
Abstract
Purpose
Raman spectroscopy is potentially an extremely useful tool for the understanding of drug-polymer interactions in solid dispersions. This is examined and demonstrated for the case of solid dispersions of nifedipine in a polymeric substrate.
Methods
Solid dispersions consisting of nifedipine and polyvinyl caprolactam - polyvinyl acetate - polyethylene glycol graft copolymer (Soluplus®) were prepared by freeze drying, melting and solvent evaporation at drug loadings of 10, 30, 50, 70 and 90% w/w. Drug-polymer interactions in the amorphous solid dispersion were estimated by Raman spectroscopy. The correlation between the solid state stability of the drug in a solid dispersion and the extent of drug-polymer interaction was monitored by X-ray diffractometry.
Results
The miscibility limit of nifedipine-Soluplus® was found to be 30% w/w drug loading for all preparation methods. The drug was found to interact with Soluplus®, through a hydrophilic interaction identified by infrared spectroscopy and a hydrophobic interaction which could be quantified by Raman spectroscopy. The average extent of the drug-polymer interaction in the studied amorphous samples at equivalent drug loading was similar, regardless of the preparation method. Inhomogeneities in samples prepared by melting contributed to a wider variation in drug-polymer interaction and poorer solid state stability, in terms of its crystallization tendency.
Conclusions
Raman spectroscopy was shown to be a useful technique in classifying miscibility levels based on the hydrophobic interaction between the drug and the polymer. Different drug loadings showed varying degrees of drug-polymer interaction, and hence variable solid state stability of the solid dispersion.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Solid dispersion of a drug molecule within a polymer is one approach to enhance oral bioavailability of poorly water-soluble drugs. The method of preparing the solid dispersion allows for the conversion of the crystalline drug to its amorphous counterpart resulting in higher solubility and faster dissolution (1). The development of a solid dispersion in which the dispersed drug is physically stable is, however, still a challenge because the drug in the amorphous state is not at equilibrium and tends to recrystallize, leading to a decrease in solubility and dissolution rate. Recrystallization in solid dispersions has been shown to be delayed and/or minimized when the amorphous drug is homogeneously and molecularly dispersed in the polymer as a single phase amorphous system, so-called “miscible dispersions” (2–4). One of the underpinning mechanisms which helps prevent the drug molecule from recrystallizing is the molecular interaction between drug and polymer. The intermolecular interactions (which may be specific such as hydrogen bonding and/or non-specific such as van der Waals force (5)) increase kinetic barriers by lowering molecular mobility (6,7). The strength of intermolecular interaction is a key factor governing the physical stabilization of solid dispersions and it may be related to the miscibility of the drug in the polymer matrix (7). To date, there have been several attempts to apply analytical methods to characterize drug-polymer miscibility, e.g., differential scanning calorimetry (DSC) (3,8–16), infrared spectroscopy (IR) (8–13,17–19), solid state-nuclear magnetic resonance (ss-NMR) (14,16), X-ray powder diffractometry (XRD) (10), X-ray photoelectron spectroscopy (XPS) (14) and Raman spectroscopy (15,19–23). The strength of intermolecular hydrogen bonding between drug and polymer in a solid dispersion has been investigated by IR based on the peak position (12,13,18) and peak height ratio (13,17). The strength of drug-polymer hydrogen bonding has also been studied by XPS (14) and ss-NMR (16) based on the shift in peak position. Recently, Raman spectroscopy has been widely utilized to reveal the occurrence of specific interaction (5,15,19,21,23). As with IR, changes in the chemical environment of the drug will affect the Raman spectral pattern in terms of peak position (15,19–24), peak shape (22) or new peaks (5,24). The presence of a single peak indicates a molecular dispersion of the drug and suggests the presence of a drug-polymer interaction. A shift in a drug related peak position to lower wavenumber indicates stronger drug-polymer interaction than drug-drug interaction and vice versa (23). The stronger drug-polymer interaction results in a weakening of drug–drug interactions, causing a shift to lower frequency. Since the principle of Raman spectroscopy is based on light scattering due to changes in polarization (24), it may be more useful than IR for determining non-specific interactions such as the hydrophobic interaction in a drug-polymer mixture which is rich in aromatic conjugated systems (5).
Within the miscible drug-polymer mixture, the drug may be present in two distinct states, namely, “monomolecularly” and “molecularly” dispersed states, depending on its content. “Monomolecularly dispersed state” here implies that all drug molecules are completely distributed in the polymer and only drug–polymer interactions are present (no drug–drug interactions). This state is obtained when the dispersed drug content in solid dispersion is much lower than its saturated concentration, allowing the drug-drug interaction to be minimized. On the other hand, a “molecularly dispersed state” is obtained with a concentrated or saturated concentration of the drug in the polymer; that is, the drug content in the solid dispersion is significantly higher than that in a monomolecularly dispersed mixture. In this regard, there is a balance between drug-drug interactions and drug-polymer interactions, yet there is no phase separation (4,11) as the conceptual models illustrates in Fig. 1a and b. The relative amount of the drug distributed between the two states and amorphous state would suggest the degree of drug-polymer interaction. In this study, Raman spectroscopy was employed to investigate the miscibility of nifedipine in solid dispersions with polyvinyl caprolactam - polyvinyl acetate - polyethylene glycol graft copolymer (Soluplus®). Nifedipine, a calcium channel blocker for treatment of cardiovascular disease, contains hydrogen bond acceptors including C=O and NO2, and hydrogen bond donors including NH which might form hydrogen bonds with the OH and C=O groups of Soluplus®, respectively. The non-specific van der Waals interactions between nifedipine and Soluplus® might also occur between the aromatic ring of nifedipine and the polyvinyl caprolactam segments of Soluplus® (Fig. 2). The degree of molecular interaction in the solid dispersions with different drug loadings, prepared by freeze drying (FD), melting (ME) and solvent evaporation (SE) is classified based on the amorphous state and two states of molecular dispersion as described above. The data obtained were correlated with the crystallization tendency of the dispersions.

The schematic representation of (a) monomolecularly dispersed state (b) molecularly dispersed state (c) combination of molecularly dispersed state and amorphous clusters. The dot lines between drug molecules demonstrate the balance between drug-drug interaction and drug-polymer interaction.

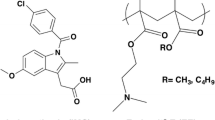
Chemical structures of (a) nifedipine and (b) Soluplus®.
Materials and Methods
Nifedipine crystalline α-form (α-NIF) was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan) and used without further purification. Soluplus® was donated from BASF SE (Ludwigshafen, Germany). Methanol, tertiary-butyl alcohol and acetonitrile were purchased from Kanto Chemical Co., Inc. (Tokyo, Japan).
Preparation of Amorphous Nifedipine
α-NIF was placed in a stainless steel beaker and heated in an oil bath containing silicone fluid oil (KF-968-100CS, Shin-Etsu Chemical Co., Ltd., Tokyo, Japan) at 182 ± 2°C for 15–30 min until completely melted. Quenching was done by removing the molten drug from the heated stainless steel beaker, and equilibrating to room temperature. The physical state of amorphous nifedipine (am-NIF) was confirmed by XRD and Raman spectroscopy.
Preparation of Nifedipine Crystalline β-Form
Nifedipine crystalline β-form (β-NIF) was prepared by a modification of the method published by Grooff et al. (25); am-NIF was placed in a stainless steel beaker and heated in an oil bath at 92 ± 1°C for 3 min, then placed at room temperature. The physical state of β-NIF was confirmed by XRD and Raman spectroscopy.
