
Abstract
Purpose
The aims of this study were twofold. First, to evaluate the effectiveness of selected polymers in inhibiting solution crystallization of celecoxib. Second, to compare the release rate and crystallization tendency of celecoxib amorphous solid dispersions (ASDs) formulated with a single polymer, or binary polymer combinations.
Methods
The effectiveness of polymers, polyvinylpyrrolidone (PVP), hydroxypropylmethyl cellulose (HPMC) or HPMC acetate succinate (HPMCAS), in maintaining supersaturation of celecoxib solutions was evaluated by performing nucleation induction time measurements. Crystallization kinetics of ASD suspensions were monitored using Raman spectroscopy. Dissolution experiments were carried out under non-sink conditions.
Results
Pure amorphous celecoxib crystallized rapidly through both matrix and solution pathways. Matrix and solution crystallization was inhibited when celecoxib was molecularly mixed with a polymer, resulting in release of the drug to form supersaturated solutions. Cellulosic polymers were more effective than PVP in maintaining supersaturation. Combining a cellulosic polymer and PVP enabled improved drug release and stability to crystallization.
Conclusions
Inclusion of an effective solution crystallization inhibitor as a minor component in ternary dispersions resulted in prolonged supersaturation following dissolution. This study shows the feasibility of formulation strategies for ASDs where a major polymer component is used to achieve one key property e.g. release, while a minor polymer component is added to prevent crystallization.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
It has been suggested that up to 80% of new molecular entities have sub-optimum aqueous solubility [1] which can lead to solubility-limited bioavailability. The low aqueous solubility of many new compounds can be attributed in part to the nature of contemporary drug discovery methodologies. Formulations containing amorphous drug are promising for the oral delivery of poorly water-soluble drugs since the amorphous form of a compound has higher free energy as compared to the crystalline counterparts, which may give rise to higher apparent solubility and dissolution rates [2]. This in turn may lead to improved drug absorption and increased bioavailability relative to the crystalline counterpart. Despite the potential benefit, the application of formulations containing pure amorphous drug remains limited primarily because of their higher instability: the thermodynamic driving force always favors a transformation towards a lower energy crystalline state, negating the solubility advantage [3].
Polymers are often employed to form amorphous solid dispersions (ASDs) with a drug to improve the physical stability during processing and storage [4]. Polymers are thought to inhibit crystallization through a number of mechanisms including reducing the drug molecular mobility, by increasing the glass transition temperature (Tg) of the system [5], and/or forming hydrogen bonds with the drug [6, 7].
While it is obviously critical to stabilize the amorphous drug in the solid state, it is of equal importance to prevent crystallization during dissolution of the ASD. Nevertheless, some of the fundamental processes underlying the concentration-time profiles attained during dissolution are poorly understood, in particular due to the tendency to run dissolution studies under sink conditions, which are unlikely to be found in vivo for many poorly water soluble compounds. A number of factors need to be considered when evaluating the dissolution behavior of ASDs including the amount of ASD introduced to the medium, the drug loading, the relative dissolution rates of the individual components, the equilibrium solubility of drug, the degree of supersaturation achieved, and the crystallization kinetics of the drug. Alonzo et. al. demonstrated two pathways through which crystallization could occur during the dissolution process [8]. Nucleation and crystal growth could commence at the surface of the amorphous solid matrix upon contact with dissolution medium due to a reduction in Tg caused by the absorbed water. In this case, only a small extent of supersaturation can be generated. Alternatively, if the dissolution rate of drug is fast relative to matrix crystallization, drug may crystallize from the supersaturated solution, resulting in desupersaturation at some point following dissolution. The more supersaturated the solution is, the more prone it will be to crystallize. While it has been demonstrated that polymers, when pre-dissolved in buffer, may inhibit either or both routes of crystallization for pure amorphous compounds [8], little is known regarding the role of polymer during dissolution of ASDs. The aims of the current study were to evaluate the crystallization propensity of the pure amorphous form of the poorly soluble anti-inflammatory agent, celecoxib (CEX), and to compare the impact of various polymers, alone and in combination, on the route and kinetics of crystallization and their impact on the dissolution rate of CEX from ASDs. It was also of interest to determine the feasibility of using polymer combinations to improve the performance of ASDs with high drug loadings, hence the dispersions studied herein had a 50% drug loading.
