
Abstract
Purpose
To understand and evaluate the stability and skin permeation profiles of fentanyl reservoir systems as a function of patch age.
Methods
Drug release and skin permeation studies were performed using a modified USP apparatus 5 with a novel sample preparation technique.
Results
The amount of fentanyl present in the EVA/adhesive layer (EAL) increased from about 17% of label claim (LC) at 5 months to 25% LC at 22 months. The increase in the drug concentration was mainly observed in the peripheral EAL. Simultaneously, the alcohol content of the patch decreased as a function of patch age. A significant effect of patch age on the drug content in the EAL and the drug release from the system was observed; however, skin permeation studies did not indicate an increase in drug delivery rate.
Conclusions
Novel sample preparation technique with USP Apparatus 5 allowed determination of in vitro skin permeation rates for fentanyl transdermal patches with different designs. Permeation rates with cadaver skin as substrate were found not to change with patch age despite changing drug concentration in the EAL.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The restrictive nature of percutaneous absorption limits the use of passive transdermal delivery route to drug molecules with low molecular weight, small size, moderate lipophilicity and high therapeutic potency. This method of administration is predominantly useful with medications that are used for relatively long-term preventive treatment or maintenance therapy of chronic conditions.
The basic components of any transdermal delivery system (patch) include the drug(s) dissolved in solution or dispersed in an inert polymer matrix that provides a platform for drug release; an outer backing film of paper, plastic, or foil; and a pressure-sensitive adhesive that anchors the patch to the skin. The adhesive is covered by a release liner, which is removed before applying the patch to the skin. While the rate-limiting step in drug delivery can be either the drug release from the delivery system or its absorption into the skin, a well-designed patch system ensures that the former is the rate-limiting step in order to provide drug uptake at a predetermined rate that is independent of skin variability. There are two basic transdermal patch designs (i) matrix or monolithic where the drug is incorporated in an inert polymer matrix which controls drug release from the device (e.g., Nitro-Dur® Patch, Nicotrol® Patch [OTC], and Vivelle® transdermal), and (ii) reservoir or membrane (Fig. 1) where a rate-controlling membrane present between the drug matrix and the adhesive layer serves as the rate-limiting barrier or provides at least partial control of drug release from the device (Transderm-Nitro® Patch, Transderm Scop® Patch, Catapres®-TTS Estraderm® transdermal, Duragesic® transdermal, Nicoderm® CQ Patch, and Androderm® transdermal system).

Schematic showing the outer and inner EAL regions of the Duragesic® FTS (patch cut along the dotted line).
In 1979, the Food and Drug Administration approved the first transdermal drug delivery system (Transderm Scop® Patch), a reservoir type design. Unlike, the drug-in-adhesive type systems, reservoir systems have several components such as the rate limiting membrane and a permeation enhancer that could contribute to variability in drug release and/or drug absorption. In addition, there are several manufacturing steps that need to be monitored in order to ensure no drug leakage occurs from the reservoir. Another important component of the reservoir system is the contact adhesive (adhesive between the rate controlling membrane and the protective liner) equilibrated with drug, which acts as the immediate release layer. Following manufacture, the drug slowly migrates into the rate-limiting membrane and contact adhesive layer. To insure product quality, reservoir patches are not distributed until the drug in the adhesive layer has reached equilibrium (Alza Corporation, personal communication). According to the manufacturer of one of the reservoir patches, “Equilibration is defined as the time period in which it takes for the drug to migrate into the adhesive/EVA layers in order to meet the filed in vitro drug release criteria. There is a universal waiting period before lot clearance testing for drug release profile is conducted. The lots are then shipped to pharmacies. A small amount of drug continues to migrate within the heatseal area (the outer edges) which is not delivered to the systemic circulation. However, this rate is understood and an appropriate expiration date has been established in order to ensure that the product will meet the drug release criteria throughout the entire shelf life” (Alza Corporation, personal communication). However, there are no studies reported in the literature that investigate the magnitude of the effect, the mechanism underlying this phenomenon of drug migration or its effects on drug dissolution and drug delivery.
