
ABSTRACT
In this review, we first summarize the structure and properties of biological membranes and the routes of passive drug transfer through physiological barriers. Lipophilicity is then introduced in terms of the intermolecular interactions it encodes. Finally, lipophilicity indices from isotropic solvent systems and from anisotropic membrane-like systems are discussed for their capacity to predict passive drug permeation across biological membranes such as the intestinal epithelium, the blood-brain barrier (BBB) or the skin. The broad evidence presented here shows that beyond the predictive power of lipophilicity parameters, the various intermolecular forces they encode allow a mechanistic interpretation of passive drug permeation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
DRUG DIFFUSION AND TRANSPORT ACROSS BIOLOGICAL BARRIERS
Successful drug development requires not only optimization of specific and potent recognition by its pharmacodynamic targets, but also efficient delivery to these target sites. To elicit its pharmacological and therapeutic effects, a drug has to cross various cellular barriers by passive and/or transporter-mediated uptake. Membrane permeability is a key determinant in pharmacokinetic behavior (Absorption, Distribution, Metabolism and Excretion (ADME)) of drugs and especially of absorption, distribution and excretion. In recent years, advances in automated synthesis, combinatorial chemistry and innovative high-throughput screening have led to the production of a vast number of potential drug candidates, often making delivery problems the rate-limiting step in drug research (1). In order to overcome this problem, it is necessary to have a detailed picture of the structures of pharmacokinetic barriers.
Structure of Cell Membranes
The currently accepted structure of typical membranes is a fluid-like bilayer arrangement of phospholipids (2) (Fig. 1). Proteins and other substances such as steroids and glycolipids are either associated with its surface or embedded in it to different degrees. This structure is an intermediate state between the true liquid and solid states, with the lipid and protein molecules having a limited degree of rotational and lateral movement (3). The polar heads of phospholipid molecules are orientated to form an almost continuous polar layer on both the inner and outer side of membranes. In contrast, the long hydrophobic chains of phospholipids molecules extend into the central core of the membrane.

The fluid mosaic model of membranes.
The lipid component of mammalian cell membranes is mainly composed of glycerophospholipids, sphingolipids and cholesterol, whose structure is shown in Fig. 2 (4,5). It can be seen from the structures that the lipid molecules are either negatively charged or zwitterionic (5,6) (i.e., electrically neutral due to an equal number of positive and negative charges). These lipids are distributed asymmetrically in the inner and outer leaflets in most biological membranes. The outer leaflet of the bilayer consists mainly of electrically neutral lipids, such as phosphatidylcholine (PC) and phosphatidylethanolamine (PE), while the negatively charged lipids, such as phosphatidylserine, are located in the inner layer (7). These lipid molecules are held together by weak hydrophobic bonding and van der Waals’s forces.

The chemical structures of main lipids found in membranes.
The peripheral and integral proteins located in the membranes are responsible for carrying out many of the active functions of membranes, such as acting as receptors and transportation routes for various substances in and out of cells. The formation of pores, including ion channels, is also associated with integral proteins.
Neighboring cells are linked to each other by a continuous junctional complex referred to as tight junction. It is a region where the outer leaflets of the lipid bilayer comprising the membrane of neighboring cells are fused. The interconnected monolayer of cells in the intestinal epithelium is the principle permeation barrier for oral absorption of drugs. Similarly, a special class of capillary endothelial cells interlinked by exceptionally tight junctions constitutes the main barrier for drug transport from blood to brain (8).
The Transfer of Drugs Through Cell Membranes
Passive Diffusion
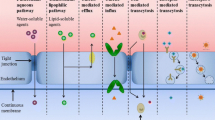
The process by which a compound moves down its concentration gradient without a membrane actively participating is termed passive diffusion. Passive diffusion across a biomembrane may occur through its lipid structures (i.e., transcellular pathway, B in Fig. 3) or through its water-filled pores or tight junctions (i.e., paracellular pathway, A in Fig. 3) (9,10). For many drugs, transport is mediated by passive transcellular diffusion through the apical cell membranes, across the cell proper and across the basolateral membrane (8). The ease of passive transcellular diffusion depends on the ability of the molecule to partition into cell membranes. In order to permeate by this route, a compound must have an optimal lipophilicity, because if the solute is too lipophilic it will remain trapped in the membrane. A measure of lipophilicity can be obtained by the partition coefficients in different systems such as n-octanol/water and liposome/water partitioning systems. The predictive value of the relations between lipophilicity and membrane permeation depends on the relevance of the partitioning systems as models of biomembranes (11).

Pathways of transport across the intestinal mucosa. A: Passive diffusion via paracellular route. B: Passive diffusion via transcellular route. C: Transporter-facilitated pathway. D: Transport-restricted pathway (e.g., by efflux transporters). Modified from Ref. (9).
Intestinal absorption via the paracellular route is relevant for hydrophilic compounds with molecular weights lower than about 200 Da (12). Since the paracellular pathway is an aqueous extracellular route across the intestinal epithelium, sufficient hydrophilicity is the most important prerequisite for a drug to traverse the cell barrier via this pathway (13). In addition, the size and charge of a drug are also crucial molecular characteristics for this route. It has been reported that tight junctions are impermeable to molecules with radii larger than 10–15 Å (14) or 4 Å in more recent studies (15,16) and that an optimal net charge is very important for the efficient transport through intestinal epithelium via this route (17).
In contrast, paracellular absorption is negligible at the blood-brain barrier (BBB) due to the occlusive network of tight junctions of the brain capillary endothelial cells (18).
Active Transport
Active transport processes lie outside the scope of this review but are mentioned here for contextual reasons. Some hydrophilic drugs whose chemical structure mimics various nutrients can be transported across the membranes by carrier-facilitated transport (C in Fig. 3). Such processes usually operate against a concentration gradient and are fairly substrate-specific. Different carriers and transporters have been described in various types of cells. They have been identified mostly as integral membrane proteins (19).
Contrary to carrier-facilitated transport, efflux systems (D in Fig. 3) present in the membranes create a major barrier to the absorption of a wide variety of xenobiotics. Although these efflux systems are most commonly observed in tumor cells, they are also known to be present in normal intestinal epithelia and at the BBB (20). These efflux systems are related to P-glycoprotein, the principal component of multidrug resistance in a variety of cell types. P-glycoprotein is a 170–180 kDa membrane glycoprotein that acts as an ATP-dependent efflux pump, thereby reducing the transcellular flux of a wide variety of drugs (21).
The Main Physiological Barriers
Intestinal Epithelium
Because the majority of marketed medicines (about 90%) are administered orally, the main physiological barrier drugs have to pass to enter the body is the intestinal epithelial membrane. The human small intestine membrane has a fractal-like structure showing ridges (oriented circumferentially around the lumen), villi and microvilli (22). The membrane surface is expanded approximately up to 600-fold by the villi and microvilli. Due to this large surface area, the small intestine is the main site of drug absorption (23). The intestinal membrane has the mucus layer on the villi, which is thought to maintain the unstirred water layer (UWL). The UWL is also considered a significant barrier to the passive diffusion of lipophilic drugs (24).
The permeation of drugs across the intestinal epithelial membrane can occur via the passive transcellular pathway, the paracellular pathway, or active transport, depending on the physicochemical properties and structural characteristics of the compounds.
