
ABSTRACT
Purpose
To investigate the effect of crystal size on the dissolution and oral absorption of nitrendipine, a poorly soluble drug, in rats.
Methods
Five types of nitrendipine crystal suspensions with different particle sizes (200 nm, 620 nm, 2.7 μm, 4.1 μm, 20.2 μm) were prepared either by the precipitation-ultrasonication or the anti-solvent precipitation method. The simulated intestinal fluid in the fasted state (FaSSIF) was selected as the dissolution medium, and the dissolution behaviors of different nitrendipine crystals were simulated based on a Noyes-Whitney type equation. The in vivo absorption and the absolute bioavailability of the different nitrendipine crystals were evaluated in Wistar rats.
Results
The dissolution rate of nitrendipine was significantly increased by a reduction in particle size. The dissolution test in FaSSIF could discriminate between the differences in the dissolution rates of the different particle sizes, and the simulated results were in agreement with the observed dissolution curves. From the simulated T50% values (50% dissolution time), the dissolution rates of crystals with particle sizes of 200 nm, 620 nm, 2.7 μm, 4.1 μm and 20.2 μm were calculated to be 5.1 × 104, 1.0 × 104, 237, 64 and 11-fold greater than that of the raw crystals and resulted in absolute bioavailability of 61.4% 51.5%, 29.4%, 26.7%, 24.7%, respectively. The reduction in the drug particle size correlated well with incremental improvements in oral absorption. A good linear relationship was observed between the Log (T50%) and the absolute bioavailability of nitrendipine.
Conclusions
The dissolution rate and the oral bioavailability of nitrendipine were significantly affected by the crystal size, and the oral bioavailability could be improved significantly by preparing it as nanocrystals. FaSSIF can be used to predict differences in oral absorption of crystals with different particle sizes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Bioavailability is a measurement of the rate and amount of a therapeutically active drug that reaches the systemic circulation and is available at the site of action (1). Among the many factors that influence oral drug absorption, solubility in water and permeability of the gastrointestinal membrane are the most important factors affecting the fraction of the dose absorbed (2). Amidon et al. have defined three parameters for drug absorption, i.e. absorption number, dose number, and dissolution number. They also proposed a biopharmaceutics classification system (BCS) in which drugs are classified into four groups according to their permeability and solubility (3,4).
For oral administration of class II drugs (poorly soluble and highly permeable), the rate of dissolution is the key to the evaluation of bioavailability (5,6). Many poorly soluble drugs fail to be completely absorbed after oral administration due to their inappropriately slow dissolution from the administered dosage form. In the case of dissolution rate-limited absorption, it is well known that the dissolution rate of a drug is proportional to the surface area available for dissolution as described by the Noyes-Whitney equation (7). Thus, the particle size and resulting surface area could have a significant effect on the dissolution rate over the time interval during which gastrointestinal absorption occurs and, therefore, affect their bioavailability (8,9). The particle size of a poorly soluble drug has attracted the attention of many investigators involved in biopharmaceutical research. Many studies of particle size-dependent absorption have been published (8,10–15), and many powerful commercial, software packages, such as GastroPlus™ and PK-Sim® have been used to predict the effects of particle size on the dissolution and absorption of poorly soluble drugs (12,16). However, few in vivo studies have been carried out so far to fully investigate the absorption of poorly soluble drug crystals with different particle sizes ranging from nanometers to micrometers.
Nitrendipine is a dihydropyridine calcium channel antagonist with a very low solubility (2.0 μg/ml at 37°C) in water. Its octanol-water distribution coefficient (log P oct) is 3.59 (17), and it is classified as a Class II active pharmaceutical ingredient (API) by the BCS. The absolute oral bioavailability of this drug is reported to range from about 10% to 20%, depending in part on the dosage form (18,19). The dissolution is the rate-limiting factor for absorption. In this paper, nitrendipine crystal suspensions with five different particle sizes were prepared either by the precipitation-ultrasonication (200 nm, 620 nm, and 2.7 μm) or the anti-solvent precipitation (4.1 μm, 20.2 μm) method. The effects of particle size on the in vitro dissolution and the extent of in vivo absorption of nitrendipine were fully investigated.
MATERIALS AND METHODS
Materials
Nitrendipine was purchased from Nanjing Pharmaceutical Factory (China), and polyvinyl alcohol (PVA, Mw 30–70 kDa, 88% alcoholysis) was generously supplied by Shin-Etsu Chemical Ind. Co. Ltd. (Japan). PEG 200 and acetone of analytical grade were purchased from Ruijingte Chemical Agent Company (Tianjing, China). Cyclohexane, isopropyl alcohol, methanol and acetonitrile (chromatographic grade) were purchased from Concord Chemical Agent Company (China). Tween 80 was obtained from Yili Chemical Agent Company (Beijing, China).