A fentanyl transdermal system (Duragesic®) was used as a model system for this study because of its reservoir design and the narrow therapeutic index of fentanyl. Several deaths have also been reported with fentanyl transdermal systems (1). Fentanyl is a potent synthetic opioid agonist used for the palliative treatment of late stage cancer and chronic pain. Low molecular weight (336 g/mole), high octanol: water partition coefficient (860:1), solubility in both lipids and water, short half life and a high metabolic clearance make fentanyl, an ideal candidate for transdermal delivery (2). In 1991, Janssen’s Duragesic® fentanyl transdermal system (D-FTS), a reservoir-based transdermal system with rate limiting membrane, was approved for the treatment of cancer and other chronic pains. The rate limiting membrane of D-FTS provides continuous, systemic delivery of fentanyl for 72 h from one application. Patches are available in five strengths—12.5, 25, 50, 75 and 100 mcg/h. The amount of fentanyl released per hour from each system is proportional to the surface area (25 mcg/h per 10 cm2). The composition per unit area is identical for all dosage strengths. The Duragesic patches contain 0.1 mL of alcohol per 10 cm2 as a penetration enhancer (3).
The current manuscript focuses on the evaluation of the effect of patch age on the amount of drug in the EVA/adhesive layers (EAL), drug release from the patch and skin permeation. Typically Franz diffusion cells are used to study skin permeation from TDDS with large patches being cut to fit the cells. However, because D-FTS (large reservoir type TDDS) cannot be cut and still retain their integrity, a new method was developed for the study of skin permeation characteristics. In addition, different synthetic membranes will be evaluated to identify a suitable membrane that can be used as an alternative for skin.
MATERIALS
Multiple lots of Duragesic® fentanyl transdermal systems (25 mcg/h strength containing 2.50 mg of fentanyl per patch) (expiry dates, Jul 09, April 09, Jan 09, Dec 07) were purchased from local suppliers. Silicone rubber sheet, Medical Grade (0.005 inches thick, Dimensions 12 × 12 in.)—(Previously sold by Dow Corning as SILASTIC®) was purchased from Specialty Manufacturers, Inc., Saginaw, MI and Dow Corning® 7-4107 silicone elastomer membrane was obtained from Dow Corning, Midland, MI. Ethylene vinyl acetate (EVA) membranes with 9% VA content (EVA-9; 3M CoTran 9702) and 19% VA content (EVA-19; 3M CoTran 9715) and 3M 927 adhesive transfer tape were obtained from 3M Pharmaceuticals, USA. Fentanyl USP reference standard (99.7% purity) was purchased from USP. Hanks balanced salts were purchased from Sigma Aldrich, St. Louis, MO and full thickness cadaver skin samples were obtained from National Disease Research Interchange, Philadelphia, PA.
METHODS
Drug Content in the EVA/Adhesive Layers (EAL) of Duragesic® FTS (D-FTS)
The EVA/adhesive layer has two distinct regions (Fig. 1)—peripheral EAL (beneath the heat sealed edges) measuring 8.9 cm2 and inner EAL (beneath the gel reservoir) measuring 10 cm2. Four different D-FTS lots with different expiry dates (patch ages 5 (two lots), 8 and 22 months) were tested to determine the amount of fentanyl in the EAL (Table I). The assay was performed on five replicates from each lot. The EAL layer was isolated from the reservoir gel and then cut to separate the peripheral and the inner EAL. The resulting peripheral and inner EAL were individually extracted along with their corresponding protective liner sections (peeled from the patch prior to extraction but extracted together) in 50 mL methanol at 70°C for 6 h. Extracted samples were analyzed using a Waters Acquity UPLC system with Acquity photo diode array detector (method validated in-house). All injections were performed using an autosampler with injection volume of 10 μL. Separation was accomplished using a Waters Acquity UPLC BEH-C18, 100 × 2.1 mm, 1.7 μm at 30°C with a mobile phase consisting of 25 mM Phosphate buffer (pH 2.3)/Methanol (55:45), at a flow rate of 0.35 mL/min and a detection wavelength of 206 nm.