Blood-Brain Barrier (BBB)
The BBB, which has an estimated surface area of 12 m2 in humans (25), is formed by the tight endothelial cell layer in the brain capillaries and controls the exchange of drugs, nutrients, hormones, metabolites and immune cells between blood and brain in both directions. The BBB endothelium forms a much tighter interface than peripheral endothelia. In the periphery, most small solutes can diffuse between the intercellular clefts of 50–200 nm width (26). In contrast, the gaps between capillary endothelial cells in most parts of the brain are sealed by tight junctions and thus have severely limited permeability (27). The paracellular pathway is negligible for most compounds under physiological conditions. Passive permeation is mainly restricted to the lipophilic compounds which are able to traverse the lipid membranes of the cells (25,28).
Skin Permeation
The transdermal route has some advantages for the systemic delivery of drugs. These include the ease of use (and withdrawal in the advent of side-effects), avoidance of first-pass metabolism, and improved patient compliance. The transdermal permeation rate of most drugs is limited by the stratum corneum (SC) (29). The SC comprises 10–15 layers of flat keratin-filled cells, closely packed in a non-polar lipid matrix, mainly composed of free fatty acids (10%–15%), cholesterol (25%), sterol esters (5%), and ceramides (50%) (30). Ceramides are categorized into nine subgroups (structures shown in Fig. 4) (31), whose headgroups can form lateral intermolecular hydrogen bonds. The phase behavior of the lammellar lipid is different from that of the lipid bilayer mainly composed of phospholipids. The thickness of the SC is different in each body part, for example about 15 μm in the abdominal skin and 10 μm in the dorsal skin. The primary transport pathway for most drugs passively traversing the SC is the intercellular lipid region (32).

The chemical structure of ceramides.
THE STRUCTURAL INFORMATION CONTENT OF LIPOPHILICITY
Lipophilicity
Lipophilicity is usually measured by the partition coefficient (log P) of a compound in a single electrical form, a molecular parameter which describes the partition equilibrium of a solute between water and an immiscible organic solvent. Partition coefficients are obtained as the logarithm of the ratio of concentrations at equilibrium:
The most commonly used measure of lipophilicity is the n-octanol/water partition coefficient (log Poct), which when written in this way refers to the neutral form of the solute.
The effectiveness of log Poct in correlating biological properties has been extensively investigated. However, the log Poct scale of lipophilicity alone is not effective in modelling the crossing of any kind of cell membranes due to large differences in their biophysical properties (33,34). Thus, four types of isotropic solvent systems (amphiprotic, inert, hydrogen-bond donor and hydrogen-bond acceptor), called the ‘critical quartet’ (e.g., n-octanol/water, alkanes/water, chloroform/water, and dibutyl ether/water), are necessary in order to cover adequately the range of biophysical properties of membranes (35,36). The ‘critical quartet’ expresses in partly overlapping and partly complementary ways the recognition forces that account for membrane partitioning and biological selectivity (37,38).
For ionizable drugs, the apparent distribution coefficient (log D) at pH 7.4 is also often used. Unlike log P, which is valid only for a single electrical species, log D refers to a pH-dependent mixture of all electrical species present at that pH. For a solute with a single ionizable group, we have
where fN and fI are the molar fractions of the neutral and ionized forms, and PN and PI are their respective partition coefficients.
The experimental techniques for lipophilicity measurement in isotropic solvent/water systems are the shake-flask method (39) and potentiometric titration (40,41), which have been thoroughly described in the literature.
Intermolecular Forces Encoded in Lipophilicity
Lipophilicity is the net result of all intermolecular forces involving a solute and the two phases between which it partitions. A highly informative interpretation of lipophilicity is based on linear solvation free-energy relationships (LSERs) (42–45). This method factorizes lipophilicity into a number of parameters: molar volume V (or van der Waal volume Vw), which encodes hydrophobic and dispersion forces, and solvatochromic parameters, defined as the solute’s H-bond donor acidity (α), H-bond acceptor basicity (β), and dipolarity/polarizability (π*), which accounts for orientation and induction forces.
For example, the n-octanol/water and heptane/water partition coefficient (log Poct and log Phep) can be expressed as (46)
where N is the number of compounds, r 2 the squared correlation coefficient, s the standard deviation, and F the Fisher’s test.
As a result of such equations, lipophilicity can be factorized into two sets of terms (Eq. 5), namely hydrophobicity, which accounts for hydrophobic and dispersion forces, and polar terms, which account for hydrogen bonds, and orientation and induction forces (47):
Equation 3 shows that V and β are the most important structural descriptors contributing to log Poct, while π* is of secondary importance, and α has no statistical significance in the n-octanol/water system. A markedly different partitioning mechanism exists in heptane/water system (Eq. 4), where V, α and β are the most important structural descriptors contributing to log Phep, while π* is of lesser importance. The compared information content of Eqs. 3 and 4 is best appreciated through the Δlog P parameter, as discussed below.
Lipophilicity as a molecular parameter encodes different intermolecular forces, as schematized in Fig. 5. When expressed by logP measured in isotropic organic solvent/water systems, lipophilicity fails to encode some important recognition forces in biochemistry and molecular pharmacology, most notably ionic bonds, which are of particular importance when modeling the interaction of ionized compounds with biomembranes (46). Because the majority of the drugs are ionizable (48), any prediction of their pharmacodynamic and pharmacokinetic properties should take their ionization into account.

A comparison between the intermolecular forces that govern molecular recognition in biochemical and pharmacological processes, and those expressed in lipophilicity. Modified from Ref. (46).
To obtain lipophilicity parameters of improved pharmacokinetic and pharmacodynamic relevance, partition coefficients between artificial membranes and water (such as liposome/water) are receiving increased attention. Since artificial membranes provide the amphiphilic microenvironment of biological membranes, they should be able to take ionic bonds into account. In anisotropic membrane-like systems, the definition of “lipophilicity” must be extended to include ionic interactions (attractive and repulsive):
The Δlog P Parameters
Lipophilicity descriptors obtained in different isotropic solvent systems can be compared to derive a Δlog P parameter when the two log P values do not contain identical structural information (46). A well-known example is Δlog Poct-alk, the difference between n-octanol/water and alkane/water partition coefficients (44). This parameter is physicochemically meaningful and expresses mainly the H-bond donor acidity of solutes as shown by Eq. 7:
To overcome some experimental problems caused by the low alkane solubility of many compounds, the 1,2-dichloroethane/water (DCE/water) system was suggested to replace the alkane/water system (45). Therefore, Δlog Poct-dce is now proposed instead of Δlog Poct-alk.
The diff(log PN−I) Parameters
The difference between log P values of the neutral and ionized species of an ionizable solute, diff(log PN−I) is a parameter containing important structural information. It expresses the influence of ionization on the intermolecular forces and intramolecular interactions of a given solute (49). For a given system, diff(log PN−I) is rather constant for structurally related compounds, e.g., its value is 3–4 for monoprotic substance, depending on charge delocalization in the n-octanol/water system (50,51). Thus, an anomalous value of diff(log PN−I) of a solute suggests specific intramolecular interactions affecting either the neutral or ionized form (52).
The parameter diff(log PN−I) is also of interest in the study of zwitterions, which are receving much interest due to their peculiar partitioning and pharmacokinetic behavior (53–55). The pH-partitioning profiles of ampholites (56) can be bell-shaped or U-shaped. Zwitterions with large KZ (tautomeric equilibrium constant, defined as the ratio of concentrations of the zwitterionic and neutral tautomers) can display either behavior. Intramolecular effects play a marked role in enhancing the lipophilicity of the zwitterionic species and thus in determining the shape of the lipophilicity profile. The difference between the log P of the neutral and zwitterionic forms (diff(log PN−Z)) has been found to be useful in pointing to intramolecular effects and thus deciding the lipophilicity profiles (56). As mentioned, the diff(log PN−I) of a given solute in octanol/water ranges from 3 to 4 (50), and thus a diff(log PN−Z) of about 6 is expected for zwitterions for which internal neutralization is totally absent.