Preparation of Nitrendipine Nano- and Micro-crystals
Nitrendipine nanocrystals were prepared by the precipitation-ultrasonication method, which was described in detail in our previous work (20) (Fig. 1). Briefly, nitrendipine was dissolved in a mixed solvent of PEG 200 and acetone (1/1, v/v) to form organic solutions containing 30, 60 and 100 mg/ml of drug. Then, 2 ml of organic solution was quickly introduced into 20 ml of the precooled aqueous solution containing 0.15% PVA (w/v) (below 3°C) at a stirring speed of 400 rpm. After mixing, the samples were immediately treated with an ultrasonic probe at power input of 400 W for 15 min. During this process, the temperature was controlled using an ice-water bath. The obtained nanosuspensions were concentrated by centrifugation. After removing the organic solvent, the final drug content was adjusted to 20 mg/ml. Similar experiments were carried out involving the direct precipitation of nitrendipine organic solution in anti-solvent followed by placing the sample at 3°C for 24 h instead of ultrasonication to obtain microcrystals. The process parameters for the preparation of various nano- and micro- crystals are shown in Table I.

Flow chart of the procedure for the preparation of nitrendipine nano- and micro-crystals.
Size, Size Distribution and Morphology of Nitrendipine Crystals
The size and size distribution were determined by laser diffractometry using a Coulter LS230 instrument (Beckman-Coulter Co. Ltd., USA). The crystal size was expressed as the volume diameter. The morphology of the nanocrystals, microcrystals and raw crystals were observed by scanning electron microscopy (SEM, S-4800, Hitachi, Japan).
X-ray Powder Diffraction (XRPD) and Differential Scanning Calorimetry (DSC) Characterization
The crystal structures of the nano- and micro-crystals were analyzed using an X-ray Diffractometer (DX-2700, China) with Cu Kα radiation at a wavelength of 1.542 Å, generated at 30 mA and 40 kV. The scanning speed was 10°/min from 5° to 55° with a step size of 0.03°. The phase transition of nitrendipine crystals was analyzed by differential scanning calorimetry (DSC-60, Shimadzu Co., Japan) at a heating rate of 10°C/min from 30°C to 300°C. This was done in a dry nitrogen atmosphere, and Al2O3 was used as a reference.
Dissolution Rate and Saturation Solubility
Dissolution experiments were performed according to the Ch.P.2005 Ed paddle method (ZRD6-B, Huanghai Pharmatest Apparatus Factory, Shanghai, China). In order to select an appropriate dissolution medium for predicting in vivo dissolution, three kinds of dissolution medium were evaluated. These dissolution media were distilled water containing 0.1 M hydrochloric acid and 30% ethanol (Medium 1), distilled water containing 0.1 M hydrochloric acid and 0.1% SDS (Medium 2), and simulated intestinal fluid in the fasted state (FaSSIF). FaSSIF contains 3 mM sodium taurocholate and 0.75 mM lecithin, and the pH value was adjusted to 6.5 using KH2PO4-NaOH buffer solution (21,22). The temperature was maintained at 37 ± 0.5°C, and the paddle speed was 100 rpm. Accurately weighed samples containing the equivalent of 2.5 mg nitrendipine were dispersed in the 500 ml dissolution medium. Samples, each of 4 ml, were withdrawn at different times and passed through a 0.1 μm syringe filter (Membrane Solutions Inc, Shanghai, China). Quantification of the samples was carried out by using a HPLC (Pump model: Hitachi L7110, Japan). The analytical column was a Diamonsil™ C18 (5 μm, 200 × 4.6 mm), the mobile phase was composed of methanol and water (75/25, v/v), at a flow rate of 1.0 ml/min, and detection was performed at 236 nm using a UV-VIS detector (Model: Hitachi L7420, Japan). The measurements were repeated three times.
The saturation solubility of nitrendipine crystals with different particle size was evaluated in FaSSIF at 37°C. Samples were dispersed in 10 ml FaSSIF to obtain suspensions containing 2 mg/ml of drug. The suspensions were placed on a shaking water bath for 48 h to ensure that the solubility equilibrium had been reached. After centrifugation, the concentration of nitrendipine in the resulting supernatants was quantified by the HPLC method mentioned above. The measurements were repeated three times.
Animals and Dosing
Male Wistar rats (body weight 200 ± 20 g) were supplied by the Experimental Animal Center of Shenyang Pharmaceutical University (Shenyang, China), and all animal experiments were evaluated and approved by the Animal Ethics Committee, Shenyang Pharmaceutical University. All rats were divided randomly into six groups of five animals each and were housed in cages for at least 3 days prior to the beginning of the study and had free access to food and water. All rats were fasted for 12 h before the beginning of the experiments. Six types of nitrendipine crystal suspensions, i.e. two nanocrystal suspensions, three microcrystal suspensions and a raw crystal suspension, were orally administered to six groups of rats at a dose of 40 mg/animal. In addition, a nitrendipine solution in a mixed solvent of PEG 200, Tween 80 and water (3.5/1.0/3.5, v/v) was also intravenously administered to a group of four male Wistar rats at a dose of 4 mg/animal. Rats were allowed free access to water throughout the experiment. Blood (0.25 ml) was sampled by retro-orbital puncture at the following times: 15, 30, 45 min, 1, 1.5, 2, 3, 4, 6, 8, 10, and 12 h for the oral administration, and 5, 15, 30, 45 min, 1, 1.5, 2, 4, 6, 8, 10, and 12 h for the intravenous administration. Plasma was obtained from whole blood in heparinized tubes by centrifugation at 10,000 rpm for 5 min and was frozen at −20°C until analysis.