Preparation of Standards for Molecular Dispersion of Nifedipine in a Solid Dispersion
An accurately weighed amount of α-NIF was dissolved in tertiary-butyl alcohol, while Soluplus® was dissolved in a minimal amount of methanol, not exceeding 10% v/v of the total amount of solvent. The Soluplus® solution was added into the nifedipine solution with constant agitation. The mixtures were quenched by liquid nitrogen and placed under vacuum (FD-80, Eyela, Tokyo Rikakikai Co., Ltd., Tokyo, Japan) at approximately 0°C for 4 days. The batch sizes of the standards were 0.5 to 3 g, sufficient for characterization. The samples were further dried in a vacuum chamber containing silica gel at room temperature and 15–20% relative humidity for at least 24 h.
Preparation of Solid Dispersion Samples
Solid dispersions of nifedipine and Soluplus® were prepared with drug loadings of 10, 30, 50, 70 and 90% w/w by three different methods, i.e., FD, ME and SE as follows:
-
FD: The samples were prepared as described above for standards of molecular dispersion.
-
ME: An accurately weighed amounts of α-NIF and Soluplus® were gently mixed by a mortar and pestle about 3 min before placed in a stainless steel beaker. The mixtures were heated in an oil bath containing silicone fluid oil (KF-968-100CS, Shin-Etsu Chemical Co., Ltd., Tokyo, Japan) at 182 ± 2°C for 15–30 min until completely melted. Quenching was done by removing the molten mixture from the heated stainless steel beaker, and equilibrating to room temperature. Then, the samples were gently pulverized using a mortar and pestle.
-
SE: An accurately weighed amount of α-NIF and Soluplus® was dissolved in methanol. The solvent was removed under vacuum by rotary evaporator (Pair stirrer PS-100, Eyela, Tokyo Rikakikai Co., Ltd., Tokyo, Japan) at 50–55°C. The samples were further dried in a vacuum chamber containing silica gel at room temperature and 15–20% relative humidity for at least 24 h. Then the samples were gently pulverized using a mortar and pestle.
The batch sizes of the samples varied from 3 to 6 g, depending on the yield obtained from each preparation method. All samples were stored in amber bottles at −20°C before characterization.
Characterization of Solid Dispersions
Raman Spectroscopy
All samples were compressed at 0.5 ton/cm2 in order to obtain flat surfaces before measuring. Raman spectra were measured at room temperature by a Raman RXN2 (Kaiser Optical Systems, Inc., MI, USA), and controlled by Hologram version 4.1 (Kaiser Optical Systems, Inc., MI, USA). A 1,000 nm excitation wavelength with a 400 mW laser power source and InGaAs array detectors was used. A 50× objective (DM2500, Leica Microsystems, Tokyo, Japan) with a 145 mW laser power, and a 16-μm laser sampling diameter, was utilized to acquire a point mapping with a 20-μm step size. Each spectrum was collected from 200 to 2,400 cm−1 with a resolution of 5 cm−1 and a 5-s exposure time. Naphthalene was used to calibrate the wavenumber. The spectra of α-NIF, Soluplus®, β-NIF and am-NIF were averaged from a 100-point map of sampling area of approximately 200 × 200 μm2. The spectra of standard samples were averaged from a 1,200-point map of sampling area of approximately 800 × 600 μm2. The spectra of solid dispersion samples for qualitative analysis were averaged from a 1,200-point map. The spectra of solid dispersion samples for quantitative analysis were averaged from a 100-point map each, and six maps per sample were done.
Spectral Data Analysis
Raman spectra were preprocessed using ISys® Chemical imaging analysis version 5.0.0.11 (Malvern Instruments, Inc., MD, USA). The spectral region of 550–1800 cm−1 was extracted. The mapping spectra were averaged, then the baseline was corrected at 660 and 872 cm−1. The spectral region of 775–850 cm−1 was further analyzed to evaluate the quantity of nifedipine present as molecular states and as an amorphous state. The method of curve fitting based on Gaussian function which has been applied to both qualitatively and quantitatively determine the chemical compound in an overlapping spectrum (26–28) was used. In the present study, the experimental spectra were converted to a series of Gaussian peaks by introducing three parameters including peak intensity, peak width and peak position into each spectrum, then the linear combination of peaks was manipulated to construct the Gaussian function models using Microcal Origin® version 3.5 (OriginLab Corporation, MA, USA).
The Gaussian function models of am-NIF and Soluplus® were established based on their experimental spectrum per se. The Gaussian function models of single phase, monomolecularly and molecularly dispersed states were derived from the experimental spectrum of the chosen standard solid dispersions. The monomolecularly dispersed states were selected based on the shift in peak position of FD solid dispersions with drug loadings of 0.1, 0.5, 1, 3 and 5% w/w. The sample which demonstrated the most shifted peak position was selected as the standard for the monomolecularly dispersed state and its spectrum was used to establish a Gaussian function model.
The molecularly dispersed state was selected from FD solid dispersions with drug loadings 10, 30 and 50% w/w. To construct a Gaussian function model, the model spectrum of Soluplus® was subtracted from the experimental spectrum of the standard monomolecularly dispersed state. The resulting spectrum was then fitted with a new set of Gaussian functions and the resulting function termed “model M”, as it reflects solely the monomolecular dispersed drug with no associated polymer. After subtracting the model spectrum of am-NIF and that of model M from the experimental spectra of the solid dispersions, the resulting spectrum was used to construct further Gaussian function models.
The highest drug loading solid dispersion which possessed the greatest fractional area under the model spectrum relative to the model spectrum of am-NIF and that of model M was chosen as the standard for the molecularly dispersed state.
The Gaussian function models were utilized to estimate the proportion of drug-polymer interaction (in terms of the amorphous and the two dispersed states) in amorphous solid dispersion samples, which had been identified by XRD and confirmed with the results of Raman spectroscopy in the 775 to 850 cm−1 region. The experimental spectra of amorphous samples were fitted to each of the standard spectra obtained from the Gaussian function models of the standard states. Each standard model was adjusted by varying the coefficients so that the model spectrum gave a best fit to the experimental spectrum of solid dispersion sample as expressed in Eqs. (1)–(3).
Where f(x) is a function representing the model spectrum of a sample by summation of the model spectra of nifedipine in each state; A, B, C are the Gaussian function models of monomolecularly dispersed state, molecularly dispersed state and amorphous state, respectively; d is y-intercept of baseline; a, b, c are the coefficients of the Gaussian function models of monomolecularly dispersed state, molecularly dispersed state and amorphous state, respectively; g(x) is a series of Gaussian peaks describing the model spectrum of each state; abs is absolute; exp is exponential; P1 is the peak height; P2 is the peak width; P3 is the peak position and x is the experimental peak position.
The weight fraction of the drug distributed in each state was calculated based on the fractional area under the peak, relative to the total area which corresponded to the total drug loading. An example calculation is shown in Eq. (4), which is normalized to 100% w/w drug loading as shown in Eq. (5)
Where A is the monomolecularly dispersed state, B is the molecularly dispersed state and C is the amorphous state.
X-ray Powder Diffractometry (XRD)
XRD patterns were measured with a Bruker D8 Discovery (Bruker AXS Inc., WI, USA) controlled by GADDS version 4.1.36 (Bruker AXS Inc., WI, USA) on quartz sample holder using Cu K-α radiation. Each sample was measured for 3 min from 2θ of 3.8 to 27.5° with a step size of 0.02°, and the voltage and current was 40 kV and 40 mA, respectively.