Materials
CEX was purchased from Attix Pharmaceuticals (Toronto, Ontario, Canada). Polyvinylpyrrolidone (PVP) (Grade K12: Mw 2000–3000 g mol−1) was provided by BASF (Ludwigshafen, Germany) and polyvinylpyrrolidone (PVP) (Grade K29/32: Mw 58,000 g mol−1 ) was purchased from ISP Technologies, INC (Wayne, NJ, USA). Hydroxypropylmethylcellulose acetate succinate (HPMCAS, Type AS-MF), and hydroxypropyl methylcellulose (HPMC, Grade 606) were supplied by Shin-Etsu Chemical Co. (Tokyo, Japan). The molecular structures are shown in Fig. 1.

Chemical structures of CEX (a), PVP (b), HPMCAS (c), and HPMC (d).
Methods
Preparation of Bulk Amorphous Materials
CEX and the polymer(s) were dissolved in a 50:50 v/v solution of ethanol and dichloromethane. Solvent removal was achieved by rotary evaporation. The ASDs (Table I provides a list of various compositions that were prepared) were subsequently dried in a vacuum oven overnight to remove any residual solvent. Pure amorphous CEX was prepared by melting crystalline CEX at approximately 180°C on aluminum foil using a hot plate, followed by quench cooling. Based on analysis using high performance liquid chromatography, no degradation was observed using this procedure. Both ASDs and pure amorphous CEX were ground using a mortar and pestle and sieved to obtain particle size fraction of 106–250 μm. The solids were then stored in a desiccator containing phosphorous pentoxide at room temperature. The amorphous nature of the ASDs and the pure CEX were verified by powder X-ray diffraction prior to use.
Phase Transformation of Slurred ASDs
ASD (200 mg) was slurred in 2 mL pH 6.8 100 mM sodium phosphate buffer (SPB) in a scintillation vial equilibrated at 37°C and stirred at a constant rate. The kinetics of phase transformation was monitored using a RamanRxn-785 Raman Spectrometer (Kaiser Optical Systems, Inc., Ann Arbor, MI, USA) with a laser wavelength of 785 nm. Spectra were collected every 15 min for 7 hours. Data were analyzed with OPUS software (Version 7.2, Bruker Optics Inc, Billerica, Massachusetts, USA).
Inhibitory Effectiveness of Polymers on Crystallization from Supersaturated Solution
Two hundred seventy five microliters of a 4 mg/mL methanol solution of CEX was pipetted into 50 mL pH 6.8 100 mM SPB with and without 5 μg/mL pre-dissolved polymer. The solution was equilibrated at 37°C and stirred at 300 rpm (+shaped magnetic stirrer, 0.75 inch diameter). Solution concentrations were measured as a function of time using a SI photonics UV–vis spectrometer coupled with a fiber optic probe (SI Photonics Inc, Tuscon, Arizona, USA). Wavelength scans (200–450 nm) were performed at 1 min interval for 8 hours. The absorption peak at 249 nm was used to monitor solution concentration. Light scattering was detected by monitoring the extinction at 350 nm at which the drug has no absorbance. All measurements were performed in triplicate.
Dissolution Study of ASDs
8.8 mg crystalline or amorphous CEX or 17.6 mg of ASD was added to 400 mL pH 6.8 100 mM SPB, equilibrated at 37°C and stirred at 300 rpm with a stir bar (+shaped, 0.75 inch diameter). Solution concentration evolution as a function of time was measured using the SI photonics system. Wavelength scans (200–450 nm) were performed at 1 min time intervals for 16 hours. The absorption peak at 249 nm was used to monitor solution concentration. Calibration solutions of CEX were prepared in methanol. All measurements were performed in triplicate.