THE INFLUENCE OF LIPOPHILICITY ON DRUG PASSIVE TRANSCELLULAR PERMEATION
The relationships between the physicochemical properties of drugs and pharmacokinetic processes have been extensively studied. Some physicochemical parameters of drugs, such as lipophilicity, hydrogen-bonding capacity, molecular size and polar surface area, have proved useful for predicting passive transfer and permeation across biomembranes in ADME, but none has attracted as much interest in quantitative structure-permeation relationship (QSPeR) studies as lipophilicity.
Lipophilicity from Isotropic Solvent Systems and Its Relationship with Drug Transcellular Permeation
In numerous studies on drug permeation through biological membranes (e.g., gut wall, skin, blood-brain barrier, and Caco-2 cell monolayers), relationships between permeation and lipophilicity have been developed with homologous series of compounds of a diverse nature (acidic, alkaline and neutral) to explore the influence of lipophilicity on passive diffusion.
Thus, linear relationships were found between absorption rate constants from rat stomach and a) partition coefficient in CCl4/water for 16 barbiturates (57), b) partition coefficient in n-octanol/water for 11 carbamates (58). A bilinear correlation was found between in situ gastric absorption rate constants determined in rats and lipophilicity indices (measured in RP-HPLC) for a series of phenylalkylcarboxylic acids (59). Sigmoidal relationships were established between permeability coefficients in Caco-2 cell monolayers and log Doct (pH 7.4) for a set of β-blockers (60) and a series of N-acylated derivatives of 5-fluorouracil (61), respectively. The same type of relationship between permeability in the rat jejunum and log Poct was also found for seven steroids (62) and 11 β-blockers (63). Parabolic correlations were found between human skin permeability coefficients and log Poct for a homogeneous set of phenols (64), between human epidermis permeability coefficients and log Poct for 6 non-steroidal anti-inflammatory agents (65), and between corneal permeability coefficients and log Doct (pH 7.65) for a series of 12 β-blockers (66). To rationalize the observed relationships, different theoretical models for passive membrane diffusion have been derived and discussed (10,67,68).
The Δlog Poct-alk parameter, which mainly expresses H-bond donor acidity, also showed its value in predicting drug permeation. For a series of H2-receptor antagonists, brain penetration (logarithm of brain/blood ratios) in the rat was inversely correlated with the Δlog Poct-cyc parameter (cyc = cyclohexane) (69). Similar correlations were found between permeability coefficients across excised rabbit cornea and Δlog Poct-cyc for a set of drugs including β-blockers and steroids (70), between oral absorption and Δlog Poct-cyc for a family of azole endothelin antagonists (71), and between human skin permeability coefficients and Δlog Poct-hep for a set of compounds including alcohols and steroids (64). These examples imply that the H-bond donor acidity is very important in drug design to improve the pharmacokinetic profile of drugs.
Lipophilicity from Anisotropic Biomembrane-Like Systems and Its Relationship with Drug Transcellular Permeation
The mechanisms of drug-biomembrane interactions involve partitioning (into the hydrophobic core of the membrane) and/or binding (to the headgroup region on the surface of the membrane) (72,73). Therefore, lipophilicity from anisotropic biomembrane-like systems represents a border case between partitioning and specific binding. Because of the poor degree of specificity involved, it is suggested that anisotropic lipophilicity is better described as a partitioning than a binding process and can be modeled by molecular descriptors such as log D and log P (72). It should be pointed out that partition measured in biomembrane-like systems may not always reflect transmembrane permeation, as solutes may associate with the membrane interface without entering the bilayer interior (74).
Liposome/Water Partitioning System
Liposomes have been used for several decades as model membranes to study solute/biological membranes interactions. The partitioning behavior of solutes between liposomal membranes and aqueous phases provides information on their affinity to biological membranes and on their in vivo pharmacokinetic behavior in general. The liposomes/water partitioning system is increasingly employed as an alternative to n-octanol/water for the estimation of pharmacokinetic behavior of drugs (72).
Liposomes can be prepared from a variety of lipids and mixtures of them. Phospholipids are frequently used to obtain standardized and easily reproducible systems (75). Liposomes have a spherical shape and are composed of one to several hundred concentric bilayers. Their size ranges from 20 nm to several dozens micrometers, whereas the thickness of the bilayer membrane is approximately 4–7 nm (5). Depending on the number of bilayer sheets, liposomes can be classified into large multilamellar vesicles (LMVs), large unilamellar vesicles (LUVs) and small unilamellar vesicles (SUVs, diameter smaller than 100 nm). Liposomes for partition studies are ideally unilamellar, since these have a reproducible size distribution.
Liposomes of various compositions have been shown to yield better correlations with pharmacokinetic behavior or biological activities than the n-octanol/water system (76–79). Thus, for a series of nine nitroimidazole drugs a liposomal partitioning system (LMVs) composed of L-α-dimyristoylphosphatidylcholine (DMPC) provided a much better prediction of plasmatic clearance rate than the n-octanol/water partitioning system (77). For a set of 21 structurally diverse ionizable compounds, the phosphatidylcholine liposomes (SUVs)/saline system revealed a better correlation between passive intestinal absorption in humans and a so-called absorption potential (AP) (Fig. 6) than n-octanol/water partition system. The high absorption of compounds 1 (aspirin) and 3 (allopurinol) in Fig. 6 may be explained by their particularly low molecular weight, enabling additional paracellular permeation through membranes. The AP is calculated from log D at pH 6.8 (pH of the fasted small intestine), solubility, mean single oral dose, and intestinal fluid volumes (78). For a set of 10 imidazolidines, liposomal partitioning systems composed of DMPC or DMPC/CHOL(chlosterol)/DCP (dicetylphosphate) showed advantages over the n-octanol/water system to predict α2-adrenergic potencies (79).

The correlation between human passive intestinal absorption and APSUV derived from log D from phosphatidylcholine liposomes (SUV’s)/saline system. Redrawn from Ref. (78).
The most popular methods to investigate lipophilicity in liposome/water systems are potentiometric titration (80) and equilibrium dialysis (81). However, these two methods are very labor-intensive. In recent years, high-throughput chromatographic techniques, including immobilized artificial membrane (IAM) HPLC-columns (82), immobilized liposome chromatography (83), and liposome (or microemulsion) electrokinetic chromatography (84) have been developed for the determination of drug partitioning into biomembranes.
Immobilized Artificial Membrane (IAM) Chromatography
The main reason to use a chromatographic system to determine lipophilicity is to conveniently model drug transport processes across biological membranes. Hence, the components of the chromatographic and biological systems should be comparable. An HPLC system used to model transport of a drug through biological membranes should be composed of an aqueous phase and an organized phospholipid layer. Immobilized artificial membranes (IAMs) consist of monolayers of phospholipids covalently immobilized on a silica surface, thus mimicking the lipid environment of a fluid cell membrane on a solid matrix (85,86). The structures of some commercially available immobilized artificial membranes are shown in Fig. 7.

Structures of some commercially available immobilized artificial membranes.
The lipophilicity index obtained from IAM chromatography is the capacity factor log kIAMw at 100% aqueous phase. For hydrophilic compounds, log kIAMw can be determined directly by using the aqueous mobile phase. For lipophilic drugs, it is necessary to add an organic modifier (methanol or acetonitrile) to the mobile phase to accelerate the elution. The log kIAMw value is then extrapolated from isocratic capacity factors (log k) at different ratios of organic modifier (φ) using Eq. 8.