Differential Scanning Calorimetry (DSC)
Thermal analysis was performed by a DSC 8500 (PerkinElmer, Inc., MA, USA) equipped with Intracooler 2P (PerkinElmer, Inc., MA, USA), and controlled by Pyris version 11.1.0.0488 (PerkinElmer, Inc., MA, USA). Indium was used to calibrate temperature and enthalpy. Samples of 4–15 mg were placed in a crimped standard aluminium pan (PerkinElmer, Inc., MA, USA). Nitrogen at a flow rate of 20 ml/min was used as the purge gas. am-NIF for thermal analysis was formed in situ in the DSC pan by heating α-NIF to 178°C with a heating rate of 10°C/min, holding at 178°C for 15 min and cooling to 0°C with a cooling rate of 50°C/min. β-NIF for thermal analysis was also formed in situ in the DSC pan by a modified method of Grooff et al. (25); am-NIF was heated to 120°C at a heating rate of 10°C/min followed immediately by quenching to 0°C. The thermal transitions of am-NIF, β-NIF and α-NIF were measured from 0 to 200°C with a heating rate of 10°C/min. For Soluplus® and solid dispersions, samples were heated from 0 to 80°C with a heating rate of 10°C/min and held at 80°C for 20 min, followed by cooling to 0°C with a cooling rate of 50°C/min. The second heating run was carried out from 0 to 200°C with a heating rate of 10°C/min. Glass transition temperature (Tg), crystallization temperature and melting temperature were determined during the second heating run to reduce the effects of thermal history and plasticizing solvents on Tg (11). The values reported are midpoint Tg, onset crystallization temperature and onset melting temperature. A theoretical Tg was calculated based on Gordon-Taylor equation as shown in Eqs. (6) and (7) (29).
Here w and ρ are the weight fractions and true densities, respectively, of each component and Tg is the glass transition temperature. The true density of Soluplus® was determined by pycnometer (Quantachrome instruments, FL, USA), and that of am-NIF was reported by Forster et al. (29).
Fourier Transform-Infrared Spectroscopy (FT-IR)
IR spectra were examined using FT/IR-4100 equipped with attenuated total reflection (ATR) PRO410-S (JASCO International Co., Ltd., Tokyo, Japan) controlled by Spectra manager software version 2.2.9.1 (JASCO International Co., Ltd., Tokyo, Japan). Each spectrum was collected for 64 scans, and measured from 400 to 4,000 cm−1 with a resolution of 4 cm−1.
Solid State Nuclear Magnetic Resonance (ss-NMR)
ss-NMR was performed by a Bruker Avance II 300 (Bruker AXS Inc., WI, USA), and controlled by Topspin software version 3.1 (Bruker AXS Inc., WI, USA). The instrument was operated on 13C cross-polarization/magic angle spinning (CPMAS) with a transmitter frequency of 75 MHz. The experiment was run using a standard bore MAS probe (Bruker AXS Inc., WI, USA) at temperature of 300 K with CPMAS of 5 kHz for 13C and 62 kHz for 1H. Samples were placed in MAS 7-mm zirconia rotors and covered with Kel-F caps (Bruker AXS Inc., WI, USA). The spinning rate was set at 5 kHz. The contact time was 1.75 milliseconds and the amplitude ramp on 1H channel was approximately 49 Watts (from 50 to 100%) with two pulse phase modulation (TPPM) 15 decoupling. Glycine was used as an external standard to calibrate the chemical shift with a carbonyl peak referenced to 176.03 ppm.
High Performance Liquid Chromatography (HPLC)
The chemical stability of am-NIF and solid dispersion samples were determined by an HPLC method according to the United States Pharmacopeia 36 (USP 36) (30). An HPLC comprising of an SCL-10A system controller, a DGU-3A degasser, an LC-10AD liquid chromatograph, a CTO-10A column oven and a SPD-10A UV spectrophotometric detector (Shimadzu Corporation, Kyoto, Japan) was used to determine the nifedipine content. The separation was carried out in a C18 Inertsil® ODS-3, 5-μm particle size, 4.6 mm × 250 mm column (GL Sciences Inc., Tokyo, Japan) at room temperature with 25-μL injection volume and 265-nm UV detector. The mobile phase consisted of water, acetonitrile and methanol at a ratio of 50:25:25 (v/v/v), respectively and the flow rate was 1 ml/min. The peak areas were integrated by CDS Lite version 5.0 (DataApex, Prague, Czech Republic) software. Standard samples were prepared by dissolving in a small amount of methanol and diluting with mobile phase to obtain a nifedipine concentration of 0.1 mg/ml.
Solid State Stability
The solid state stability of nifedipine in solid dispersions was determined by the onset time for observable crystallization. The study was carried out at an elevated temperature and relative times to detectable crystallization were compared. Samples with drug loadings of 50 and 70% which were supposed to exhibit rapid crystallization were observed in real time by variable-temperature X-ray powder diffraction (VT-XRD) (RINT 2000 Ultima+, Riguku Corporation, Tokyo, Japan) controlled by Windmax software (Riguku Corporation, Tokyo, Japan). The samples were placed on aluminium holders (Riguku Corporation, Tokyo, Japan) and heated with X-ray powder diffraction-differential scanning calorimetry (XRD-DSC II, Riguku Corporation, Tokyo, Japan) from room temperature to 98 ± 2οC with a heating rate of 10οC/min and nitrogen flow rate of 100 ml/min controlled by ThermoPlus 2 version 4.208 (Riguku Corporation, Tokyo, Japan). The onset time for observable crystallization was determined after the maximum temperature was reached. The solid dispersion with 30% w/w nifedipine which was supposed to exhibit slow crystallization was heated in a hot air oven (WFO-450ND, Eyela, Tokyo Rikakikai Co., Ltd., Tokyo, Japan) at 98 ± 2οC. XRD patterns were collected using Cu K-α radiation from 2θ of 5.0 to 30.0° with a step size of 0.02° and a scan speed of 20°/min. The voltage and current was 40 kV and 40 mA, respectively. The measurements were done in triplicate.
Results
The physical forms of commercially available α-NIF, prepared β-NIF, and prepared am-NIF were confirmed by XRD and Raman spectroscopy (25,31). The chemical stability of nifedipine in am-NIF, and solid dispersion samples prepared by FD, ME and SE was proved and the remaining nifedipine content was between 90.0 and 110.0%. A slight degradation was observed but the Raman spectra of degraded products showing the characteristic peaks of nitroso-derivative and nitro-derivative at approximately 1729 cm−1 and 1728 cm−1, respectively did not interfere with the results of the present study.
DSC
Thermal events of α-NIF, β-NIF, am-NIF, Soluplus® and solid dispersions composed of nifedipine and Soluplus® with drug loadings of 10, 30, 50, 70 and 90% w/w prepared by FD, ME and SE are presented in Table I. α-NIF exhibited a melting event at 172°C. β-NIF presented an endothermic transformation to another metastable form at 61°C, followed by an exothermic transformation to α-NIF at 119°C and a subsequent melting event of α-NIF at 170°C. am-NIF showed a Tg at 45°C followed by exothermic crystallization to a metastable form at 96°C, which was then exothermically transformed to α-NIF at 119°C followed by melting at 170°C. This corresponds to the values reported by Grooff et al. (25,32). The Tg of Soluplus® was observed at 76°C. The single Tg of the samples shifted downwards towards the value for nifedipine with increasing weight fractions of the drug. FD samples with 10, 30 and 50% w/w drug loadings exhibited a single Tg at 73°C, 66°C and 58°C, respectively. ME samples with 10 and 30% w/w drug loadings showed a single Tg at 70 and 67οC, respectively, and SE samples with 10 and 30% drug loadings showed a single Tg at 72 and 65οC, respectively. Crystallization and melting of nifedipine was not observed in these samples. With a drug loading of 50% w/w, ME and SE samples demonstrated Tg at 59 and 60οC, respectively and melted at 158οC and 144οC, respectively. The presence of a melting event indicated phase separation which was not found in the FD sample at an equivalent drug loading. The FD sample with 70% w/w drug loading showed a Tg at 47°C and melting at 158°C. The ME sample with 70% w/w drug loading exhibited Tg and melting at 48 and 166οC, respectively; whereas, for SE samples with equivalent drug loadings the values were 51 and 160οC, respectively. At drug loading of 70% w/w, the Tg of the SE sample was higher than that of FD and ME samples. This could possibly be explained by the conversion of some am-NIF fraction to α-NIF during the SE process. As a consequence, the remaining portion of am-NIF in the SE sample was lower than those in the FD and ME samples, and affected the shift in Tg to higher temperature. The FD sample with 90% w/w drug loading did not show a glass transition but demonstrated an endothermic event at 58°C which could be attributed to the transformation of β-NIF to a metastable form, which was converted to α-NIF at 135°C, followed by melting at 170°C. Nifedipine in ME samples with a drug loading of 90% w/w was present as amorphous clusters as indicated by a Tg of 47οC followed by melting at 171οC. Whereas, at equivalent drug loading, nifedipine in the SE sample was present as α-NIF as indicated by the detection of a melting event at 171οC without the presence of a glass transition.