Effect of Polymer on the Equilibrium Solubility of CEX
The equilibrium solubility of CEX was determined by adding an excess amount of crystalline CEX to 20 mL pH 6.8 100 mM SPB with the absence/presence of 22 μg/ml pre-dissolved polymer in scintillation vials. The vials were equilibrated at 37°C for 48 h in an agitating water bath (Dubnoff metallic shaking incubator; PGC Scientific, Palm Desert, CA, USA). Samples were then subject to ultracentrifugation to separate excess crystalline CEX particles from the supernatant. Ultracentrifugation was performed at 35,000 rpm for 30 min in an Optima L-100XP ultracentrifuge equipped with Swinging-Bucket Rotor SW 41 Ti (Beckman Coulter, Inc., Brea, CA, USA). HPLC analyses were carried out with an Agilent HPLC 1260 Infinity system (Agilent Technologies, Santa Clara, California, USA). The chromatographic separation was performed by an XTerra Shield RP18 Column (125 Å, 3.5 μm, 3.9 mm X 100 mm) (Waters Cooperation, Milford, MA, USA). Water (20%) and acetonitrile (80%) mixture was used as mobile phase and the flow rate was 0.25 mL/min. The ultraviolet detection wavelength was 250 nm. All measurements were performed in triplicate at room temperature.
Polarized Light Microscopy
The crystallization behavior of the pure amorphous CEX was observed using a Nikon Eclipse E600 Pol microscope with 10x objective (Nikon Company, Tokyo, Japan). The pulverized samples (106-250um) were placed on a microscope slide containing a depression. 3–4 drops of pH 6.8 100 mM SPB with or without a pre-dissolved polymer were then added to the particles. Images were processed by NIS-Elements software package (Version 2.3; Nikon Company, Tokyo, Japan).
Results
Nucleation Induction Time
The effectiveness of the polymers in inhibiting crystallization from supersaturated solution was assessed by performing nucleation induction time measurements. The experimental nucleation induction time, t ind , can be defined as the sum of the time for critical nucleus formation (true nucleation time, t n ), and growth to detectable size, t g .[9]
The initial solution concentration generated was 22 μg/mL, which was approximately the calculated “amorphous solubility” of CEX [10, 11], and therefore the maximum theoretical concentration of free drug that can be achieved by dissolving an ASD. The onset of crystallization was determined as a sudden increase of light scattering at 350 nm concomitant with a rapid decrease in absorbance at 249 nm [12]. As shown in Fig. 2, in the absence of any polymer, the drug concentration decreased rapidly and crystallization commenced within 5 min. The solution became increasingly turbid with the development of macroscopic crystals. Nucleation induction times extended to approximately 1 hr in the presence of PVP-K12 and 2 hrs with PVP-K29/32. It was noteworthy that the rate of desupersaturation following nucleation induction was slower in the presence of PVP-K29/32 than in the presence of PVP-K12 and slower in the presence of both of these polymers relative to in the absence of a polymer. In the case of HPMCAS and HPMC, however, no substantial nucleation/crystal growth occurred and the initial level of supersaturation was maintained for more than 8 hours. Furthermore, when combinations of HPMCAS with PVP-K12 or PVP-K29 were evaluated, no significant desupersaturation occurred, indicating that the effectiveness of HPMCAS in maintaining supersaturation was not impaired by the presence of PVP.

Induction time measurements of CEX in the presence and absence of different polymers.
Crystallization Kinetics of Slurred ASDs
The tendency of pulverized amorphous CEX and ASDs formulated with a 50% drug loading to crystallize when added to aqueous media (SPB at 37°C) was evaluated using Raman spectroscopy and results for the various systems summarized in Fig. 4. The crystalline and amorphous CEX reference spectra show distinct differences in peak position, intensity and width, which can be used to monitor the phase transformation from the amorphous to the crystalline form. For example, the crystalline form spectrum has a peak at 1614 cm−1 with a small shoulder at 1596 cm−1, whereas the reference amorphous CEX spectrum shows a peak at 1611 cm−1 with a much more pronounced shoulder at approximately 1598 cm−1 (Fig. 3).

Raman spectra of pure crystalline (top) and amorphous (bottom) CEX over the wavenumber range 1700–1000 cm-1.