Alcohol Content Determination
The alcohol content in the patches (5 and 22 month old lots) was determined by extraction of the alcohol from the patch into water at 50°C for 16 h. The method was validated (99.5% recovery) for loss of alcohol during the extraction procedure using a solution of known concentration. The extracted samples were analyzed by HPLC using refractive detection (4). Analytical grade water adjusted to pH 3.0 was used as the mobile phase with a flow rate of 0.8 ml/min. The injection volume used was 50 μl. Four replicate assays were performed for each patch type.
Solubility Determination
Equilibrium or saturation solubility of fentanyl was determined in water and Hanks balanced salts solution (HBSS) buffered at different pHs. Excess drug was placed in either 5 ml deionized water or HBSS at different pHs in 10 ml glass vials and sealed. Vials were placed in a shaker water bath at 32°C and agitated at 60 oscillations per minute. Samples were withdrawn at 1, 24, 48 and 184 h, filtered, diluted appropriately and assayed for drug content using the UPLC method described above. All solubility determinations were carried out in duplicate.
In Vitro Release of Fentanyl from Transdermal Systems
Apparatus
USP dissolution apparatus 5 consisting of a Distek 2100A Dissolution Apparatus with a Distek DS 4300 Dissolution Sampler was used for determining the drug release. The paddle rotation speed was 50 rpm with 600 mL of 0.1 M phosphate buffer (32 ± 0.5 C, pH 3.5) and degassed (5) serving as the dissolution medium.
Sample Preparation
Adhesive Transfer tape (6.25 × 6.25 cm) was applied to each of six stainless steel disks (Diameter = 7.5 cm). The transdermal system was applied to the adhesive on the disk such that the backing film of the patch faced the adhesive and the clear drug containing side with release liner faced up. The release liner was peeled off just before the disk assembly was introduced into the dissolution vessel. The disk assembly was carefully placed in the dissolution vessel parallel to the paddle blade with the sample side up. The bottom of the paddle was lowered to 25 ± 1 mm from the surface of the disk assembly. The dissolution vessels were protected from light using aluminum foil and black polyethylene covers. Data analysis was based on three replicates. Samples were withdrawn at predetermined time intervals (0.5, 1, 2, 3.5 5, 8, 12, 18, 24, 32, 40, 48, 56, 64 and 72 h) and assayed using the UPLC method discussed earlier.
In Vitro Drug Permeation
In Vitro drug permeation studies were performed using an Ethylene Vinyl Acetate (EVA-9) membrane (2.0 mil thickness) with 9% Vinyl Acetate (VA) content (which is equivalent to the EVA membrane used in the D-FTS) or heat separated epidermis as barriers. For D-FTS, the drug release rate across the EVA membrane is approximately equal to the average rate of drug uptake by the skin (about 4 mcg/h/cm2) (6). Three to six replicates were performed for each experiment. Full thickness human skin samples excised within 24 h postmortem were obtained and stored at −20°C and used within 3 months from procurement. Prior to analysis, the skin samples (6 × 6 cm) were thawed for 2 h at room temperature followed by heating at 60°C for 90 s. The epidermis was carefully separated using forceps, soaked in Hanks balanced salts solution for 30 min and then dried by placing between Kimwipes. The heat separated epidermis was then tested for barrier integrity by measuring TransEpidermal Water Loss (Delfin Vapometer, Delfin Technologies, Kuopio, Finland).
A USP dissolution apparatus 5 as described above was used for determining drug permeation. The dissolution media consisted of 600 mL of either 0.1 M phosphate buffer (for membrane permeation study) or Hank’s Balanced Salts Solution (for skin permeation study) with other parameters as described above.