Based on DSC findings of a single Tg without a melting event, samples prepared by FD with drug loadings of 10, 30 and 50% w/w were miscible, while the miscible samples prepared by ME and SE were found at drug loadings of 10 and 30% w/w. In Fig. 3 the observed Tg is plotted against polymer content and compared with the theoretical Tg calculated from the Gordon-Taylor equation using the true densities of 1.36 g/cm3 (29) and 1.16 g/cm3 for am-NIF and Soluplus®, respectively. The plot (Fig. 3) shows minor negative deviations of samples from ideality.

Theoretical and observed Tgs of FD, ME and SE samples with Soluplus® content of 0–100% w/w. Tg of FD and SE samples with Soluplus® content of 10% w/w could not be determined (n = 3).
IR
Visual inspection of IR spectra of FD samples with drug loadings of 10, 30, 50, 70 and 90% w/w showed a subtle change in peak positions in the spectral region between 1450 and 1600 cm−1. This spectral region corresponds to the asymmetric vibration of NO2 of nifedipine (31) which might form hydrogen bonds with the OH groups of Soluplus®. The observation of the shift in peak position assigned to OH stretching vibration of Soluplus® was limited due to the broad IR pattern over the spectral region between 3300 and 3650 cm−1 (data not shown). α-NIF and am-NIF exhibited peak positions at 1526.4 and 1527.4 cm−1, respectively as shown in Fig. 4. Peak positions of nifedipine in FD samples with drug loadings of 10, 30 and 50% w/w occurred at 1531.2, 1530.2 and 1530.2 cm−1, respectively. This peak was slightly shifted to 1529.3 cm−1 as the drug loading increased to 70% w/w. The FD sample with a drug loading of 90% w/w showed a peak position at 1528.3 cm−1 which did not corresponded to am-NIF, β-NIF or α-NIF.

IR spectra over the spectral region of 1450 to 1600 cm−1 of (a) Soluplus®; FD samples with drug loadings of (b) 10, (c) 30, (d) 50, (e) 70, (f) 90% w/w; (g) am-NIF; (h) β–NIF and (i) α-NIF.
Similar IR results were found for ME and SE samples over the spectral region between 1450 and 1600 cm−1. Nifedipine’s characteristic peak in ME samples with 10, 30, 50 and 70% w/w appeared at 1531.2, 1530.2, 1529.3 and 1528.3 cm−1, respectively, and then shifted to the characteristic peak of am-NIF at 1527.4 cm−1 as the drug loading increased to 90% w/w. SE samples containing up to 70% w/w drug presented the same peak positions as ME samples. The peak position of the sample with 90% w/w drug was at 1526.4 cm−1, similar to the characteristic peak of α-NIF. Generally, there appeared to be certain shifts for the samples prepared by three different methods. Insignificant changes in peak position was observed in other IR spectral regions.
ss-NMR
Similar findings to IR results were evident from the ss-NMR results which showed shifts from the am-NIF peak position at 147.5 ppm depending on the drug loading as can be seen in Fig. 5. This chemical shift region is assigned to C-12 carbon at -C-NO2 of aromatic rings (33,34). FD samples with 70, 50, 30 and 10% w/w drug loadings exhibited chemical shifts at 147.6, 147.8, 147.9 and 147.9 ppm, respectively. FD sample with 90% w/w drug loading gave chemical shifts at 148.0 and 146.6 ppm corresponding to the characteristic peak of β-NIF; while this could not be identified by IR. ME samples with drug loadings of 90, 70, 50, 30 and 10% w/w exhibited peak positions at 147.6, 147.6, 147.7, 147.8 and 147.9 ppm, respectively. There was no consistent shift in SE samples. SE samples with 30 and 10% w/w drug possessed a peak position at 147.8 ppm. SE samples with drug loadings of 90, 70 and 50% w/w presented peak positions at 148.3, 148.1 and 148.1 ppm, and these spectra were similar to the spectral pattern of α-NIF. No significant change in chemical shift was observed in other spectral regions.

ss-NMR spectra over the chemical shift of 155 to 140 ppm of (a) Soluplus®; FD samples with drug loadings of (b) 10, (c) 30, (d) 50, (e) 70, (f) 90% w/w; (g) am-NIF; (h) β–NIF and (i) α-NIF.
Raman Spectroscopy
The Raman spectra of solid dispersions over the spectral region of 550 to 1800 cm−1 were investigated by considering the shift in the nifedipine peak position with respect to the drug content. Shifts in peak positions as detected by IR over the spectral region between 1450 and 1600 cm−1 was not observed. However, systematic shifts in peak positions with respect to drug/ polymer ratios were found between 790 and 820 cm−1. The characteristic peaks of α-NIF, β-NIF and am-NIF were at 810.6, 806.7 and 807.3 cm−1, respectively and agreed with the values reported by Chan et al. (31), while the characteristic peak of Soluplus® was at 795.6 cm−1 (Fig. 6). This region is assigned to the vibrational mode of deformation of di-substituted aromatic compounds as reported by Rawlinson et al. (5). Standard samples of nifedipine in the two molecular states were prepared by the FD method, which due to the fast solidification should provide a homogeneous product. The development of the distinct shoulder (due to molecular nifedipine) on increasing the drug loading was used to aid the assignment of the monomolecularly dispersed standard. A drug loading of 0.1% w/w exhibited a similar pattern to Soluplus® and no distinct shoulder that could be resolved from Soluplus® spectrum. FD samples containing 0.5 and 1% w/w drug had Soluplus® peak overlapped with the shoulder of 798.6 cm−1. As the position of nifedipine peak was not further shifted to lower wavenumber when drug loadings decreased from 1 to 0.5% w/w, this suggested that the molecular environment of nifedipine in solid dispersions composed of drug 0.5 and 1% w/w was identical, and it could be assumed that only drug-polymer interaction occurred in these solid dispersions. On the contrary, when the drug loading was increased from 1 to 3 and 5% w/w, the peak position was shifted toward that of am-NIF. They also showed that the Soluplus® peak overlapped with the shoulder of 800.1 (3% w/w) and 801.6 cm−1 (5% w/w), respectively as shown in Fig. 7a. This revealed that the molecular environment of nifedipine in the solid dispersions composed of 1 and 3% w/w drug was different. The shift in peak position of the 3% w/w sample toward the peak position of am-NIF implied the presence of drug-drug interaction. A solid dispersion with 0.5% w/w drug was, therefore, selected to be the monomolecular standard because it showed sufficient shift in the peak position to enable it to be differentiated from Soluplus®’s characteristic peak and, in addition, it contained lower drug content compared to the 1% w/w drug loading sample. Hence, there is a greater opportunity for all drug molecules to be in the monomolecular state.

Raman spectra over the spectral region of 790 to 820 cm−1 of α-NIF, β-NIF, am-NIF and Soluplus®. The characteristic peaks of α-NIF, β-NIF, am-NIF and Soluplus® were at 810.6, 806.7, 807.3 and 795.6 cm−1, respectively. Intensity was normalized and re-scaled to compare peak positions.

Raman spectra over the region of 790 to 820 cm−1of (a) FD samples with drug loadings of 0.1% (red), 0.5% (purple), 1% (green), 3% (blue) and 5% (brown), and (b) FD samples with drug loadings of 10% (red), 30% (purple), 50% (green) and 70% (blue) w/w. Intensity was normalized and re-scaled to compare peak positions.