By monitoring the decrease in intensity of the 1598 cm−1 shoulder as a function of time, it can be seen that the CEX:PVP-K12 ASD crystallized within 4 hours, as shown in Fig. 4. When a small amount of the PVP was replaced with HPMCAS, to form a ternary dispersion (consisting of 50% CEX, 40% PVP-K12 and 10% HPMCAS), the dispersion remained amorphous for the duration of the experiment (7 hours). Increasing the molecular weight grade of PVP used to form the dispersion, improved resistance to crystallization; the shoulder at 1598 cm−1 (indicative of the amorphous form) persisted for 7 hours when CEX:PVP-K29/32 ASDs were slurried. Both the CEX: HPMCAS ASD and the CEX: PVP-K29/32: HPMCAS ternary ASD remained amorphous over this time frame. Pure amorphous CEX crystallized rapidly during slurrying, whereby crystallization was complete within an hour. Furthermore, it was found that the polymer needed to be present in the dispersion to be an effective crystallization inhibitor; adding the polymer in pre-dissolved form (22 μg/ml) to the buffer did not substantially retard the crystallization kinetics of the pure amorphous CEX (data not shown).

Normalized intensities (y-axis) of CEX peaks in ASDs slurried in buffer for different time periods: (a) CEX: PVP-K12 5:5 (b) CEX: PVP-K12: HPMCAS 5:4:1, (c) CEX: PVP-K29/32 5:5, (d) CEX: HPMCAS 5:5, (e) CEX-PVP-K29/32: HPMCAS 5:4:1. All ratios are on a weight basis. The wavenumber range shown 1640–1540 cm−1(x-axis). From top to bottom: unexposed ASD, and after 0.5 hr, 2.5 hrs, 4 hrs, and 7 hrs exposure to buffer, respectively.
Dissolution Behavior of CEX ASDs
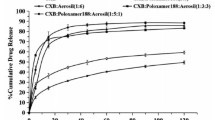
Figures 5, 6 and 7 show the dissolution profiles of ASDs of CEX formulated with different polymers and polymer combinations at a drug loading of 50% (w/w). The maximum apparent solution concentration of CEX achieved by dissolving the ASDs was between 21 and 22 μg/ml in all cases except for the dispersions that contained HPMC, which equals 100% release of the CEX in the ASDs; the amount of ASD added was selected so that theoretical concentration of CEX was equivalent to the reported amorphous solubility [10, 11]. Thus, the dissolution conditions are at-sink with respect to amorphous solubility and non-sink with respect to crystalline solubility. While the maximum concentration achieved was the same for these dispersions, considerable differences in the time to achieve the maximum concentration and the longevity of the achieved supersaturation can be noted between the different formulations.

Dissolution profiles of binary ASDs.

Dissolution profiles of binary and ternary ASDs

Dissolution profiles of CEX: PVP-K12 ASDs in pure SPB with and without a pre-dissolved polymer.
Binary dispersions with either PVP-K12 or PVP-K29/32 showed faster dissolution rates than the corresponding dispersions with HPMCAS or HPMC (Fig. 5). However, PVP-K12 was the least effective polymer at maintaining the generated supersaturation, with desupersaturation being observed about 2 hrs after all the CEX had dissolved. Interestingly, the peak CEX concentration achieved by dissolving the PVP-K29/32 ASD was maintained for approximately twice as long as that of the PVP-K12 ASD, although the initial dissolution rates for the two systems were very similar. Dispersions with HPMCAS dissolved more slowly, but did not desupersaturate over the experimental timeframe. Dispersions with HPMC exhibited the slowest dissolution rate, the maximum concentration achieved was only 16 μg/ml and was still increasing by the end of the experiment.
Replacing 20% of the PVP-K12 with HPMCAS led to two major changes in the dissolution behavior of the dispersion (Fig. 6). First, the dissolution rate was much slower, being more similar in profile to that of the binary dispersion containing HPMCAS. Second, no desupersaturation was observed and thus the ternary ASD has improved stability against crystallization relative to the binary dispersion with PVP-K12. A similar profile was observed for a ternary dispersion with PVP-K29/32 and HPMCAS. Replacing 20% of the PVP-K12 or PVP-K29/32 with HPMC also resulted slower dissolution rates relative to the PVP only dispersion, albeit to different extents depending on the grade of PVP. Again, no desupersaturation was observed in either case. Interestingly, the dissolution rate of the ternary dispersion containing PVP-K12 with HPMC was much slower than for PVP-K29; this difference between the PVP grades was not observed with the other ternary dispersions.