No significant difference between methanol or acetonitrile was observed for the linearily extrapolated log kIAMw values. However, methanol is more appropriate for charged compounds (87). Furthermore, mobile phases containing more than 30% acetonitrile (w/w) must be avoided because their microheterogeneity disrupts the structure of water (88). It was reported that IAM stationary phases deteriorate after 3 months of use (89); thus, measures should be taken to check the decrease in capacity factor over time.
The interaction of drugs with phospholipids has been investigated by IAM-HPLC for different sets of neutral and charged compounds such as β-blockers (90), non-steroidal anti-inflammatory drugs (NSAIDs) (91), dihydropyridine (DHPs) calcium channel blockers (92), and for a set of compounds with a wide structural diversity (93). It was found that log kIAMw values from IAM columns correlate with log Poct only for neutral and structurally related compounds and that electrostatic interactions between the charged solutes and the polar heads of phospholipid play a vital role in IAMs.
The relationship between IAM retention and partitioning in phosphatidylcholine (PC) liposome/water was also studied (94). No correlation existed between log k 7.0IAMw and log D7.0 in egg PC liposome/water for a noncongeneric set of compounds. One possible reason for the difference between the lipophilicity indices from these two anisotropic chromatographic systems is the different density of the polar phospholipid head-groups, which were shown to play an important role in membrane partitioning studies (95).
The log kIAMw determined on IAM columns appears to be a better predictor of permeation than log Poct for drugs of different chemical nature. The log kIAMw values obtained on this type of column were reported to be well correlated with human skin permeation for a set of steroids and phenols (96). The oral absorption of cephalosporin prodrugs in mice could be better predicted by log kIAMw than by analogous parameters determined on traditional ODS reversed phases (95). The log k 7.0IAMw (measured at pH 7.0) values of 12 structurally diverse compounds were determined on an IAM.PC.DD2 phase, and their permeabilities (Pa) were measured through rat everted gut sacs for passively diffused compounds or through noneverted sacs for actively transported molecules. The log k 7.0IAMw correlated better with Pa than with log Poct. The addition of molar volume (Vx) as a second independent variable slightly improved the correlation (97). For a series of (NSAIDs), a parabolic relationship was obtained between log kIAMw and the capacity of diffusion across the rat blood-brain barier (BBB) (98), suggesting an optimum lipophilicity of drugs in the transport to brain. Compared to conventional RP-HPLC columns (XTerra MSC18 and RP18), the IAM.PC.DD2 column showed a closer similarity to human skin partition and blood-brain permeability (99), demonstrating its potential usage as a model for these biological processes. For a set of structurally diverse drugs, log kIAMw from IAM.PC.DD2 in conjunction with polar surface area (PSA) gave a good prediction of human oral absorption (100). One study showed that the IAM capacity factor together with the half-life of hepatic microsomal biotransformation may be useful in identifying compounds with high oral absorption potential in early drug discovery processes (101).
Immobilized Liposome Chromatography (ILC)
ILC is a simple and fast tool to investigate solute-membrane interactions (102). In ILC, phospholipid-based liposomes are noncovalently immobilized to gel beads (e.g., Superdex 200) as a stationary phase (103). The main advantages of this method are that phases of different chemical composition can easily and reversibly be immobilized on suitable gel supports. Phosphatidylcholine (PC), mixtures of PC/PS (phosphatidylserine) and PC/PE (phosphatidylethanolamine), egg phosphatidylcholine (EPC), lipids extracted from human red cells have been used for ILC (83,104). In addition, chromatographic retention on phospholipids is devoid of any effect caused by the presence of a silica matrix.
Various techniques have been developed to immobilize liposomes. In some studies, liposomes are mixed with dry gel beads and immobilized by gel bead swelling followed by freezing-thawing to induce liposome fusion (83,103,104). During the freezing-thawing process, liposomes grow in size and are entrapped in the gel beads pore. The immobilized liposome has a multilamellar structure (105) which should not affect the partitioning of the majority of amphiphilic drugs because they pass the bilayers fast enough during the chromatographic run (104). In other studies, unilamellar liposomes were immobilized in the gel pores of gel beads by avidin-biotin binding (106,107). The liposome immobilized gel was then packed into a column and capacity factors (log Ks) measured by HPLC using an aqueous buffer eluent as the lipophilicity index (Eq. 9):
where VR and V0 are the retention volumes of drug and unretained compound, respectively, and A is the amount of immobilized phospholipids.
No significant correlation was found between log Ks and log D from liposome/water partitioning system, log kIAMw or log Doct for structurally unrelated compounds (104,108).
A hyperbolic relationship between oral absorption in human and log Ks from EPC liposomes was established for a set of 12 structurally unrelated drugs (103). In another study by the same group, it was further observed that drugs with log Ks values (from human red blood extract) between 1.5 and 2.5 were nearly 100% absorbed (83), and the results showed that the essential feature of a good biomembrane model for drug partitioning analyses is a bilayer heterogeneity mimicking that of natural membranes. A sigmoidal relationship was found between log Ks values from EPC-PS-PE-Chol (cholesterol) liposomes and intestinal absorption for a set of 29 structurally diverse drugs (107).
Liposome Electrokinetic Chromatography (LEKC)
LEKC is a capillary electrophoresis method where liposomes are incorporated in the buffer as a pseudo-stationary phase for the determination of drug-membrane interactions (109–111). The LEKC method was proved to be fast and effective for the study of interactions between local anesthetics and liposome dispersions in order to develop a sensitive, efficient and harmless lipid-based formulation to specifically trap harmful substances (112). The antibiotic fusidic acid was shown in a LEKC study to have strong interactions with negatively charged lipid membranes despite its negative surface charge (113).
In LEKC, the composition of the lipid bilayer pseudo-phase can be carefully controlled to nearly mimic the properties of the natural membranes through adjustment of the type and mole fractions of phospholipids as well as incorporating “additives” such as cholesterol and even proteins. Figure 8 illustrates the mechanism of migration and separation of two uncharged solutes, S1 and S2 in LEKC based on negatively charged liposomes. The electrophoretic migration of the liposomes is toward the anode, but the stronger electroosmotic flow carries the liposomes and the solutes toward the cathode where they are detected. Neutral solutes are separated according to their differences in liposome/water partition.

Schematic of the migration pattern in LEKC with a negatively charged liposome. S1 and S2 represent two solutes that partition into the liposome. μLip and μEOF are the mobilities of a liposome and the ElectroOsmotic Flow (EOF), respectively. Finally, teo, tr1, tr2, and tlip are the retention times of the unretained marker (methanol), two solutes, and liposome marker (decanophenone), respectively. Modified from Ref. (109).
The retention factor log k of neutral solutes is the lipophilicity index and is calculated from LEKC data by Eq. 10, where tR, teo, and tpsp are the retention times of the solute, methanol and the marker of the pseudo-stationary phases (e.g., n-decanophenone), respectively. Charged solutes display their own electrophoretic mobility in the aqueous phase in addition to partitioning into the liposomes and migrating with them. As a result, the migration of solutes in the bulk aqueous (t0) needs to be included in the calculation of retention factors as shown in Eq. 11 (114).
There are significant advantages in using LEKC to assess drug-membrane interactions over the existing models such as n-octanol/water, IAM or ILC. Liposomes are spherical lipid bilayer microstructures that are made of phospholipids and closely resemble biological membranes; they are thus more suitable models of the dynamic and fluid lipid bilayer environment of cell membranes than other models. Also, using LEKC, it is possible to establish universal and consistent lipophilicity scales for drug-membranes interaction studies for interlaboratory use. On the contrary, RP-HPLC and IAM method lack a universal scale due to the difference of HPLC columns.