The spectral region used to construct the Gaussian function models was extended to the region from 775 to 850 cm−1 in order to prevent confusion between am-NIF and β-NIF which have peak positions nearby, between 790 and 820 cm−1. Analysis of the extended region showed that am-NIF has a sharp peak at 831.9 cm−1 compared to a broad spectral pattern of β-NIF from 825 to 835 cm−1.
The molecularly dispersed standard was investigated using the FD samples with higher drug loadings of 10, 30 and 50% w/w. Their Raman spectra in the spectral region of 790 to 820 cm−1 showed characteristic peaks at 802.5, 804.0 and 804.9 cm−1, respectively (Fig. 7b). The molecularly dispersed state model was obtained from the remaining spectrum, resulting from subtracting the am-NIF model and model M spectra from the experimental spectra of the FD samples, as described in spectral data analysis section. The remaining spectra of the FD samples with drug loading of 10 and 30% w/w could be completely fitted with the model spectrum composed of five Gaussian peaks; whereas, that of the FD sample with drug loading of 50% w/w could be fitted with the model spectrum composed of five Gaussian peaks, combining with approximately 10% of am-NIF area. The sample with a drug loading of 30% w/w was used as the molecularly dispersed standard since it contained neither am-NIF nor model M. Furthermore, this sample should be relatively closer to the saturated concentration, compared to the sample composed of 10% w/w nifedipine. The experimental spectra of the two molecular states and amorphous nifedipine could be fitted with seven, five and three Gaussian peaks, respectively. The model spectra obtained from the summation of the Gaussian peaks was almost identical to the experimental spectrum, as shown in Fig. 8. These Gaussian function models were used to estimate the nature of nifedipine in the samples of amorphous solid dispersions prepared by FD, ME and SE.

A series of Gaussian peaks fitted to the measured Raman spectra of (a) monomolecular, (b) molecular and (c) amorphous nifedipine.
XRD results showed that the FD sample with a drug loading of 90% w/w possessed diffraction peaks at 7.8, 9.5, 11.1, 12.6, 17.2 and 24.3° which was the characteristic of β-NIF as reported by Grooff et al. (25); whereas, other samples had X-ray halo patterns (Fig. 9a). All ME samples exhibited halo patterns which revealed their amorphous character (Fig. 9b). SE samples with the 10 and 30% w/w drug loadings demonstrated halo patterns; whereas, the samples with drug loadings of 50, 70 and 90% w/w possessed diffraction peaks at 8.4, 10.7, 12.0, 16.4, 19.9 and 24.9° which corresponded to α-NIF as reported by Grooff et al. (25) (Fig. 9c). These findings were consistent with results obtained from Raman spectroscopy. The FD sample with a drug loading of 90% w/w exhibited a Raman peak at 807.6 cm−1 coupled with a broad spectral pattern over the region of approximately 825 to 835 cm−1 which was similar to the characteristic pattern of β-NIF; whereas, other FD samples demonstrated the patterns of neither α-NIF nor β-NIF. The Raman peak position of the FD sample containing 70% w/w drug was at 805.8 cm−1, and the peak was systemically shifted to 804.9, 804.0 and 802.5 as the drug content was decreased to 50, 30 and 10% w/w, respectively. For all ME samples, no peak pattern for α-NIF or β-NIF was observed. ME samples with drug loadings of 90, 70, 50, 30 and 10% w/w showed characteristic peaks at 807.0, 805.5, 804.9, 804.0 and 802.8 cm−1, respectively. For SE samples, it was found that the samples comprised of drug 90, 70, 50, 30 and 10% w/w presented characteristic peaks at 810.9, 810.9, 810.6, 804.0 and 802.5 cm−1, respectively. The peak positions of samples composed of 50, 70 and 90% w/w drug which dramatically shifted to approximately 810 cm−1 suggested the presence of α-NIF in these samples. Overall Raman peak positions of samples prepared by FD, ME and SE methods at drug loadings of 10, 30, 50, 70 and 90% w/w over the spectral region of 795 to 815 cm−1 are illustrated in Fig. 10, suggesting uniform peak shifting for the samples with drug loadings of 10 and 30% w/w for three preparation methods. The deviation in peak positions in FD sample with 90% w/w drug loading was caused by β-NIF and that in SE samples with 50, 70 and 90% w/w/ drug loadings was due to α-NIF.

XRD diffractograms of (a) Soluplus®; solid dispersion samples with drug loadings of (b) 10, (c) 30, (d) 50, (e) 70, (f) 90% w/w; (g) am-NIF; (h) β–NIF and (i) α-NIF. The samples prepared by (a) FD, (b) ME and (c) SE methods.

The shifts in Raman peak positions over the spectral region of 795 to 815 cm−1 of solid dispersion samples with drug loadings of 10, 30, 50, 70 and 90% w/w prepared by FD, ME and SE methods.
Accordingly X-ray amorphous samples (FD samples composed of 10, 30, 50 and 70% w/w drug, ME samples composed of 10, 30, 50, 70 and 90% w/w drug, and SE samples composed of 10 and 30% w/w drug) were further investigated to determine the relative proportions of monomolecular, molecular and amorphous nifedipine. The results are depicted in Fig. 11. In general, the amounts of each state varied depending on the drug concentration and preparation method. No amorphous drug was observed in any sample with a drug loading of 10% w/w. The majority of nifedipine was present in its monomolecularly dispersed state, and any remaining was present in its molecularly dispersed state. Almost all nifedipine in the FD sample containing 30% w/w drug was in the molecularly dispersed state. At the same drug loading, in the ME sample, approximately 5% w/w based on nifedipine content was amorphous, while a trace of the amorphous state of approximately 1% w/w could be detected in the SE sample. An increase in the drug loadings from 50% w/w to 90% w/w showed an increase in the amorphous drug content over molecular and monomolecular states, indicating a reduction in drug-polymer interaction in the samples. A similar trend has been reported by Huang et al. (8). At drug loadings of 70 and 90% w/w nifedipine was present mainly in its amorphous state. This implied that drug-drug interactions or the cohesive force between drug molecules was predominant relative to drug-polymer interaction. These findings suggested that the maximum concentration of nifedipine in Soluplus® which provided a miscible mixture was 30% w/w for samples prepared by FD and SE. However, the ME sample with 30% w/w drug loading contained trace amount of the amorphous state as described earlier, so cannot be unambiguously described as miscible.

The content of miscible nifedipine presenting as monomolecularly and molecularly dispersed states and immiscible nifedipine presenting as amorphous clusters in the studied X-ray amorphous samples prepared by FD, ME and SE methods. The content of each state in different drug loading samples was normalized to 100% w/w (n = 6).
Solid State Stability
The average time to detectable crystallization from three measurements of an FD sample containing 50% w/w drug was approximately 10 h (Fig. 12a); and 6 min for an FD sample with drug loading of 70% w/w (Fig. 12b). Interestingly, although the samples with drug loading of 50% w/w prepared by FD and ME showed similar drug-polymer interaction, recrystallization of nifedipine in the ME sample took approximately 11 min, which was dramatically more rapid than that in FD sample (Fig. 12c). This phenomenon was attributed to inhomogeneity in the ME sample as there may be large amorphous clusters inducing recrystallization within a shorter detectable time at the elevated temperature. Similar to that of the FD sample, the ME sample containing 70% w/w drug showed an observable crystallization time of approximately 7 min (Fig. 12d). Despite monitoring for 28 days, no sign of recrystallization could be detected in any sample with a drug loading of 30% w/w. These observations suggest the influence of drug-polymer interaction in lowering molecular mobility and thereby retarding recrystallization. It should be noted that, after 28-days, the remaining drug content in the stressed samples with drug loading of 30% w/w prepared by FD, ME and SE had decreased to 86.7, 78.4 and 76.2% of the drug content, respectively due to chemical degradation. It was possible that crystallinity in these samples was not observed owing to the detection limits of XRD, particularly in the presence of extensive amorphous background. It was also possible that with 15–25% degradation over 28 days, failure to detect crystallinity might be due to the fact that the degradation product did not crystallize under the conditions of the experiment.