Additionally, it was found that pre-dissolved cellulosic polymers present in the dissolution medium slightly reduced the dissolution rate of the CEX: PVP-K12 ASD, resulting in a longer time period to achieve complete release, but were able to prevent desupersaturation (Fig. 7).
In contrast to the dispersions, the solution concentration time profile achieved by dissolving amorphous CEX was similar to that obtained by dissolving crystalline CEX, despite a faster initial release rate. Dissolution of amorphous CEX into buffer containing 5 μg/ml pre-dissolved HPMCAS or HPMC resulted in only a slightly higher final concentration, as shown in Fig. 8.

Dissolution profiles of crystalline and amorphous CEX in SPB with and without a pre-dissolved polymer.
Polarized Light Microscopy
Figure 9 shows that when neat amorphous CEX was exposed to phosphate buffer, the material appears to undergo rapid crystallization from the amorphous particles. In addition, some crystallization from the solution phase was also observed. When a cellulosic polymer was present in the buffer, solution crystallization was not observed whereas matrix crystallization was still evident, albeit proceeding at a much slower rate (Figs. 10 and 11).

Polarized light microscope images of neat amorphous CEX exposed to pure SPB. (a) Unexposed, (b) 2 mins, (c) 10 mins.

Polarized light microscope images of neat amorphous CEX exposed to SPB with 5 μg/mL pre-dissolved HPMC. (a) Unexposed, (b) 20 mins, and (c) 60 mins.

Polarized light microscope images of neat amorphous CEX exposed to SPB with 5 μg/mL pre-dissolved HPMCAS. (a) unexposed, (b) 5 mins, and (c) 60 mins.
Equilibrium Solubility of CEX
The measured equilibrium solubility of crystalline CEX was 1.5 μg/mL. Polymers, when present at a concentration of 22 μg/ml in SPB, did not substantially impact the crystalline solubility of CEX, as demonstrated by the results summarized in Table II.
Discussion
For ASD formulations, it is essential to prevent crystallization (either nucleation and/or growth) both in the solid formulation during storage, as well as during the dissolution process, either from the dissolving amorphous matrix, or from the supersaturated solution generated by dissolution under non-sink conditions. However, the polymer that is the best crystallization inhibitor in the solid formulation, may not be effective in preventing crystallization from aqueous solution and vice versa. For example, it was observed that polyacrylic acid was very effective at inhibiting the crystal growth of acetaminophen from supercooled liquids [13], and hence retarded crystallization from amorphous solid dispersions, but was ineffective in preventing either nucleation or crystal growth from aqueous solutions [14]. Other systems show similar types of behavior [6, 15–17]. Therefore, it may be appropriate to use combinations of polymers [18] to ensure adequate stability during storage and optimum performance during dissolution. Although ternary dispersions have been used to improve dissolution rates, [19, 20], as well as to improve physical stability in the solid dispersion [21], there is relatively little work evaluating the impact of binary polymer combinations on crystallization kinetics during ASD dissolution.
Crystallization during dissolution of amorphous materials can occur through surface/bulk crystallization of the solid matrix upon contact with the dissolution medium or from the supersaturated solution generated upon dissolution [8, 22]. The former process impacts the achievable extent of supersaturation, because any crystals formed will act as seeds, growing rapidly and depleting the supersaturation. If no crystallization from the matrix occurs, solution crystallization will govern the longevity of the supersaturated solution formed by dissolution of amorphous material; once nucleation from the solution phase commences, desupersaturation will be observed shortly thereafter due to growth of the nuclei. Ideally, matrix crystallization should be inhibited during the dissolution process, in order to achieve a higher level of supersaturation [8].
The microscope images and the Raman spectra revealed that pure amorphous CEX crystallized rapidly and primarily via the matrix route upon contact with buffer. Absorption of water will decrease the glass transition temperature, leading to increased molecular mobility and hence rapid crystallization. Polymers, when pre-dissolved in buffer, were not able to substantially impede the onset of crystallization, although their presence did extend the time needed to complete crystallization during the slurry experiment. Correspondingly, little supersaturation was generated during the dissolution of pure amorphous CEX in buffer or buffer containing a pre-dissolved polymer (Fig. 8). This indicates either that complete matrix crystallization is rapid, or that the seed crystals formed grow rapidly under these conditions, depleting the supersaturation produced by dissolution of any residual amorphous material. Furthermore, polymers dissolved in solution are clearly unable to inhibit the formation of crystals in the amorphous matrix. These observations confirm the need to prevent matrix crystallization in order generate supersaturated solutions.