Unilamellar liposomes with a narrow size distribution are most often used as pseudo-stationary phases. With multilamellar liposomes, size can range from approximately 100 nm to 1 μm due to a large number of bilayers; such liposomes are not suitable as carriers in LEKC because the background in the electrophorogram is noisy and the sensitivity low (115).
A few examples will serve to illustrate the correlations found for nonhomogenous drugs of different chemical nature. A good correlation was shown between log k values from LEKC based on POPC-PS liposomes and Caco-2 cell monolayer permeability (84). A sigmoidal relationship was reported between log k values from LEKC based on EPC-PS liposomes and human intestinal drug absorption (Fig. 9), whereas log Poct did not show a good predicting power (116). As shown in Fig. 9, the behavior of the majority of drugs would be correctly predicted with respect to in vivo passive transcellular absorption by log k. Moreover, the drugs (MW < 200 Da, open triangles) via the paracellular diffusion route and the actively transported drugs (filled triangles) were identified as outliers. Log k from LEKC using PC-PS liposomes gave the better correlation with drug penetration across BBB than log D 7.4oct , Clog P or PSA (117). Recently, it was demonstrated that LEKC is a promising method to predict drug penetration through the skin. Quantitative retention-permeability relationships (QRPeRs) were successfully constructed between skin permeability coefficients (log Kp) and retention values (log k) (118).

Relationship between log k on LEKC based on egg PC/PS and drug oral fraction absorbed (Fa%) in human. Redrawn from Ref. (116).
CONCLUSION
In recent years, advances in automated synthesis and combinatorial chemistry have led to the preparation of a vast number of potential drug candidates, often making delivery problems the rate-limiting step in drug research. In order to solve this problem, a thorough understanding of the structure and characteristics of physiological barriers and of the mechanisms of drug transport is necessary.
Lipophilicity is a molecular parameter encoding different intermolecular forces. In isotropic organic solvent/water systems, lipophilicity mainly expresses the balance of hydrophobic and polar interactions. In anisotropic membrane-like systems, lipophilicity can even take ionic bonds into account, showing a closer analogy with the intermolecular recognition forces operating in molecular pharmacology and biochemistry.
Although traditional partitioning systems and high-throughput chromatographic systems are available to derive lipophilicity scales, a comparative study of the described lipophilicity parameters is still missing, and it is difficult to judge which lipophilicity scale has the best and broadest predictive capacity. Comprehensive investigations are needed based on large, structurally diverse sets of compounds to reach sound conclusions.
Numerous significant correlations between lipophilicity and drug passive permeation have been established. This implies that both terms are mechanistically related, meaning that they result from a comparable balance of intermolecular forces (hydrophobic, polar and/or ionic interactions). As this balance changes with different membranes, the physicochemical meaningful lipophilicity-derived parameters (e.g. Δlog P) may be more informative and yield better correlations, as seen with skin and BBB permeation.
The predictive value of the relations between lipophilicity and passive membrane permeation depends on the relevance of the physical systems as models of biomembranes. Therefore, new lipophilicity-derived model systems should be checked for their adequacy in mimicking the permeation properties of natural membranes.
A further complication arises from the fact that different modes of interaction between drugs and membranes exist and coexist. Indeed, the process of membrane permeation proper may be preceded by or be in competition with binding to the membrane surface. Both processes involve hydrophobic and electrostatic forces, but their relative contributions differ from one interaction mode to the other. Thus, electrostatic and mainly ionic bonds often predominate in binding to membrane surfaces (119). But there is more, since the relative contribution of non-covalent forces also differs from one solute to the other. As a consequence and as a matter of principle, a perfect correlation with maximal predictive capacity is impossible to obtain, implying that the best quantitative permeation models one can hope to achieve will remain a mere compromise, best balancing the various and varying contributions.
Abbreviations
- ADME:
-
absorption, distribution, metabolism and excretion
- BBB:
-
blood-brain barrier
- CHOL:
-
cholesterol
- cyc:
-
cyclohexane
- DCE:
-
the 1,2-dichloroethane/water system
- DCP:
-
dicetylphosphate
- DMPC:
-
L-α-dimyristoylphosphatidylcholine
- EPC:
-
egg phosphatidylcholine
- IAM:
-
immobilized artificial membrane
- ILC:
-
immobilized liposome chromatography
- LEKC:
-
liposome electrokinetic chromatography
- LMVs:
-
large multilamellar vesicles
- log D:
-
distribution coefficient
- log k:
-
capacity factor
- log Kp:
-
permeability coefficients
- log P:
-
partition coefficient
- LUVs:
-
large unilamellar vesicles
- PC:
-
phosphatidylcholine
- PE:
-
phosphatidylethanolamine
- PS:
-
phosphatidylserine
- SC:
-
stratum corneum
- SUVs:
-
small unilamellar vesicles
- UWL:
-
unstirred water layer
REFERENCES
Stenberg P, Norinder U, Luthman K, Artursson P. Experimental and computational screening models for the prediction of intestinal drug absorption. J Med Chem. 2001;44(12):1927–37.
Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175(23):720–31.
Thomas G. Medicinal chemistry. An introduction. Chichester: Wiley; 2000.
Delattre J, Couvreur P, Puisieux F, Philippot JR, Schuber F. Les liposomes aspects technologiques, biologiques et pharmacologiques. Paris: Les Editions INSERM; 1993.
Lasic DD. Liposomes: from physics to applications. Amsterdam: Elsevier; 1993.
Tsui FC, Ojcius DM, Hubbell WL. The intrinsic pKa values for phosphatidylserine and phosphatidylethanolamine in phosphatidylcholine host bilayers. Biophys J. 1986;49(2):459–68.
Albert A. Selective toxicity. The physico-chemical basis of therapy. London: Chapman and Hall; 1985.
Conradi RA, Burton PS, Borchardt RT. Physico-chemical and biological factors that influence a drug’s cellular permeability by passive diffusion. In: Pliska V, Testa B, van de Waterbeemd H, editors. Lipophilicity in drug action and toxicology. Weinheim: VCH Publishers; 1996. p. 233–52.
Borchardt RT. Biological models to acess drug bioavailability. In: Testa B, van de Waterbeemd H, Folkers G, Guy RH, editors. Pharmacokinetic optimization in drug research: biological, physicochemical and computational strategies. Zurich: Wiley-VHCA; 2001. p. 117–26.
Camenisch G, Folkers G, van de Waterbeemd H. Review of theoretical passive drug absorption models: historical background, recent developments and limitations. Pharm Acta Helv. 1996;71(5):309–27.
Smith DA, Jones BC, Walker DK. Design of drugs involving the concepts and theories of drug metabolism and pharmacokinetics. Med Res Rev. 1996;16(3):243–66.
Lennernas H. Does fluid flow across the intestinal mucosa affect quantitative oral drug absorption? Is it time for a reevaluation? Pharm Res. 1995;12(11):1573–82.
Pauletti GM, Gangwar S, Siahaan TJ, Abé J, Borchardt RT. Improvement of oral peptide bioavailability: peptidomimetics and prodrug strategies. Adv Drug Deliv Rev. 1997;27:235–56.
Nellans HN. Mechanisms of peptide and protein absorption. Paracellular intestinal transport: modulation of absorption. Adv Drug Deliv Rev. 1991;7:339–64.
Van Itallie CM, Holmes J, Bridges A, Gookin JL, Coccaro MR, Proctor W, et al. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci. 2008;121(3):298–305.
Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9(11):799–809.
Pauletti GM, Okumu FW, Borchardt RT. Effect of size and charge on the passive diffusion of peptides across Caco-2 cell monolayers via the paracellular pathway. Pharm Res. 1997;14(2):164–8.
Brightman MW, Tao-Chen JH. Tight junctions of brain endothelium and epithelium. In: Pardridge WM, editor. The blood–brain barrier cellular and molecular biology. New York: Raven Press; 1993. p. 107–25.
Wunderli-Allenspach H. Methodologies in cell culture. In: Testa B, van de Waterbeemd H, Folkers G, Guy RH, editors. Pharmacokinetic optimization in drug research: biological, physicochemical and computational strategies. Zurich: Wiley-VHCA; 2001. p. 99–116.
Peters WH, Boon CE, Roelofs HM, Wobbes T, Nagengast FM, Kremers PG. Expression of drug-metabolizing enzymes and P-170 glycoprotein in colorectal carcinoma and normal mucosa. Gastroenterology. 1992;103(2):448–55.
Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62:385–427.
Avdeef A. Physicochemical profiling (solubility, permeability and charge state). Curr Top Med Chem. 2001;1(4):277–351.
Balimane PV, Chong S. Cell culture-based models for intestinal permeability: a critique. Drug Discov Today. 2005;10(5):335–43.
Audus KL, Bartel RL, Hidalgo IJ, Borchardt RT. The use of cultured epithelial and endothelial cells for drug transport and metabolism studies. Pharm Res. 1990;7(5):435–51.
Kramer SD, Abbott NJ, Begley DJA. Biological models to study blood brain barrier permeation. In: Testa B, van de Waterbeemd H, Folkers G, Guy RH, editors. Pharmacokinetic optimization in drug research: biological, physicochemical and computational strategies. Zurich: Wiley-VHCA; 2001. p. 127–53.
de Boer AG, Breimer DD. Reconstitution of the blood-brain barrier in cell culture for studies of drug transport and metabolism. Adv Drug Deliv Rev. 1996;22:251–64.
van Bree JB, de Boer AG, Danhof M, Breimer DD. Drug transport across the blood-brain barrier. I. Anatomical and physiological aspects. Pharm Weekbl Sci. 1992;14:305–10.
Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37(1):13–25.
Hadgraft J. Skin deep. Eur J Pharm Biopharm. 2004;58(2):291–9.
Hadgraft J, Pugh WJ. The selection and design of topical and transdermal agents: a review. J Investig Dermatol Symp Proc. 1998;3(2):131–5.
de Jager MW, Gooris GS, Dolbnya IP, Ponec M, Bouwstra JA. Modelling the stratum corneum lipid organisation with synthetic lipid mixtures: the importance of synthetic ceramide composition. Biochim Biophys Acta. 2004;1664(2):132–40.
Heisig M, Lieckfeldt R, Wittum G, Mazurkevich G, Lee G. Non steady-state descriptions of drug permeation through stratum corneum. I. The biphasic brick-and-mortar model. Pharm Res. 1996;13(3):421–6.
Roberts MS, Pugh WJ, Hadgraft J, Watkinson AC. Epidermal permeability penetrant structure relationships.1. An analysis of methods of predicting penetration of monofunctional solutes from aqueous solutions. Int J Pharm. 1995;126(1–2):219–33.
Roberts MS, Pugh WJ, Hadgraft J. Epidermal permeability: penetrant structure relationships.2. The effect of H-bonding groups in penetrants on their diffusion through the stratum corneum. Int J Pharm. 1996;132(1–2):23–32.
Leahy DE, Morris JJ, Taylor PJ, Wait AR. Model solvent systems for qsar.3. An lser analysis of the critical quartet—new light on hydrogen-bond strength and directionality. J Chem Soc Perk Trans 2. 1992;4:705–22.
Leahy DE, Morris JJ, Taylor PJ, Wait AR. Model solvent systems for qsar.2. Fragment values (F-Values) for the critical quartet. J Chem Soc Perk Trans 2. 1992;4:723–31.
Leahy DE, Taylor PJ, Wait AR. Model solvent systems for qsar,1, Propylene-Glycol Dipelargonate (Pgdp)—a new standard solvent for use in partition-coefficient determination. Quant Struct-Act Relat. 1989;8(1):17–31.
Pagliara A, Caron G, Lisa G, Fan W, Gaillard P, Carrupt PA, et al. Solvatochromic analysis of di-n-butyl ether/water partition coefficients as compared to other solvent systems. J Chem Soc, Perkin Trans 1997;2639–43.
Dearden JC, Bresnen GM. The measurement of partition-coefficients. Quant Struct-Act Relat. 1988;7(3):133–44.
Avdeef A. pH-metric log P. II: refinement of partition coefficients and ionization constants of multiprotic substances. J Pharm Sci. 1993;82(2):183–90.
Scherrer RA, Donovan SF. Automated potentiometric titrations in KCl/water-saturated octanol: method for quantifying factors influencing ion-pair partitioning. Anal Chem. 2009;81(7):2768–78.
Kamlet MJ, Doherty RM, Abraham MH, Marcus Y, Taft RW. Linear solvation energy relationships.46. An improved equation for correlation and prediction of octanol water partition-coefficients of organic nonelectrolytes (Including strong hydrogen-bond donor solutes). J Phys Chem-Us. 1988;92(18):5244–55.
Abraham MH, Chadha HS. Application of a solvation equation to drug transport properties. In: Pliska V, Testa B, van de Waterbeemd H, editors. Lipophilicity in drug action and toxicology. Weinheim: VCH Publishers; 1996. p. 311–37.
El Tayar N, Tsai RS, Testa B, Carrupt PA, Leo A. Partitioning of solutes in different solvent systems: the contribution of hydrogen-bonding capacity and polarity. J Pharm Sci. 1991;80(6):590–8.
Steyaert G, Lisa G, Gaillard P, Boss G, Reymond F, Girault HH, et al. Intermolecular forces expressed in 1, 2-dichloroethane-water partition coefficients—a solvatochromic analysis. J Chem Soc Faraday Trans. 1997;93(3):401–6.
Testa B, Crivori P, Reist M, Carrupt PA. The influence of lipophilicity on the pharmacokinetic behavior of drugs: concepts and examples. Perspect Drug Discov. 2000;19(1):179–211.
van de Waterbeemd H, Testa B. The parametrization of lipophilicity and other structural properties in drug design. In: Testa B, editor. Advances in drug research, vol. 16. London: Academic; 1987. p. 87–227.
Comer J, Tam K. Lipophilicity profiles: theory and measurement. In: Testa B, van de Waterbeemd H, Folkers G, Guy RH, editors. Pharmacokinetic optimization in drug research: biological, physicochemical and computational strategies. Zurich: Wiley-VHCA; 2001. p. 275–304.
Caron G, Gaillard P, Carrupt PA, Testa B. Lipophilicity behavior of model and medicinal compounds containing a sulfide, sulfoxide, or sulfone moiety. Helv Chim Acta. 1997;80(2):449–62.
Kubinyi H. QSAR: Hansch analysis and related approaches. Weinheim: VCH Publisher; 1993.
Rolando B, Lazzarato L, Di Stilo A, Fruttero R, Carrupt PA, Martel S, et al. Physicochemical profile and in vitro permeation behavior of a new class of non-steroidal anti-inflammatory drug candidates. Eur J Pharm Sci. 2010;40(3):217–21.