Nitrendipine in Plasma
The concentration of nitrendipine in plasma was determined by HPLC (L7110 pump equipped with a L7420 UV-VIS detector, Hitachi, Japan). The analytical column was a Diamonsil™ C18 (5 μm, 200 × 4.6 mm), the mobile phase was a mixture of methanol, acetonitrile and water (50/20/30, v/v), and the flow rate was 1.2 ml/min. UV-detection was performed at 236 nm. Nimodipine was employed as the internal standard. One-hundred microlitres of plasma were homogenized and extracted with 0.5 ml mixed organic solvent (cyclohexane and isopropyl alcohol (50/2, v/v)) on a vortex mixer (XW-80A, Shanghai, China) for 5 min. After centrifugation at 10,000 rpm for 5 min, the organic layer was transferred to another clean tube and evaporated under nitrogen gas. Then, the residue was redissolved in 30 μl mobile phase, and 20 μl of the sample was injected into the HPLC for analysis.
Data Analysis
Standard pharmacokinetic (PK) parameters (±S.D.) of nitrendipine were derived from plasma concentration-time curves using a non-compartmental model with WinNonlin® Professional Version 3.1 (Pharsight Corp., Mountain View, CA). The PK parameters that were analyzed included the maximum peak concentration of the drug in plasma (Cmax), the time to reach maximum concentration (Tmax), and the area under the curve (AUC0→12 and \( {\hbox{AU}}{{\hbox{C}}_{0 \to \infty }} \)). The absolute bioavailability (F abs ) was calculated based on the following equation:
The in vivo absorption rate of nitrendipine in 2 h after oral administration was estimated using a numerical deconvolution method (23,24). The mean plasma drug concentration data from the oral administration study were assigned as a response function, while the data from the intravenous administration study were used as a weight function. For computation, a time step of 15 min was used, and some nodes were interpolated based on the information from neighboring nodes.
Simulation of In Vitro Dissolution
In vitro dissolution of nitrendipine crystals in FaSSIF was simulated using the method reported by Johnson et al. (9,25). The authors derived a Noyes-Whitney-type equation to simulate dissolution and absorption of monodisperse powders to polydisperse mixtures of a drug under non-sink conditions in terms of the spherical particle geometry, the time-dependent diffusion layer thickness and the constant number of particles with time. The particle size distribution of a polydisperse power was simulated by treating it as several discrete monodisperse size fractions i to n, covering the range of particle sizes obtained by the laser diffractometry using a Coulter LS230 instrument. The rate of change in the mass of dissolved drug is given by Eq. 2:
where X di is the amount of dissolved drug for the size fraction i at any time t, X si is the amount of solid drug for the size fraction i at any time t, D is the drug diffusion coefficient, X oi is the initial amount of solid drug for size fraction i, ρ is the drug density, h i is the diffusion layer thickness of the size fraction i, r oi is the initial particle radius for size fraction i, C s is the drug solubility, X dT is the total amount of dissolved drug at any time t, and V is the volume of the dissolution medium. In this simulation, the diffusion layer thickness h i was set at 30 μm for a particle size r it larger than 30 μm and equal to r it for r it less than 30 μm, where r it is the radius of size fraction i at time t. In the case of r it less than 30 μm, the rate of change in the mass of dissolved drug is given by Eq. 3:
The total amount of dissolved drug X dT at any time t for the size fractions i = 1 to n is given by Eq. 4:
Equations 2 and 3 were solved by the 4th Runge-Kutta numerical integration method using Microsoft Excel with a self-designed Visual Basic Macro to calculate X dT . The true density ρ of nitrendipine was determined using the liquid-displacement method. The diffusion coefficient was estimated using the Nakanishi Correlation (Eq. 5), which refers to the empirical parameters denoted as A, I, and S in order to take into account the interactions between the solvent and the solute (26–28). The Nakanishi Correlation is given by
where A B = 2.8 and S B = 1 for water and I A = S A = 1 for nitrendipine, and the viscosity of solvent η B was determined using a U-tube Viscometer (Pinkevitch type). The molar volume V B and V A for water and nitrendipine, respectively, were estimated using Schroeder’s method (28). The parameters used for the simulation are listed in Table II.