XRD diffractograms of (a) FD sample with 50% w/w drug, (b) FD sample with 70% w/w drug, (c) ME sample with 50% w/w drug and (d) ME sample with 70% w/w drug stored at 98οC showing onset of crystallization.
Discussion
Miscibility of the studied samples was classified by DSC and XRD together with Raman spectroscopy. XRD and DSC have been useful in identifying the amorphous solid dispersion. An X-ray halo indicates the absence of crystallinity. As such it is unable to distinguish between a miscible mixture and a physical mixture of two amorphous solids. DSC is more useful in this case because when there is a single Tg detected between the Tg of the drug and the polymer, the amorphous drug is considered to be miscible with the polymer (10). In contrast, when there are two separated Tgs, this suggests that the drug stays as a separated phase of amorphous clusters. The DSC results may help in estimating drug-polymer miscibility. However, this technique is unable to reveal whether the drug is dispersed in its monomolecularly or molecularly dispersed states.
The XRD data indicated that the drug was present in its amorphous form for maximum drug loadings of 70%, 90% and 30% w/w in the samples prepared by FD, ME and SE, respectively. At a drug loading of 90% w/w in an FD sample, metastable β-NIF was present. While, at 50% w/w drug loading and above, α-NIF was found in SE samples. The drug in all ME samples remained amorphous.
The DSC results further elucidate the miscibility of the amorphous drug in the samples. A single Tg was found between that of the drug and the polymer in FD and SE samples with up to 70% w/w drug loading and in the ME sample with 90% w/w drug loading. This primarily indicated that the binary mixture was miscible. However, melting events (suggesting crystallization) of ME and SE samples with drug loadings above 30% w/w and of an FD sample with 70% w/w were observed during the second heating cycle. This phenomenon suggested that there might be traces of amorphous clusters within the samples leading to crystallization after heating. A melting event without the observation of a crystallization event has been reported by Yuan et al. (16). This was probably due to the limitation of this instrument and/or the inappropriate conditions for DSC to detect crystallization event. Each preparation method used different rates of cooling and it is known that slower cooling rates can give rise to lower Tgs (35). However, the Tgs of all samples at the same drug loading were found to be very similar, irrespective of the preparation method. The plot of observed Tg of FD, ME and SE samples against the polymer content also demonstrated negative deviations from ideality. This indicates that nifedipine and Soluplus® interacted more weakly than nifedipine with itself (11,36).
The IR results reflected the change in molecular surrounding of nifedipine at the nitro group (31) as small shifts in nifedipine peak positions were seen from am-NIF at 1527.4 cm−1 to 1530.2 cm−1 for drug loadings up to 30% w/w (seen in the samples prepared by all three methods). The systematic shifts in peak positions over this region could not be observed by Raman spectroscopy. This suggested that the nitro group of nifedipine might form a hydrogen bond with the OH of Soluplus®. The upward shifts of peak positions on decreasing the drug loading resulted from a change in the micro-environment of nifedipine and suggested weaker drug-polymer interactions compared to drug-drug interactions (12,23,37). There was no significant change in nifedipine peak position over the spectral region of aromatic hydrocarbon between 775 and 850 cm−1 (data not shown). This result revealed that the hydrophobic interaction between nifedipine and Soluplus® might not exhibit a change in dipole moment. Thus, IR spectroscopy was not able to detect this interaction.
A similar finding was observed by ss-NMR; FD samples showed a change in chemical shift of C-12 carbon at -C-NO2 from the am-NIF peak position at 147.6 ppm towards 147.9 ppm as the weight fraction of the drug decreased from 70 to 10%. A consistent trend in drug-polymer interaction was also provided by ME samples which showed shifts from 147.6 ppm toward 147.9 ppm as the weight fraction of the drug decreased from 90 to 10%. SE samples with 10 and 30% w/w drug possessed peak positions at 147.8 ppm. It has been reported that strong molecular interactions i.e., hydrogen bonding between two components in solid dispersions demonstrated a chemical shift alteration of approximately 1–2 ppm (33,38). In this study, the downfield shifts of the characteristic peak of am-NIF from 147.5 ppm to 147.9 ppm as drug loadings decreased to 10% w/w was approximately 0.4 ppm which indicated weak hydrogen bonding. These results suggested the possibility of other molecular interactions (the hydrophobic interaction for example) as being important for determining the miscibility of nifedipine and Soluplus® at a molecular level. However, it was not possible to use ss-NMR to study this as there was no significant change in nifedipine peak position over other chemical shift regions.
Raman spectroscopy was used to determine the hydrophobic interaction in the binary mixture of nifedipine and Soluplus® as it could detect the shifts in peak positions of nifedipine arising in the spectral region of 790 to 820 cm−1, a characteristic region for the vibration of aromatic hydrocarbon (5). There was a possibility that nifedipine, containing aromatic hydrocarbon, could form weak van der Waals interactions with the cyclic amide presenting in polyvinyl caprolactam segment of Soluplus®. The shifts in the extended spectral region covering 775 to 850 cm−1 confirmed the presence of β-NIF and α-NIF in FD with a drug loading of 90% w/w and in SE samples with a drug loading above 30%, which corresponded to conclusions from a study of their X-ray patterns.
Raman spectroscopy is also a useful tool to provide insight into the content of drug-polymer interaction and amorphous cluster in the X-ray amorphous samples. Peak positions and shapes were unique for each sample and influenced by the strength of the drug-polymer interaction (22). This implies that the difference in the sample spectral patterns resulted from the particular combination of monomolecularly dispersed state, molecularly dispersed state and amorphous state coexisting within samples. Theoretically, the degree of shift in peak position indicates the strength of the drug-polymer interaction; a larger shift reveals a stronger drug-polymer interaction (15).
Raman peak positions of nifedipine generally shifted to lower wavenumber upon lowering the drug to polymer ratios from 70 to 10% w/w in FD samples, 90 to 10% w/w in ME samples and 30 to 10% w/w in SE samples. This suggested that the drug-polymer interaction was stronger than the drug-drug interaction. In the amorphous samples, the strength of drug-polymer interaction (indicated by the shift), was inversely correlated with nifedipine weight ratio as can be seen in Fig. 10. It has been reported that more molecular dispersions were obtained from the solid dispersion of nifedipine in ethylcellulose and/or Eudragit RL, prepared by the method with faster solidification rate (8). However, in the present study, the samples prepared by FD, ME and SE methods at equivalent drug loadings of 10 and 30% w/w exhibited shifts of peaks to similar frequencies (around 802–804 cm−1). This revealed that the strength of the drug-polymer interaction at equivalent drug loading was similar regardless of the preparation method. The Gaussian function models constructed from FD samples were applied to ME and SE samples; the model spectra of ME and SE samples were well fitted in the FD models. The average proportion of nifedipine in each state was consistent for all three preparation methods at equivalent drug loading as shown in Fig. 11. This result agreed with the similar degree of shifting in nifedipine peak positions for the three preparation methods. At each level of 10 and 30% w/w drug loading, the strength of established drug-polymer interaction in FD, ME and SE samples were similar due to the comparative amounts of monomolecularly and molecularly dispersed states. The shift to lower wavenumber found for the samples with 10% w/w drug loading corresponded to relatively higher amounts of monomolecularly dispersed state, suggesting the occurrence of stronger adhesive force, comparing with the samples with 30% w/w drug loading. However, the amount of each state present in all ME samples showed a relatively high variation, having greater standard deviation in Fig. 11. It may be due to poor mixing; either between drug and polymer powders or in the molten state during the preparation of dispersion. All samples composed of 10% w/w drug showed a large proportion of the monomolecularly dispersed state with less of the molecularly dispersed state. Trace amounts of the amorphous state (approximately 5% w/w based on nifedipine content) could be found in an ME sample with drug 30% w/w. At 50% w/w drug loading, the monomolecularly dispersed state could not be detected in all samples and the amorphous state became more significant. The proportion of the amorphous state became greater than molecularly dispersed state in FD and ME sample with 70% w/w drug loading and almost all drug molecules exhibited as amorphous state in ME sample with 90% w/w drug loading.