Caron G, Steyaert G, Pagliara A, Reymond F, Crivori P, Gaillard P, et al. Structure-lipophilicity relationships of neutral and protonated beta-blockers Part I Intra- and intermolecular effects in isotropic solvent systems. Helv Chim Acta. 1999;82(8):1211–22.
Caron G, Pagliara A, Gaillard P, Carrupt PA, Testa B. Ionization and partitioning profiles of zwitterions: the case of the anti-inflammatory drug azapropazone. Helv Chim Acta. 1996;79(6):1683–95.
Tsai RS, Testa B, El Tayar N, Carrupt PA. Structure lipophilicity relationships of zwitterionic amino-acids. J Chem Soc Perk Trans 2. 1991;11:1797–802.
Tam KY, Avdeef A, Tsinman O, Sun N. The permeation of amphoteric drugs through artificial membranes—an in combo absorption model based on paracellular and transmembrane permeability. J Med Chem. 2010;53(1):392–401.
Pagliara A, Carrupt PA, Caron G, Gaillard P, Testa B. Lipophilicity profiles of ampholytes. Chem Rev. 1997;97(8):3385–400.
Kakemi K, Arita T, Hori R, Konishi R, Nishimura K. Absorption and exretion of drugs. XXXIV. An aspect of the mechanism of drug absorption from the intestinal tract in rats. Chem Pharm Bull (Tokyo). 1969;17(2):255–61.
Houston JB, Upshall DG, Bridges JW. A re-evaluation of the importance of partition coefficients in the gastrointestinal absorption of anutrients. J Pharmacol Exp Ther. 1974;189(1):244–54.
Garrigues TM, Perezvarona AT, Climent E, Bermejo MV, Martinvillodre A, Pladelfina JM. Gastric absorption of acidic xenobiotics in the rat—biophysical interpretation of an apparently atypical behavior. Int J Pharm. 1990;64(2–3):127–38.
Murthy KS, Zografi G. Oil-water partitioning of chlorpromazine and other phenothiazine derivatives using dodecane and normal-octanol. J Pharm Sci. 1970;59(9):1281–5.
Buur A, Trier L, Magnusson C, Artursson P. Permeability of 5-fluorouracil and prodrugs in Caco-2 cell monolayers. Int J Pharm. 1996;129(1–2):223–31.
Komiya I, Park JY, Kamani A, Ho NFH, Higuchi WI. Quantitative mechanistic studies in simultaneous fluid-flow and intestinal-absorption using steroids as model solutes. Int J Pharm. 1980;4(3):249–62.
Taylor DC, Pownall R, Burke W. The absorption of beta-adrenoceptor antagonists in rat in-situ small intestine; the effect of lipophilicity. J Pharm Pharmacol. 1985;37(4):280–3.
El Tayar N, Tsai RS, Testa B, Carrupt PA, Hansch C, Leo A. Percutaneous penetration of drugs: a quantitative structure-permeability relationship study. J Pharm Sci. 1991;80(8):744–9.
Singh P, Roberts MS. Skin permeability and local tissue concentrations of nonsteroidal anti-inflammatory drugs after topical application. J Pharmacol Exp Ther. 1984;268:144–51.
Schoenwald RD, Huang HS. Corneal penetration behavior of beta-blocking agents I: physiochemical factors. J Pharm Sci. 1983;72(11):1266–72.
Dearden JC. Molecular structure and drug transport. In: Ramsden CA, Hansch C, Sammer PG, Taylor JB, editors. Comprehensive medicinal chemistry. The rational design, mechanistic study & therapeutic applications of chemical compounds, vol. 4. Oxford: Pergamon; 1990. p. 375–411.
Camenisch G, Folkers G, van de Waterbeemd H. Shapes of membrane permeability-lipophilicity curves: extension of theoretical models with an aqueous pore pathway. Eur J Pharm Sci. 1998;6(4):321–9.
Young RC, Mitchell RC, Brown TH, Ganellin CR, Griffiths R, Jones M, et al. Development of a new physicochemical model for brain penetration and its application to the design of centrally acting H2 receptor histamine antagonists. J Med Chem. 1988;31(3):656–71.
Yoshida F, Topliss JG. Unified model for the corneal permeability of related and diverse compounds with respect to their physicochemical properties. J Pharm Sci. 1996;85(8):819–23.
von Geldern TW, Hoffman DJ, Kester JA, Nellans HN, Dayton BD, Calzadilla SV, et al. Azole endothelin antagonists. 3. Using delta log P as a tool to improve absorption. J Med Chem. 1996;39(4):982–91.
Plemper van Balen G, Martinet CM, Caron G, Bouchard G, Reist M, Carrupt PA, et al. Liposome/water lipophilicity: methods, information content, and pharmaceutical applications. Med Res Rev. 2004;24(3):299–324.
Seydel JK, Velasco MA, Coats EA, Cordes HP, Kunz B, Wiese M. The importance of drug-membrane interaction in drug research-and-development. Quant Struct-Act Relat. 1992;11(2):205–10.
Malkia A, Murtomaki L, Urtti A, Kontturi K. Drug permeation in biomembranes: in vitro and in silico prediction and influence of physicochemical properties. Eur J Pharm Sci. 2004;23(1):13–47.
New RRC. Liposomes. A practical approach. Oxford: IRL Press; 1990.
Betageri GV, Rogers JA. Thermodynamics of partitioning of beta-blockers in the normal-octanol-buffer and liposome systems. Int J Pharm. 1987;36(2–3):165–73.
Betageri GV, Rogers JA. Correlation of partitioning of nitroimidazoles in the n-octanol/saline and liposome systems with pharmacokinetic parameters and quantitative structure-activity relationships (QSAR). Pharm Res. 1989;6(5):399–403.
Balon K, Riebesehl BU, Muller BW. Drug liposome partitioning as a tool for the prediction of human passive intestinal absorption. Pharm Res. 1999;16(6):882–8.
Rogers JA, Choi YW. The liposome partitioning system for correlating biological activities of imidazolidine derivatives. Pharm Res. 1993;10(6):913–7.
Avdeef A, Box KJ, Comer JE, Hibbert C, Tam KY. pH-metric logP 10. Determination of liposomal membrane-water partition coefficients of ionizable drugs. Pharm Res. 1998;15(2):209–15.
Pauletti GM, Wunderliallenspach H. Partition-coefficients in-vitro—artificial membranes as a standardized distribution model. Eur J Pharm Sci. 1994;1(5):273–82.
Ong S, Cai SJ, Bernal C, Rhee D, Qiu X, Pidgeon C. Phospholipid immobilization on solid surfaces. Anal Chem. 1994;66(6):782–92.
Beigi F, Gottschalk I, Hagglund CL, Haneskog L, Brekkan E, Zhang YX, et al. Immobilized liposome and biomembrane partitioning chromatography of drugs for prediction of drug transport. Int J Pharm. 1998;164(1–2):129–37.
Ornskov E, Gottfries J, Erickson M, Folestad S. Experimental modelling of drug membrane permeability by capillary electrophoresis using liposomes, micelles and microemulsions. J Pharm Pharmacol. 2005;57(4):435–42.
Pidgeon C, Ong S, Liu H, Qiu X, Pidgeon M, Dantzig AH, et al. IAM chromatography: an in vitro screen for predicting drug membrane permeability. J Med Chem. 1995;38(4):590–4.
Liu H, Ong S, Glunz L, Pidgeon C. Predicting drug-membrane interactions by HPLC: structural requirements of chromatographic surfaces. Anal Chem. 1995;67(19):3550–7.
Taillardat-Bertschinger A, Carrupt PA, Barbato F, Testa B. Immobilized artificial membrane HPLC in drug research. J Med Chem. 2003;46(5):655–65.