RESULTS AND DISCUSSION
Particle Size and Morphology
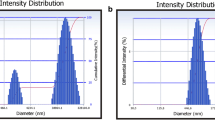
When the precipitation-ultrasonication method was used, the particle size was mainly affected by the drug concentration in organic phase. By adjusting the drug concentration, nitrendipine crystals with different particle sizes were obtained. However, if the samples were not subjected to ultrasonic treatment, the newly precipitated particles grow to the micrometer range by incorporation of molecules into the crystal lattice, agglomeration or the Ostwald-ripening mechanism after keeping the sample at 3°C for 24 h (20). The PVA, which contains an abundance of hydroxyl groups, can form hydrogen bonds with drug molecules and, thus, inhibit crystal growth. The crystal size was influenced by the PVA concentration. This will be discussed in a forthcoming publication. The particle size distribution profiles of each crystal suspension are shown in Fig. 2, and their morphology is shown in Fig. 3. From the SEM observations, it was found that the crystals which were subjected to ultrasonication had a flaky shape, while the crystals not subjected to ultrasonication had a spherical shape.

Particle size distribution profiles of nitrendipine crystals.

SEM images of nitrendipine crystals: 200 nm (a), 620 nm (b), 2.7 μm (c), 4.1 μm (d), 20.2 μm (e) and raw crystals (f). ((a) and (f) was reprinted from reference 20 with the permission of Elsevier).
XRPD and DSC Analysis
X-ray diffraction has been used to analyze potential changes in the inner structure of nitrendipine crystals. According to the X-ray diffraction patterns, as shown in Fig. 4a, it was confirmed that no substantial crystalline change was found in the nanocrystals and spherical microcrystals compared with raw crystals. However, the differences in the relative intensities of their peaks might be attributed to differences in the degree of crystallinity of the samples. DSC was also performed to analyze the different samples (Fig. 4b). In all cases, the DSC scanning of each sample showed a single sharp endothermic peak ascribed to the melting of the drug, which also indicated that there was no substantial crystalline change.

XRPD (a) and DSC (b) curves of nitrendipine crystals with different particle sizes.
Dissolution Rate and Saturation Solubility
The dissolution profiles of different nitrendipine crystals in three types of dissolution medium are shown in Fig. 5. It can be seen that the dissolution of nitrendipine was significantly improved by a reduction in particle size. At least 90% of the nanocrystals with a particle size of 200 nm dissolved in 2 min no matter what kind of dissolution medium was used. However, in the case of raw crystals, less than 10% dissolved in 2 min, and complete dissolution was not achieved until the end of the whole test period of 60 min in all types of dissolution medium. This enhanced dissolution rate can be attributed to the tremendous increase in surface area of nanocrystals available for dissolution and the reduced diffusion layer thickness (9).

Dissolution profiles of nitrendipine crystals with different particle sizes (Medium 1 (a), Medium 2 (b), FaSSIF (c). Keys: +, 200 nm; △, 620 nm; □, 2.7 μm; ♦, 4.1 μm; ▲, 20.2 μm; ■, Raw crystals; the red points represent the simulated data) (n = 3).
The dissolution rate was also affected by the type of dissolution medium. As illustrated in Fig. 5a, no significant difference in dissolution profiles was observed between crystals with a particle size ranging from 200 nm to 4.1 μm using Medium 1. The solubility of nitrendipine in Medium 1 was 30 μg/ml, and so-called sink conditions were met. The term sink conditions is defined as the volume of medium at least three times greater than that required to form a saturated solution of drug substance (29). When Medium 2 was used, the dissolution profiles of 200 nm, 620 nm and 2.7 μm still appeared to be identical, while large differences could be observed among 4.1 μm, 20.2 μm and raw crystals (Fig. 5b). The solubility of nitrendipine in Medium 2 was about 14 μg/ml. However, when FaSSIF was selected as the dissolution medium, as demonstrated in Fig. 5c, marked differences in dissolution profiles among various particle sizes were observed. The crystals dissolved more slowly in FaSSIF compared with Medium 1 and Medium 2. The solubility of nitrendipine in FaSSIF was about 5 μg/ml. The volume of dissolution medium was calculated to be larger than 1500 ml to provide sink conditions. Therefore, the sink conditions were not fully met using FaSSIF as the dissolution medium in this study, although FaSSIF had a discriminatory power superior to Medium 1 and Medium 2.
The simulation study, as indicated in Fig. 5c, showed that the simulated results were in agreement with the actually observed dissolution curves, which suggested that the dissolution of nitrendipine crystals followed the Noyes-Whitney type equation. The results also demonstrated that the assumptions of the model and the choice of parameters were appropriate. Based on the simulated results, the T 50% values (50% dissolution time) for the 200 nm and 620 nm, as shown in Table III, were calculated to be 0.9 s and 4.5 s, respectively, indicating that their dissolution rates were 5.1 × 104-fold and 1.0 × 104-fold greater than that of raw crystals, respectively. The dissolution rates of crystals with diameters of 2.7 μm, 4.1 μm and 20.2 μm were also, respectively, 237-fold, 64-fold and 11-fold greater than that of the raw crystals.