Based on the Raman results, the samples with 10% drug loading, prepared by all three methods, were miscible because no amorphous drug was found. The miscibility limits of binary mixtures of nifedipine and Soluplus® prepared by FD and SE were approximately 30% w/w drug loading and these samples were also classified as miscible mixtures. This agreed with the miscibility results classified by the presence of a single Tg, with no melting event, in these samples. Although the ME sample with 30% w/w drug loading was also classified as a miscible mixture by DSC, there appeared to be 5% w/w of amorphous drug. The samples prepared by all three methods with drug loadings of 50, 70 and 90% w/w were clearly classified as immiscible mixtures as amorphous and/or crystalline drugs were determined. DSC demonstrated the limitation of multiple Tgs detection, commonly used to indicate phase separation in immiscible samples such as the FD sample with drug loading of 50% w/w and the ME sample with drug loading of 30% w/w. This was probably due to the presence of amorphous clusters in the bulk samples which were smaller than the detection limit of DSC. It has been reported that amorphous clusters smaller than 30 nm, even tens of microns (39), generally could not be distinguished by DSC and resulted in misleading conclusions as to the nature of a molecularly dispersed drug (16). These conclusions highlight the significance of Raman spectroscopy as a technique to examine the hydrophobic interactions between the drug and polymeric matrix. A further possible reason for the disagreement between DSC and Raman spectroscopy in determining the miscibility limit could be poor uniformity of mixing as ME samples show a relatively higher variation in amorphous content as shown in Fig. 11.
Despite this, among preparation methods, ME was demonstrated to be the most effective technique at providing drug-polymer interaction in amorphous samples over the entire range of drug loadings including 10, 30, 50, 70 and 90% w/w. Recrystallization was apparent in the FD sample consisting of 90% w/w drug and SE samples consisting of 50, 70 and 90% w/w drug. The recrystallization of the FD and SE samples with relatively high drug loadings could be due to the higher molecular mobility of the drug before the removal of solvent. This would allow the drug molecules to rearrange into a more thermodynamically stable form. For SE samples, this recrystallization phenomenon to the more stable α-NIF form occurred at lower drug loadings because of slower entrapment rate of the drug molecules into the polymeric matrix. ME samples showed the highest variation throughout all drug loadings; whereas, distribution of drug-polymer interaction was completely homogeneous in FD and SE samples. However, when comparing among X-ray amorphous samples, the average degree of drug-polymer interaction was independent of the preparation method.
The solid state stability of amorphous pharmaceuticals is affected by both drug content (the thermodynamic factor) and preparation method (the kinetic factor) (4,6). In this study, the samples were stored at much higher temperature than their Tgs to accelerate crystallization. At this temperature, the samples were in their rubbery state and molecular mobility was high. Consequently, the effect of the preparation method, i.e., cooling rates, on Tg and molecular mobility in the glassy samples might be ignored. Crystallization only results from nucleation and crystal growth during aging. If there were substantial drug-polymer interactions in the samples, the polymer would help delay crystallization of the drug in the samples stored even at the elevated temperature. The correlation between the degree of drug-polymer interaction and the onset for observable crystallization was elucidated. Onset times for observable crystallization of FD and ME samples consisting of 70% w/w drug, which possessed separate amorphous phases, were approximately 6–7 min. The existence of numerous amorphous clusters in the samples substantially induced nucleation. However, an FD sample with 50% w/w drug loading had a detectable crystallization time of 10 h. Even though this sample contained a significantly higher proportion of monomolecular and molecular dispersed drug than amorphous state, the greater thermodynamic driving force for the amorphous cluster to crystallize was able to overcome the kinetic barrier provided by the drug-polymer interaction, leading to crystallization within a relatively short onset time. Interestingly, although solid dispersions with drug loading of 50% w/w prepared by FD and ME methods exhibited similar distributions of dispersed and amorphous drug, the onset time for observable crystallization of the ME sample (approximately 11 min) was dramatically more rapid than the FD sample at an equivalent drug loading. This phenomenon was attributed to the inhomogeneous nature of ME sample. Despite measuring for 28 days, no sign of crystallization could be detected in FD, ME and SE samples with drug loading of 30% w/w. Although ME sample containing 30% w/w drug demonstrated amorphous fraction of about 5% w/w based on nifedipine content, this trace amorphous state might insufficient to induce nucleation. Therefore, the ME sample with drug loading of 30% w/w could be categorized as a miscible mixture. The solid state stability of the miscible mixture at the elevated temperature was attributed to the lower molecular mobility due to the drug-polymer interaction detected at ambient temperature, and the reduced thermodynamic driving force as drug content was within miscibility limit. The drug-polymer interaction occurred through hydrophobic interaction as suggested by Raman shifts and hydrogen bonding as suggested by typically shifting in IR peak position for these samples. However, the study assumed that higher temperatures did not increase the saturated concentration of the drug. Also, it must be addressed here that the studied temperature was much higher than the ambient and accelerated temperatures used in a conventional stability study. These results did not suggest how distinguishable the physical stabilities would be at ambient conditions but revealed that the crystallization tendency of homogenous samples was inversely proportional to the extent of drug-polymer interaction.
Conclusions
The study demonstrated the use of Raman spectroscopy to qualitatively examine the miscibility level in binary mixture, and to quantitatively determine the degree of hydrophobic interaction between drug and polymer. The miscibility limit was estimated to be 30% w/w in which all drug molecules were interacted with polymer regardless of preparation method. The drug loading had an impact on miscibility level, while the preparation methods rather influenced the homogeneity of the drug-polymer interaction, which eventually led to differences in solid state stability. This high selectivity technique might be helpful providing the supportive data to justify an ambiguous result obtained from other methods e.g., DSC.
Abbreviations
- am-NIF:
-
Amorphous nifedipine
- DSC:
-
Differential scanning calorimetry
- FD:
-
Freeze drying
- HPLC:
-
High performance liquid chromatography
- IR:
-
Infrared spectroscopy
- MAS:
-
Magic angle spinning
- ME:
-
Melting
- Min:
-
Minute
- SE:
-
Solvent evaporation
- ss-NMR:
-
Solid state-nuclear magnetic resonance
- Tg :
-
Glass transition temperature
- XPS:
-
X-ray photoelectron spectroscopy
- XRD:
-
X-ray powder diffraction; X-ray powder diffractometry
- α-NIF:
-
Nifedipine crystalline α-form
- β-NIF:
-
Nifedipine crystalline β-form
References
Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50(1):47–60.
Ivanisevic I. Physical stability studies of miscible amorphous solid dispersions. J Pharm Sci. 2010;99(9):4005–12.
Marsac PJ, Shamblin SL, Taylor LS. Theoretical and practical approaches for prediction of drug-polymer miscibility and solubility. Pharm Res. 2006;23(10):2417–26.
Qian F, Huang J, Hussain MA. Drug-polymer solubility and miscibility: stability consideration and practical challenges in amorphous solid dispersion development. J Pharm Sci. 2010;99(7):2941–7.
Rawlinson CF, Williams AC, Timmins P, Grimsey I. Polymer-mediated disruption of drug crystallinity. Int J Pharm. 2007;336(1):42–8.
Bhugra C, Pikal MJ. Role of thermodynamic, molecular, and kinetic factors in crystallization from the amorphous state. J Pharm Sci. 2008;97(4):1329–49.
Kestur US, Van Eerdenbrugh B, Taylor LS. Influence of polymer chemistry on crystal growth inhibition of two chemically diverse organic molecules. CrystEngComm. 2011;13(22):6712–8.