Marcus Y, Migron Y. Polarity, hydrogen-bonding, and structure of mixtures of water and cyanomethane. J Phys Chem-Us. 1991;95(1):400–6.
Hanna M, de Biasi V, Bond B, Camilleri P, Hutt A. Biomembrane lipids as components of chromatographic phases: comparative chromatography on coated and bonded phases. Chromatographia. 2000;52:710–20.
Barbato F, di Martino G, Grumetto L, La Rotonda MI. Can protonated beta-blockers interact with biomembranes stronger than neutral isolipophilic compounds? A chromatographic study on three different phospholipid stationary phases (IAM-HPLC). Eur J Pharm Sci. 2005;25(4–5):379–86.
Barbato F, La Rotonda MI, Quaglia F. Interactions of nonsteroidal antiinflammatory drugs with phospholipids: comparison between octanol/buffer partition coefficients and chromatographic indexes on immobilized artificial membranes. J Pharm Sci. 1997;86(2):225–9.
Barbato F, La Rotonda MI, Quaglia F. Chromatographic indices determined on an immobilized artificial membrane (IAM) column as descriptors of lipophilic and polar interactions of 4-phenyldihydropyridine calcium-channel blockers with biomembranes. Eur J Med Chem. 1996;31(4):311–8.
Liu X, Hefesha H, Scriba G, Fahr A. Retention behavior of neutral, positively and negatively charged solutes on immobilized artificial membrane (IAM) stationary. Helv Chim Acta. 2008;91:1505–12.
Taillardat-Bertschinger A, Martinet CA, Carrupt PA, Reist M, Caron G, Fruttero R, et al. Molecular factors influencing retention on immobilized artifical membranes (IAM) compared to partitioning in liposomes and n-octanol. Pharm Res. 2002;19(6):729–37.
Ong S, Liu H, Pidgeon C. Immobilized-artificial-membrane chromatography: measurements of membrane partition coefficient and predicting drug membrane permeability. J Chromatogr A. 1996;728(1–2):113–28.
Nasal A, Sznitowska M, Bucinski A, Kaliszan R. Hydrophobicity parameter from high-performance liquid-chromatography on an immobilized artificial membrane column and its relationship to bioactivity. J Chromatogr A. 1995;692(1–2):83–9.
Genty M, Gonzalez G, Clere C, Desangle-Gouty V, Legendre JY. Determination of the passive absorption through the rat intestine using chromatographic indices and molar volume. Eur J Pharm Sci. 2001;12(3):223–9.
Pehourcq F, Matoga M, Bannwarth B. Diffusion of arylpropionate non-steroidal anti-inflammatory drugs into the cerebrospinal fluid: a quantitative structure-activity relationship approach. Fundam Clin Pharmacol. 2004;18(1):65–70.
Lazaro E, Rafols C, Abraham MH, Roses M. Chromatographic estimation of drug disposition properties by means of immobilized artificial membranes (IAM) and C18 columns. J Med Chem. 2006;49(16):4861–70.
Kotecha J, Shah S, Rathod I, Subbaiah G. Prediction of oral absorption in humans by experimental immobilized artificial membrane chromatography indices and physicochemical descriptors. Int J Pharm. 2008;360(1–2):96–106.
Shin BS, Yoon CH, Balthasar JP, Choi BY, Hong SH, Kim HJ, et al. Prediction of drug bioavailability in humans using immobilized artificial membrane phosphatidylcholine column chromatography and in vitro hepatic metabolic clearance. Biomed Chromatogr. 2009;23(7):764–9.
Boija E, Johansson G. Interactions between model membranes and lignin-related compounds studied by immobilized liposome chromatography. Biochim Biophys Acta. 2006;1758(5):620–6.
Beigi F, Yang Q, Lundahl P. Immobilized-liposome chromatographic analysis of drug partitioning into lipid bilayers. J Chromatogr A. 1995;704(2):315–21.
Osterberg T, Svensson M, Lundahl P. Chromatographic retention of drug molecules on immobilised liposomes prepared from egg phospholipids and from chemically pure phospholipids. Eur J Pharm Sci. 2001;12(4):427–39.
Wiedmer SK, Jussila MS, Riekkola ML. Phospholipids and liposomes in liquid chromatographic and capillary electromigration techniques. TrAC Trends Anal Chem. 2004;23(8):562–82.
Yang Q, Liu XY, Ajiki S, Hara M, Lundahl P, Miyake J. Avidin-biotin immobilization of unilamellar liposomes in gel beads for chromatographic analysis of drug-membrane partitioning. J Chromatogr B Biomed Sci Appl. 1998;707(1–2):131–41.
Liu XY, Nakamura C, Yang Q, Kamo N, Miyake J. Immobilized liposome chromatography to study drug-membrane interactions. Correlation with drug absorption in humans. J Chromatogr A. 2002;961(1):113–8.
Liu X, Fan P, Chen M, Scriba G, Gabel D, Fahr A. Drug-membrane interaction on immobilized liposome chromatography compared to immobilized artificial membrane (IAM), liposome/water and n-octanol/water systems. Helv Chim Acta. 2010;93:203–11.
Burns ST, Khaledi MG. Rapid determination of liposome-water partition coefficients (Klw) using liposome electrokinetic chromatography (LEKC). J Pharm Sci. 2002;91(7):1601–12.
Zhang Y, Zhang R, Hjerten S, Lundahl P. Liposome capillary electrophoresis for analysis of interactions between lipid bilayers and solutes. Electrophoresis. 1995;16(8):1519–23.
Wiedmer SK, Shimmo R. Liposomes in capillary electromigration techniques. Electrophoresis. 2009;30:S240–57.
Muhonen J, Holopainen JM, Wiedmer SK. Interactions between local anesthetics and lipid dispersions studied with liposome electrokinetic capillary chromatography. J Chromatogr A. 2009;1216(15):3392–7.
Helle A, Makitalo J, Huhtanen J, Holopainen JM, Wiedmer SK. Antibiotic fusidic acid has strong interactions with negatively charged lipid membranes: an electrokinetic capillary chromatographic study. Biochim Biophys Acta. 2008;1778(11):2640–7.
Carrozzino JM, Khaledi MG. Interaction of basic drugs with lipid bilayers using liposome electrokinetic chromatography. Pharm Res. 2004;21(12):2327–35.
Wiedmer SK, Holopainen JM, Mustakangas P, Kinnunen PK, Riekkola ML. Liposomes as carriers in electrokinetic capillary chromatography. Electrophoresis. 2000;21(15):3191–8.
Wang YJ, Sun J, Liu HZ, Wang YJ, He ZG. Prediction of human drug absorption using liposome electrokinetic chromatography. Chromatographia. 2007;65(3–4):173–7.
Wang YJ, Sun J, Liu HZ, He ZG. Rapidly profiling blood-brain barrier penetration with liposome EKC. Electrophoresis. 2007;28(14):2391–5.
Wang YJ, Sun J, Liu HZ, Liu JF, Zhang LQ, Liu K, et al. Predicting skin permeability using liposome electrokinetic chromatography. Analyst. 2009;134(2):267–72.
Ermondi G, Caron G. Recognition forces in ligand-protein complexes: blending information from different sources. Biochem Pharmacol. 2006;72(12):1633–45.
ACKNOWLEDGMENT
We would like to thank Dr. Sylvio May, North Dakota State University for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, X., Testa, B. & Fahr, A. Lipophilicity and Its Relationship with Passive Drug Permeation. Pharm Res 28, 962–977 (2011). https://doi.org/10.1007/s11095-010-0303-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-010-0303-7