The drug solubility in the dissolution medium has a very important effect on the dissolution rate. A higher solubility may reduce the discriminative power of the dissolution medium between the different forms of poorly water-soluble drug products. The drug dissolution slows down as the concentration of dissolved drug approaches the saturation solubility in the dissolution medium. This can be explained by the previously stated Noyes-Whitney type equation (Eqs. 2 and 3), where Cs-X dT /V is the driving force of dissolution, which is influenced by the solubility and the concentration of drug in solution (X dT /V). Under non-sink conditions, X dT /V cannot be ignored.
Although the saturation solubility is normally considered a physicochemical constant, as indicated in Table II, the saturation solubility of nitrendipine increased when the particle size was reduced to the nanometer range. This increase in saturation solubility can be explained by the Freundlich-Ostwald equation (30):
where S is the saturation solubility of the nano-sized crystals, \( {S_\infty } \) is the saturation solubility of infinitely large crystals, γ is the crystal-medium interfacial tension, M is the compound molecular weight, r is the particle radius, ρ is the density, R is the gas constant and T is the temperature. Compared with large particles, nanoparticles in general possess a greater surface curvature, which produces more dissolution pressure with a corresponding increase in saturation solubility (31). The solubility of the drug is not only a function of particle size but also its crystallinity (32). The solubility of nitrendipine spherical crystals also slightly increased compared with raw crystals due to the reduction in the degree of crystallinity.
Bioavailability Study
The HPLC method and extraction process were fully validated. The plasma drug concentration-time profiles of nitrendipine resulting from the intravenous administration of nitrendipine solution and oral administration of suspensions with different particle sizes in Wistar rats are presented in Figs. 6 and 7, respectively. The pharmacokinetic parameters are shown in Table IV.

Average plasma drug concentration versus time profile after intravenous administration of nitrendipine in rats (n = 4, means ± S.D.).

Average plasma drug concentration versus time profiles after oral administration of each formulations (n = 5, means ± S.D.).
Analysis of the pharmacokinetic parameters shows several exciting and interesting phenomena. It is clear that the reduction in drug particle size from 36.6 μm to 200 nm correlates well with incremental improvements in the oral absorption of nitrendipine. There were marked differences in the pharmacokinetic profiles between microcrystals and nanocrystals. All parameters which are indicative of the rate and extent of absorption, i.e. Cmax, Tmax and AUC, showed that the oral drug absorption of nanocrystals was better than microcrystals. Student’s t-test between groups was performed using Microsoft Excel, which showed that the AUC and Cmax of crystals with a particle size in the range 200 nm to 20.2 μm were significantly increased (P < 0.01) compared with the corresponding parameters of raw crystals. The Tmax of nanocrystals was 1 h; however, when the particle size increased to 20.2 μm, the Tmax was extended to more than 3 h. The absorption rate was greatly slowed down due to the significant decrease in dissolution rate. Figure 8 shows a significant increase in the drug absolute bioavailability of nanocrystals compared with microcrystals. The absolute bioavailability of Size 1 (200 nm) was 61.4%; the AUC0→12 and Cmax values were approximately 6.2-fold and 8.7-fold greater than that of raw crystals, respectively. The absolute bioavailability of raw crystals was only 9.9%. These data provide strong confirmation that the absorption of nitrendipine is dissolution rate-limited. As for Size 2 (620 nm), the absolute bioavailability was 51.5%, still lower than that of Size 1. For microcrystals, their AUC values suggested that there was a slight increase in the fraction dose absorbed. The absolute bioavailability only increased modestly from 24.7% to 26.7% and 29.4% as the particle size decreased from 20.2 μm to 4.1 μm and 2.7 μm, respectively, although a large difference in dissolution rate can be observed in FaSSIF. It is well known that there is a critical particle size in the drug absorption for the Class II drugs, further decreasing of the particle size has no influence on the drug absorption, and the critical particle size is usually in the micrometer range (33). The original intention of this study was to find out the so-called critical particle size for nitrendipine, but the results showed that no critical particle size was observed. It seems that the absolute bioavailability of nitrendipine increased exponentially as the particle size decreased to the nanometer range, although little difference in dissolution rate was observed between nanocrystals. A similar phenomenon has also been found for the drug Aprepitant, which has a very poor aqueous solubility (16). The authors used software called GastroPlus™ to simulate the effect of the particle size of Aprepitant on the fraction dose absorbed. The simulation showed a significant deviation from the observed data for a particle size below 1 μm. Specifically, the software predicted a slight increase in absorption when the particle size changed from 5.49 to 1.9 μm. No further increase in absorption was predicted for submicron particles, which was contrary to the in vivo data. They concluded that the nanosuspension absorption could only be accurately simulated assuming a much higher solubility for the nanosuspension. From the in vivo absorption rate estimation of nitrendipine, as illustrated in Fig. 9, an absorption plateau was reached 2 h after administration of nanocrystals. However, in the case of the microcrystals, almost no absorption was observed 1 h after administration. It appears that the in vivo oral absorption of microcrystals is mainly dependent on the initial concentration of the dissolved drug in the GI tract. A linear regression analysis was performed on the Log (T50%) calculated by the dissolution simulation with the absolute bioavailability, and a good linear relationship (r 2 = 0.9695) was observed between the Log (T50%) and absolute bioavailability, as illustrated in Fig. 10.