When formulated as an ASD, even at drug loadings as high as 50%, CEX stability to crystallization upon exposure to buffer is considerably improved, although some differences are seen depending on the polymer used to form the ASD. The Raman data indicate that the CEX: PVP-K12 ASD is more prone to crystallization in the slurry experiments as compared to other ASDs which didn’t crystallize over the timeframe of the experiment. Raman spectroscopy alone does not enable us to identify the pathway through which crystallization occurred. However, since all binary and ternary ASDs gave complete release of the drug during the dissolution experiments, achieving the 21–22 μg/ml targeted CEX concentration and yielding supersaturated solutions, it is apparent that matrix crystallization was completely inhibited. Therefore, any difference in dissolution performance for the different ASDs can be attributed to the impact of a specific polymer or polymer combination on crystallization from the solution phase.
The overall desupersaturation profile observed following complete release of the drug will depend on the impact of the polymer on both nucleation and growth kinetics from the solution phase. Since growth cannot occur until nuclei have formed, the impact of polymers on crystal nucleation is of paramount importance. Nucleation kinetics are often inferred from measurement of the induction time, which is defined as the time required for stable nuclei to form and grow to a detectable size (Eq. 1). If it is assumed that steady-state nucleation is achieved quickly and that t n > > t g , then the induction time for the formation of a critical nucleus [9] can be expressed as:
Where J is the nucleation rate, which is given by classical nucleation theory (CNT) as:
Where γ is the interfacial tension, v is the molecular volume of the crystallizing solute, k is the Boltzmann constant, T is the temperature, and S is supersaturation [23]. S can be expressed [24] in terms of chemical potential differences as:
where μ is the solute chemical potential, and μ* is the chemical potential of a solute in a saturated solution, and R is the gas constant. In dilute solutions, S can be determined from the ratio, c/c*, where c is the solute concentration in the supersaturated solution and c is the crystal solubility. From Eq. 3 it is apparent that the main factors influencing the nucleation rate (and hence induction time) are temperature (a constant in our studies), supersaturation and interfacial tension. In the presence of the polymers, experimental induction times are extended indicating that the nucleation rate is decreased, with differences being observed between the polymers. PVP-K12 is the least effective polymer (Fig. 2) with desupersaturation commencing after 60 min whereas in the presence of HPMCAS or HPMC, no discernable nucleation or crystal growth can be observed for 8 hrs. These differences between polymer effectiveness cannot be explained based on changes in supersaturation (the supersaturation is the same for all solutions containing polymers based on consideration of solution concentration values and equilibrium solubilities; see Table I) or interfacial tension. Adsorption of a polymer to the nucleus would lead to a decrease in interfacial tension, leading to an expected increase in nucleation rate based on Eq. 2, rather than the decrease observed in this study. Although CNT is the main framework for understanding nucleation phenomena, it is apparent that this theory does not readily allow for obvious mechanistic insights into how polymers extend induction times. Computer simulations have suggested that nucleation occurs via a two-step process, whereby the first step is the formation of a dense liquid cluster, followed by structural rearrangement of this cluster to an ordered state [25]. Additives are proposed to interfere with the rearrangement process depending on their relative affinity for the solute cluster and the solvent, as well as some geometric considerations. The greater effectiveness of the cellulose derivatives as nucleation inhibitors in comparison to either PVP grade may therefore relate to a greater ability to interact with a dense liquid cluster of celecoxib, relative to the more hydrophilic PVP which will also have a competing interaction with the solvent phase. This is in accordance with previous studies where more hydrophilic polymers were found to have a smaller impact on nucleation induction times of relatively lipophilic drugs relative to more amphiphilic polymers (including many cellulose derivatives) which extended the longevity of supersaturated solutions to a greater extent [10, 26].