Huang J, Li Y, Wigent RJ, Malick WA, Sandhu HK, Singhal D, et al. Interplay of formulation and process methodology on the extent of nifedipine molecular dispersion in polymers. Int J Pharm. 2011;420(1):59–67.
Djuris J, Nikolakakis I, Ibric S, Djuric Z, Kachrimanis K. Preparation of carbamazepine-Soluplus® solid dispersions by hot-melt extrusion, and prediction of drug-polymer miscibility by thermodynamic model fitting. Eur J Pharm Biopharm. 2013;84(1):228–37.
Rumondor ACF, Ivanisevic I, Bates S, Alonzo DE, Taylor LS. Evaluation of drug-polymer miscibility in amorphous solid dispersion systems. Pharm Res. 2009;26(11):2523–34.
Janssens S, De Zeure A, Paudel A, Van Humbeeck J, Rombaut P, Van den Mooter G. Influence of preparation methods on solid state supersaturation of amorphous solid dispersions: a case study with itraconazole and Eudragit E100. Pharm Res. 2010;27(5):775–85.
Konno H, Taylor LS. Influence of different polymers on the crystallization tendency of molecularly dispersed amorphous felodipine. J Pharm Sci. 2006;95(12):2692–705.
Marsac PJ, Konno H, Taylor LS. A comparison of the physical stability of amorphous felodipine and nifedipine systems. Pharm Res. 2006;23(10):2306–16.
Maniruzzaman M, Morgan DJ, Mendham AP, Pang J, Snowden MJ, Douroumis D. Drug-polymer intermolecular interactions in hot-melt extruded solid dispersions. Int J Pharm. 2013;443(1–2):199–208.
Taylor LS, Zografi G. Sugar–polymer hydrogen bond interactions in lyophilized amorphous mixtures. J Pharm Sci. 1998;87(12):1615–21.
Yuan X, Sperger D, Munson EJ. Investigating miscibility and molecular mobility of nifedipine-PVP amorphous solid dispersions using solid-state NMR spectroscopy. Mol Pharmaceutics. 2014;11(1):329–37.
Yuasa H, Ozeki T, Takahashi H, Kanaya Y, Ueno M. Application of the solid dispersion method to the controlled release of medicine. VI. Release mechanism of a slightly water-soluble medicine and interaction between flurbiprofen and hydroxypropyl cellulose in solid dispersion. Chem Pharm Bull. 1994;42(2):354–8.
Li X, Jiang C, Pan L, Zhang H, Hu L, Li T, et al. Effects of preparing techniques and aging on dissolution behavior of the solid dispersions of NF/Soluplus/Kollidon SR: identification and classification by a combined analysis by FT-IR spectroscopy and computational approaches. Drug Dev Ind Pharm. 2015;41(1):2–14.
Taylor LS, Zografi G. Spectroscopic characterization of interactions between PVP and indomethacin in amorphous molecular dispersions. Pharm Res. 1997;14(12):1691–8.
Breitenbach J, Schrof W, Neumann J. Confocal Raman-spectroscopy: analytical approach to solid dispersions and mapping of drugs. Pharm Res. 1999;16(7):1109–13.
Schmidt AG, Wartewig S, Picker KM. Polyethylene oxides: protection potential against polymorphic transitions of drugs? J Raman Spectrosc. 2004;35(5):360–7.
Vajna B, Farkas I, Farkas A, Pataki H, Nagy Z, Madarász J, et al. Characterization of drug–cyclodextrin formulations using Raman mapping and multivariate curve resolution. J Pharm Biomed Anal. 2011;56(1):38–44.
Dürrigl M, Kwokal A, Hafner A, Klarić MŠ, Dumičić A, Cetina-Čižmek B, et al. Spray dried microparticles for controlled delivery of mupirocin calcium: process-tailored modulation of drug release. J Microencapsul. 2011;28(2):108–21.
Palermo RN, Anderson CA, Drennen Iii JK. Review: use of thermal, diffraction, and vibrational analytical methods to determine mechanisms of solid dispersion stability. J Pharm Innov. 2012;7(1):2–12.
Grooff D, Liebenberg W, De Villiers MM. Preparation and transformation of true nifedipine polymorphs: investigated with differential scanning calorimetry and X-ray diffraction pattern fitting methods. J Pharm Sci. 2011;100(5):1944–57.
Küpper H, Spiller M, Küpper FC. Photometric method for the quantification of chlorophylls and their derivatives in complex mixtures: fitting with Gauss-peak spectra. Anal Biochem. 2000;286:247–56.
Dong J, Ozaki Y, Nakashima K. Infrared, Raman, and near-infrared spectroscopic evidence for the coexistence of various hydrogen-bond forms in poly(acrylic acid). Macromolecules. 1997;30(4):1111–7.
Hu Y, Liu J, Li W. Resolution of overlapping spectra by curve-fitting. Anal Chim Acta. 2005;538:383–9.
Forster A, Hempenstall J, Tucker I, Rades T. The potential of small-scale fusion experiments and the Gordon-Taylor equation to predict the suitability of drug/polymer blends for melt extrusion. Drug Dev Ind Pharm. 2001;27(6):549–60.
United States Pharmacopeial Convention. United States Pharmacopeia and National Formulary (USP 36-NF31). Rockville: United States Pharmacopeial Convention; 2012.
Chan KLA, Fleming OS, Kazarian SG, Vassou D, Chryssikos GD, Gionis V. Polymorphism and devitrification of nifedipine under controlled humidity: a combined FT-Raman, IR and Raman microscopic investigation. J Raman Spectrosc. 2004;35(5):353–9.
Grooff D, De Villiers MM, Liebenberg W. Thermal methods for evaluating polymorphic transitions in nifedipine. Thermochim Acta. 2007;454(1):33–42.
Aso Y, Yoshioka S. Molecular mobility of nifedipine–PVP and phenobarbital–PVP solid dispersions as measured by 13C-NMR spin–lattice relaxation time. J Pharm Sci. 2006;95(2):318–25.
Apperley DC, Forster AH, Fournier R, Harris RK, Hodgkinson P, Lancaster RW, et al. Characterization of indomethacin and nifedipine using variable-temperature solid-state NMR. Magn Reson Chem. 2005;43:881–92.
Richardson MJ, Savill NG. Derivation of accurate glass transition temperatures by differential scanning calorimetry. Polymer. 1975;16(10):753–7.
Hancock BC, Zografi G. The relationship between the glass transition temperature and the water content of amorphous pharmaceutical solids. Pharm Res. 1994;11(4):471–7.
Tang XLC, Pikal MJ, Taylor LS. A spectroscopic investigation of hydrogen bond patterns in crystalline and amorphous phases in dihydropyridine calcium channel blockers. Pharm Res. 2002;19(4):477–83.
Yoshie N, Azuma Y, Sakurai M, Inoue Y. Crystallization and compatibility of poly(vinyl alcohol)/poly(3-hydroxybutyrate) blends: Influence of blend composition and tacticity of poly(vinyl alcohol). J Appl Polym Sci. 1995;56(1):17–24.
Qian F, Huang J, Zhu Q, Haddadin R, Gawel J, Garmise R, et al. Is a distinctive single T g a reliable indicator for the homogeneity of amorphous solid dispersion? Int J Pharm. 2010;395(1–2):232–5.
ACKNOWLEDGEMENTS AND DISCLOSURES
This research has been supported by the Ratchadaphiseksomphot Endowment Fund 2013 of Chulalongkorn University (CU-56-649-HR). The authors also wish to thank Department of Pharmaceutics, Faculty of Pharmaceutical Sciences, Toho University and Chulalongkorn University Centenary Academic Development Project for providing research facilities. Thanks also to Dr John Kendrick for editing and comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Keratichewanun, S., Yoshihashi, Y., Sutanthavibul, N. et al. An Investigation of Nifedipine Miscibility in Solid Dispersions Using Raman Spectroscopy. Pharm Res 32, 2458–2473 (2015). https://doi.org/10.1007/s11095-015-1638-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-015-1638-x