Absolute bioavailability as a function of particle sizes of crystals.

Absorption fraction-time profiles after oral administration of nitrendipine suspensions with different particle sizes.

Linear relationship between Log (T 50% ) and absolute bioavailability (F abs ).
The absorption of drugs from the GI tract involves several different processes, including dissolution of the drug in the GI fluids, possible degradation, adsorption or complex binding in the GI fluids as well as the absorptive flux across the epithelial cells of the intestinal wall (34). The dissolution of drug in the GI tract is mainly determined by the physicochemical properties of the drug administered and physiological factors in the GI tract. However, in vitro, it is difficult to simulate the effect of physiological factors such as the fluid volume, viscosity, content of amphiphilic compounds, complex motility and possible degradation and local solubility of the drug in the GI tract; thus, there are insufficient investigations to address the effect of biochemical/physiological factors on in vivo dissolution (6,35). In order to accurately predict the in vivo absorption of a drug after oral administration from in vitro data, it is essential that the limiting factors for absorption can be modeled in vitro (36). Medium 1 and Medium 2 probably overestimated the situation in GI and, thus, did not reflect what happened in vivo. A bio-relevant medium has been suggested to simulate the gastrointestinal fluid more accurately and has been widely used for the determination of bio-relevant solubility and dissolution studies (37). From the results of the dissolution test in FaSSIF, the trend in absorption directly correlated with the dissolution data. The bio-relevant dissolution medium can be used to predict the differences in bioavailability among various formulations, although it is difficult to determine the quantitative relationship between in vivo absorption and in vitro dissolution, because such a dissolution study is still not able to accurately reflect the complex situation in the GI tract.
The dose/solubility ratio indicates whether the capacity of the GI fluids is sufficient to dissolve the entire dose administered. A dose/solubility ratio greater than 250 indicates that the conditions in the GI tract are less than optimal for drug dissolution (36). For nitrendipine, with a dose of 40 mg and a solubility of about 5 μg/ml in FaSSIF at 37°C, the dose/solubility ratio could be as high as 8000. Therefore, it is probable that sink conditions do not prevail in vivo, which means that some crystals do not dissolve instantly when they reach the GI, even if their particle size is in the nanometer range.
Many reports have been published about the bioadhesion of nanoparticles to the gastrointestinal mucosa (38–42). It has been hypothesized that the mucous gel constitutes a porous rather than a smooth adsorbent, and the adsorption kinetics may be controlled in a second phase of transport by diffusion into the mucous network (a macromolecular network model where the chains are held together by permanent entanglements of a physical and chemical nature) (42,43). The adsorption of particles to the GI tract wall showed a strong dependence on particle size by analyzing the adsorption isotherms of the particles. Particles smaller than 1 μm could penetrate into the porous adsorbent (44), as illustrated in Fig. 11a, and this is supported by a confocal microscopy study by Scherrer et al. (45) showing that fluorescently labeled nanoparticles (211 nm in diameter) could penetrate to a depth of at least 60 μm into the mucus layer of rat intestine mucosal fragments. The SEM photograph made by Damgè et al. (46) also showed that nanoparticles with a particle size of 200 nm were in close contact with the mucous network a short time after oral administration to rats. As a result, the initially undissolved nanocrystals are most likely to penetrate into the deep layer of mucus, occupy the absorption site, and dissolve rapidly so that the diffusion distance is decreased, causing an increased concentration gradient, leading to a pronounced increase in absorption. It is worth noting that the slight increase in solubility as a function of particle size is also a favorable factor for drug absorption. In the case of larger particles, such as 2 μm, the particles are adsorbed in a monolayer on the adsorbent surface, which behaves like a smooth surface (44). As illustrated in Fig. 11b, the dissolved drug molecules should travel a relatively longer distance from the crystal surface to the absorption site of the GI than in the case of nanocrystals. Furthermore, the viscosity of the gastrointestinal fluid is also an unfavorable factor for the diffusion of dissolved drug molecules. This implies that a smaller particle size can provide almost instant drug release at the absorption site, resulting in a higher local concentration, so, more drug molecules can be absorbed.

Absorption models of nitrendipine crystals: a case of crystals less than 1 μm, b case of crystals larger than 1 μm (modified after the reference [42]).
To sum up, the improved oral bioavailability of nitrendipine could be explained by a combination of the following effects: first, the drug molecules were absorbed rapidly from the gastrointestinal wall due to the significantly improved dissolution rate produced by the reduced particle size with an increased surface area and reduced diffusion layer thickness. Second, an increase in the adhesion surface area between nanoparticles and the intestinal epithelium of villi provides a direct contact with the absorbing membranes of the gut, and the immediate release of drug makes it available at the site of absorption avoiding its passage to the distal segments where the drug is non-absorbable (14) or possible degradation in gut.