The induction time values are quite predictive of the dissolution performance, whereby the dispersions formulated with PVP-K12 also undergo desupersaturation at the earliest time point following dissolution. Interestingly, the higher tendency of the PVP-K12 dispersions to undergo crystallization from solution can be mitigated by adding a small amount of an effective crystallization inhibitor, either HPMCAS or HPMC, to the dispersion. Thus the cellulose polymers dissolve sufficiently rapidly from the ternary dispersion to be effective solution nucleation inhibitors and are able to inhibit CEX nucleation in the presence of PVP. This strategy might be useful in vivo to improve the performance of a dispersion, by combining a polymer that rapidly releases the drug, but is a poor solution crystallization inhibitor, with a small quantity of a polymer that is a good inhibitor. The induction time experiments (Fig. 2) confirm that the effectiveness of HPMCAS or HPMC in inhibiting solution nucleation was not impaired by the presence of PVP. These observations open up a strategy of utilizing more than one polymer in an ASD formulation in order to improve performance and also show that only very low polymer concentrations are required to inhibit solution nucleation (only 5 μg/mL of polymer was used for the induction time experiments shown in Fig. 2). In the current study, high drug loading ASDs formulated with PVP and HPMC or HPMCAS show enhanced stability against crystallization. Furthermore, the ternary CEX: PVP-K29/32: HPMC dispersions have enhanced release relative to the binary CEX: HPMC dispersions, from which the drug is released very slowly. However, it is important to note that relative to the binary CEX: PVP-K12 dispersion, somewhat surprisingly, release is delayed when a small quantity of either HPMCAS or HPMC is added to matrix. This reduced release rate clearly may not be desirable. However, based on the evidence shown in Fig. 7, it is apparent that the additional polymer added to improve stability against crystallization need not be present in the dispersion itself. Here it can be seen that trace amounts (5 μg/mL) of both HPMC and HPMCAS effectively inhibit crystallization from the CEX: PVP-K12 dispersions when pre-dissolved in buffer. Thus, they could be potentially added to the formulation as a separate component from the ASD matrix. Given the wide array of available polymers, as well as the current interest in increasing drug loading, using very hydrophilic polymers to ensure rapid drug release, combined with a small amount of an effective crystallization inhibitory polymer, is an approach that should be investigated further based on the promising results shown in this study.
Conclusions
High drug loading amorphous solid dispersions of celecoxib formulated with different polymers led to supersaturated solutions, but showed dramatically different release profiles and solution crystallization behavior. Using a combination of polymers, where an effective crystallization inhibition polymer was present as a minor component, led to the formation of supersaturated solutions upon solid dispersion dissolution that had improved stability against crystallization. These findings open up new formulation approaches for amorphous solid dispersions, whereby one polymer is included to inhibit crystallization during dissolution, whereas a second polymer is used to achieve another key property such as rapid release, or enhanced storage stability.
Abbreviations
- ASD:
-
Amorphous solid dispersion
- CEX:
-
Celecoxib
- HPLC:
-
High performance liquid chromatography
- HPMC:
-
Hydroxypropylmethyl cellulose
- HPMCAS:
-
Hydroxypropylmethyl cellulose acetate succinate
- PVP:
-
Polyvinyl pyrrolidone
- SPB:
-
Sodium phosphate buffer
- UV:
-
Ultraviolet
References
Fahr A, Liu X. Drug delivery strategies for poorly water-soluble drugs. Expert Opin Drug Deliv. 2007;4(4):403–16.
Hancock BC, Parks M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm Res. 2000;17(4):397–404.
Baird JA, Van Eerdenbrugh B, Taylor LS. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J Pharm Sci. 2010;99(9):3787–806.
Trasi NS, Taylor LS. Effect of additives on crystal growth and nucleation of amorphous flutamide. Cryst Growth Des. 2012;12(6):3221–30.
Van den Mooter G, Wuyts M, Blaton N, Busson R, Grobet P, Augustijns P, et al. Physical stabilisation of amorphous ketoconazole in solid dispersions with polyvinylpyrrolidone K25. Eur J Pharm Sci. 2001;12(3):261–9.
Kestur US, Taylor LS. Role of polymer chemistry in influencing crystal growth rates from amorphous felodipine. Crystengcomm. 2010;12(8):2390–7.
Kestur US, Van Eerdenbrugh B, Taylor LS. Influence of polymer chemistry on crystal growth inhibition of two chemically diverse organic molecules. Crystengcomm. 2011;13(22):6712–8.
