Vigna radiata L.) genome based on an SSR-enriched library. Mol Breed 35:25
Winarto H, Liew L, Gresshoff P, Scott P, Singh M, Bhalla P (2015) Isolation and characterization of circadian clock genes in the biofuel plant Pongamia (Millettia pinnata). Bioenergy Res 8(2):760–774
Wojciechowski M, Lavin M, Sanderson M (2004) A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. Am J Bot 91(11):1846–1862
Wu J, Wang L, Li L, Wang S (2014) De novo assembly of the common bean transcriptome using short reads for the discovery of drought-responsive genes. PLoS One 9(10):e109262
Sangwan S, Rao D, Sharma R (2010) A review on Pongamia pinnata (L.) Pierre: a great versatile Leguminous plant. Nat Sci 8(11):130–139
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34 (Web Server):w293–w297
Yin D, Wang Y, Zhang X, Li H, Lu X, Zhang J, Zhang W, Chen S (2013) De novo assembly of the peanut (Arachis hypogaea L.) seed transcriptome revealed candidate unigenes for oil accumulation pathways. PLoS One 8(9):e73767
Young N, Debelle F, Oldroyd G et al (2011) The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480:520–524
Zhang J, Liang S, Duan J, Wang J, Chen S, Cheng Z, Zhang Q, Liang X, Li Y (2012) De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in peanut (Arachis hypogaea L.). BMC Genom 13:90
Acknowledgments
We thank the anonymous referees and the editors for their comments and suggestions for improving the manuscript. This work was supported by the National Natural Science Foundation of China (Nos. 31300275 and 31370289), the Guangdong Innovation Research Team Fund (No. 2014ZT05S078) and the National Basic Research Program of China (No. 2012CB114200).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Jianzi Huang and Xiaohuan Guo have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Huang, J., Guo, X., Hao, X. et al. De novo sequencing and characterization of seed transcriptome of the tree legume Millettia pinnata for gene discovery and SSR marker development. Mol Breeding 36, 75 (2016). https://doi.org/10.1007/s11032-016-0503-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-016-0503-x