CONCLUSIONS
The dissolution of nitrendipine was significantly increased by a reduction in particle size. The dissolution test in FaSSIF can discriminate between differences in dissolution rates of various particle sizes, and the simulated results were in agreement with the observed dissolution curves. Thus, FaSSIF can be used to predict their different in vivo absorption. The in vivo study demonstrated that the reduction in drug particle size from 36.6 (raw crystals) μm to 200 nm correlated well with incremental improvements in oral absorption. A good linear relationship was observed between the Log (T50%) and the absolute bioavailability of nitrendipine. The absolute bioavailability of a nanosuspension with a particle size of 200 nm was 61.4%, and the AUC0→12 and Cmax values were approximately 6.2-fold and 8.7-fold greater than that of raw crystals, respectively. In conclusion, nanocrystals were found to be an efficient method of improving the oral bioavailability of nitrendipine.
REFERENCES
Shargel L, Wu-Pong S, Yu ABC. Applied biopharmaceutics & pharmacokinetics. 5th ed. New York: McGraw-Hill; 2004.
Kataoka M, Masaoka Y, Yamazaki Y, Sakane T, Sezaki H, Yamashita S. In vitro system to evaluate oral absorption of poorly water-soluble drugs: simultaneous analysis on dissolution and permeation of drugs. Pharm Res. 2003;20(10):1674–80.
Oh DM, Curl RL, Amidon GL. Estimating the fraction dose absorbed from suspensions of poorly soluble compounds in humans: a mathematical model. Pharm Res. 1993;10(2):264–70.
Amidon GL, Lennernas H, Shah VP, Crisons JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):414–20.
Kaneniwa N, Watari N. Dissolution of slightly soluble drugs. IV. Effect of particle size of Sulfonamides on in vitro dissolution rate and in vivo absorption rate, and their ralation to solubility. Chem Pharm Bull. 1977;26(3):813–26.
Hörter D, Dressman JB. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 1997;25(1):3–14.
Noyes A, Whitney W. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1897;19:930–4.
Scholz A, Abrahamsson B, Diebold SM, Kostewicz E, Polentarutti BI, Ungell A-L, et al. Influence of hydrodynamics and particle size on the absorption of felodipine in labradors. Pharm Res. 2001;19(1):42–6.
Hintz RJ, Johnson KC. The effect of particle size distribution on dissolution rate and oral absorption. Int J Pharm. 1989;51:9–17.
Jinno JI, Kamada N, Miyake M, Yamada K, Mukai T, Odomi M, et al. Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs. J Control Release. 2006;111:56–64.
Shaw TRD, Carless JE. The effect of particle size on the absorption of digoxin. Eur J Pharmacol. 1974;7:269–73.
Hanafy A, Spahn-Langguth H, Vergnault G, Grenier P, Tubic Grozdanis M, Lenhardt T, et al. Pharmacokinetic evaluation of oral fenofibrate nanosuspensions and SLN in comparison to conventional suspensions of micronized drug. Adv Drug Deliv Rev. 2007;59(6):419–26.
Liversidge GG, Conzentino P. Drug particle size reduction for decreasing gastric irritancy and enhancing absorption of naproxen in rats. Int J Pharm. 1995;125(2):309–13.
Jia L, Wong H, Cerna C, Weitman SD. Effect of nanonization on absorption of 301029: ex vivo and in vivo pharmacokinetic correlations determined by liquid chromatography/mass spectrometry. Pharm Res. 2002;19:1091–6.
Jounela AJ, Pentikäìinen PJ, Sothmann A. Effect of particle size on the bioavailability of digoxin. Eur J Clin Pharmacol. 1975;8:365–70.
Kesisoglou F, Wu Y. Understanding the effect of API properties on bioavailability through absorption modeling. AAPS J. 2008;10(4):516–25.
Avdeef A. Absorption and drug development: solubility, permeability, and charge state. Hoboken: Wiley; 2003.
Soons PA, Breimer DD. Stereoselective pharmacokinetics of oral and intravenous nitrendipine in healthy male subjects. Br J Clin Pharmacol. 1991;32:11–6.
Sweetman SC, Blake PS. Martindale: the complete drug reference. 36th ed. London·Chicago: Pharmaceutical Press; 2009.
Xia D, Quan P, Piao H, Piao H, Sun S, Yin Y, et al. Preparation of stable nitrendipine nanosuspensions using the precipitation-ultrasonication method for enhancement of dissolution and oral bioavailability. Eur J Pharm Sci. 2010;40(4):325–34.
Löbenberg R, Krämer J, Shah VP, Amidon GL, Dressman JB. Dissolution testing as a prognostic tool for oral drug absorption: dissolution behavior of glibenclamide. Pharm Res. 2000;17(4):439–44.
Dressman JB, Lennernäs H. Oral drug absorption: prediction and assessment. New York·Basel: Marcel Dekker, Inc; 2000.