Alonzo DE, Zhang GGZ, Zhou DL, Gao Y, Taylor LS. Understanding the behavior of amorphous pharmaceutical systems during dissolution. Pharm Res. 2010;27(4):608–18.
Sohnel O, Mullin JW. Interpretation of crystallization induction periods. J Colloid Interf Sci. 1988;123(1):43–50.
Ilevbare GA, Liu HY, Edgar KJ, Taylor LS. Maintaining supersaturation in aqueous drug solutions: impact of different polymers on induction times. Cryst Growth Des. 2013;13(2):740–51.
Abu-Diak OA, Jones DS, Andrews GP. An investigation into the dissolution properties of celecoxib melt extrudates: understanding the role of polymer type and concentration in stabilizing supersaturated drug concentrations. Mol Pharmaceut. 2011;8(4):1362–71.
Chen J, Ormes JD, Higgins JD, Taylor LS. Impact of surfactants on the crystallization of aqueous suspensions of celecoxib amorphous solid dispersion spray dried particles. Mol Pharmaceut. 2015;12(2):533–41.
Trasi NS, Taylor LS. Effect of polymers on nucleation and crystal growth of amorphous acetaminophen. Crystengcomm. 2012;14(16):5188–97.
Trasi NS, Oucherif KA, Litster JD, Taylor LS. Evaluating the influence of polymers on nucleation and growth in supersaturated solutions of acetaminophen. Crystengcomm. 2015;17(6):1242–8.
Konno H, Handa T, Alonzo DE, Taylor LS. Effect of polymer type on the dissolution profile of amorphous solid dispersions containing felodipine. Eur J Pharm Biopharm. 2008;70(2):493–9.
Konno H, Taylor LS. Influence of different polymers on the crystallization tendency of molecularly dispersed amorphous felodipine. J Pharm Sci-Us. 2006;95(12):2692–705.
Rumondor ACF, Stanford LA, Taylor LS. Effects of polymer type and storage relative humidity on the kinetics of felodipine crystallization from amorphous solid dispersions. Pharm Res. 2009;26(12):2599–606.
Marks JA, Wegiel LA, Taylor LS, Edgar KJ. Pairwise polymer blends for oral drug delivery. J Pharm Sci. 2014;103(9):2871–83.
Goddeeris C, Willems T, Van den Mooter G. Formulation of fast disintegrating tablets of ternary solid dispersions consisting of TPGS 1000 and HPMC 2910 or PVPVA 64 to improve the dissolution of the anti-HIV drug UC 781. Eur J Pharm Sci. 2008;34(4–5):293–302.
Janssens S, De Armas HN, Roberts CJ, Van Den Mooter G. Characterization of ternary solid dispersions of itraconazole, PEG 6000, and HPMC 2910 E5. J Pharm Sci. 2008;97(6):2110–20.
Al-Obaidi H, Ke P, Brocchini S, Buckton G. Characterization and stability of ternary solid dispersions with PVP and PHPMA. Int J Pharm. 2011;419(1–2):20–7.
Raina SA, Alonzo DE, Zhang GGZ, Gao Y, Taylor LS. Impact of polymers on the crystallization and phase transition kinetics of amorphous nifedipine during dissolution in aqueous media. Mol Pharmaceut. 2014;11(10):3565–76.
Mullin JW. Crystallization: Butterworth-Heinemann; 2001.
Myerson A. Handbook of industrial crystallization: Butterworth-Heinemann; 2002.
Anwar J, Boateng PK, Tamaki R, Odedra S. Mode of action and design rules for additives that modulate crystal nucleation. Angew Chem Int Ed. 2009;48(9):1596–600.
Warren DB, Benameur H, Porter CJH, Pouton CW. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: a mechanistic basis for utility. J Drug Target. 2010;18(10):704–31.
ACKNOWLEDGMENTS AND DISCLOSURES
The Dane O. Kildsig Center for Pharmaceutical Processing Research is acknowledged for providing funding for this project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xie, T., Taylor, L.S. Dissolution Performance of High Drug Loading Celecoxib Amorphous Solid Dispersions Formulated with Polymer Combinations. Pharm Res 33, 739–750 (2016). https://doi.org/10.1007/s11095-015-1823-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-015-1823-y