Langenbucher F. Handling of computational in vitro/in vivo correlation problems by Microsoft Excel: III. Convolution and deconvolution. Eur J Pharm Biopharm. 2003;56(3):429–37.
Perrier D, Gibaldi M. Pharmacokinetics. 2nd ed. New York: Marcel Dekker Inc; 1982.
Lu AT, Frisella ME, Johnson KC. Dissolution modeling: factors affecting the dissolution rates of polydisperse powders. Pharm Res. 1993;10(9):1308–14.
Nakanishi K. Prediction of diffusion coefficient of nonelectrolytes in dilute solution based on generalized hammond-stokes plot. Ind Eng Chem Fundam. 1978;17(4):253–6.
Cantrel L. Diffusion coefficients of molecular iodine in aqueous solutions. J Chem Eng Data. 1997;42(1):216–20.
Poling BE, Prausnitz JM, O’Connell JP. Properties of gases and liquids. 5th ed. New York: McGraw-Hill; 2000.
Brown CK, Chokshi HP, Nickerson B, Reed RA, Rohrs BR, Shah PA. Acceptable analytical practices for dissolution testing of poorly soluble compounds. Pharm Technol. 2004;28(12):56–65.
Kesisoglou F, Panmai S, Wu Y. Nanosizing-oral formulation development and biopharmaceutical evaluation. Adv Drug Deliv Rev. 2007;59:631–44.
Müller RH, Benita S, Böhm BHL. Emulsions and nanosuspensions for the formulation of poorly soluble drugs. Stuttgart: Medpharm Scientific Publishers; 1998.
Qiu Y, Chen Y, Zhang GGZ. Developing solid oral dosage forms: pharmaceutical theory and practice. Burlington: Academic; 2009.
Kaneniwa N, Watari N, Iijima H. Dissolution of slightly soluble drugs.V. Effect of particle size on gastrointestinal drug absorption and its relation to solubility. Chem Pharm Bull. 1978;26(9):2603–14.
Ehrhardt C, Kim K-J. Drug absorption studies: in situ, in vitro and in silico models. New York: Springer; 2008.
Bønløkke L, Hovgaard L, Kristensen HG, Knutson L, Lennernäs H. Direct estimation of the in vivo dissolution of spironolactone, in two particle size ranges, using the single-pass perfusion technique (Loc-I-Gut®) in humans. Eur J Pharm Sci. 2001;12(3):239–50.
Dressman JB, Reppas C. In vitro-in vivo correlations for lipophilic, poorly water-soluble drugs. Eur J Pharm Sci. 2000;11(Supplement 2):S73–80.
Augustijns P, Brewster ME. Solvent systems and their selection in pharmaceutics and biopharmaceutics. New York: Springer; 2007.
Lamprecht A, Schafer U, Lehr CM. Size-dependent bioadhesion of micro- and nanoparticulate carriers to the inflamed colonic mucosa. Pharm Res. 2001;18(6):788–93.
Ponchel G, Irache J-M. Specific and non-specific bioadhesive particulate systems for oral delivery to the gastrointestinal tract. Adv Drug Deliv Rev. 1998;34(2–3):191–219.
Yin Y, Chen D, Qiao M, Wei X, Hu H. Lectin-conjugated PLGA nanoparticles loaded with thymopentin: ex vivo bioadhesion and in vivo biodistribution. J Control Release. 2007;123(1):27–38.
Arangoa MA, Ponchel G, Orecchioni AM, Renedo MJ, Duchêne D, Irache JM. Bioadhesive potential of gliadin nanoparticulate systems. Eur J Pharm Sci. 2000;11(4):333–41.
Ponchel G, Montisci M-J, Dembri A, Durrer C, Duchêne D. Mucoadhesion of colloidal particulate systems in the gastro-intestinal tract. Eur J Pharm Biopharm. 1997;44(1):25–31.
Peppas NA, Hansen PJ, Buri PA. A theory of molecular diffusion in the intestinal mucus. Int J Pharm. 1984;20(1–2):107–18.
Durrer C, Irache JM, Puisieux F, Duchêne D, Ponchel G. Mucoadhesion of latexes. II. Adsorption isotherms and desorption studies. Pharm Res. 1994;11(5):680–3.
Scherrer D, Mooren FC, Kinne RKH, Kreuter J. In vitro permeability of PBCA nanoparticles through porcine small intestine. J Drug Target. 1993;1(1):21–8.
Damgè C, Michel C, Aprahamian M, Couvreur P, Devissaguet JP. Nanocapsules as carriers for oral peptide delivery. J Control Release. 1990;13:233–9.
ACKNOWLEDGEMENTS
We are grateful for financial support from the National Natural Science Foundation (No. 30873182) and National Basic Research Program of China (973 Program) (No. 2009CB930302).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xia, D., Cui, F., Piao, H. et al. Effect of Crystal Size on the In Vitro Dissolution and Oral Absorption of Nitrendipine in Rats. Pharm Res 27, 1965–1976 (2010). https://doi.org/10.1007/s11095-010-0200-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-010-0200-0