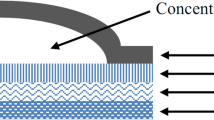
After removal of the protective liner, either the synthetic membrane or heat separated epidermis was adhered to the top of the patch (drug releasing side) (Fig. 2) such that there is no air entrapped between the patch and the membrane/skin. In addition, the membrane/skin are first adhered at one end of the disk (the other free end of the skin/membrane is held at 90° during the process) and slowly proceeding to the other end while gently pressing on the skin to prevent air entrapment. The adhesive transfer tape was sized such that it provided adhesion to the patch and the edges of the synthetic membrane/skin in order to assure no diffusion media entered the interface between the patch and the membrane/skin.

Schematic of USP dissolution apparatus 5 and Stainless steel disk assembly.
Calculations
The drug release and permeation data were best fitted according to Higuchi’s equation (7).
where Q is the amount of drug released after time t per unit exposed area. The Higuchi release or permeation rate constant, K H (for release from D-FTS or D-FTS+skin, respectively) is calculated from the slope of the linear region of the Q versus t 1/2 release profile and represented as mcg/cm2/h1/2. Higuchi release constant was calculated for each replicate and then average value calculated (The representative Higuchi plots indicate the release rate/permeation rate constant for average of all the replicates).
The average permeation rate for the time interval t y to t x (PR x→y ) are calculated from Eq. 2.
where q is the amount of drug released, and t x and t y are the times of samples x and y during the diffusion or dissolution study. In plots showing permeation versus time, PRx →y is plotted at the midpoint of t x and t y . According to the full prescribing information for Duragesic, the nominal drug delivery rate (the dose strength of the patch) reported is the average amount of drug delivered to the systemic circulation per hour across average skin (3). Hence, it would be more appropriate to calculate the average drug delivery rate in mcg/h and use that data to compare with the nominal 25 mcg/h drug delivery rate of the patches. For this reason, the release rates for predefined intervals (0–24, 24–48, 48–72, 0–72 h) (Eq. 2) were calculated for use in comparing treatments.
Statistical Analysis
Statistical Analysis was performed using JMP (version 6.0, SAS Institute) software. All Pairs, Tukey-Kramer HSD (honestly significant difference) test was used to compare skin permeation and release rates between patches of different ages. Differences were considered statistically significant when p < 0.05.
RESULTS
Drug Content in the EAL
The amount of fentanyl present in the EAL increased with time (age of the patch) until approximately its shelf life of 24 months (Table I). The values are reported in terms of % label claim or the amount of drug per square cm (represented as concentration). The fentanyl content in the EAL (inner + peripheral) increased from about 17% of label claim (LC) or 23 mcg/cm2 at 5 months to 25% LC or 33 mcg/cm2 at 22 months. During this time period (5–22 months) the drug content of the inner EAL region (directly below the gel reservoir) remained constant at ∼10% LC or ∼25 mcg/cm2 while the drug content in the peripheral EAL (heat seal area) continuously increased from 7% LC or 23 mcg/cm2 to 14% LC or 40 mcg/cm2.
Alcohol Content in the D-FTS
A validated method was used to extract alcohol from the patch. The patch was extracted in 50 mL of water for 16 h. HPLC with refractive index detection was used to measure the alcohol content in the whole D-FTS patches. For the oldest patches (22 months) the alcohol content was determined to be 47.8 ± 2.5 mg which is significantly lower (p < 0.05) than that determined for the newest patches (5 months) which contained 51.0 ± 0.3 mg.
Saturation Solubility Determination
The saturation solubility of fentanyl was determined in water and in HBSS at different pHs in order to establish an appropriate diffusion media so that sink conditions are maintained during the diffusion study; the measured values are shown in Table II. Fentanyl has a very low water solubility of 32 mcg/mL (pH 5.9), and pH has a significant effect on the solubility of fentanyl in HBSS. Since fentanyl is a weak base (pKa of 8.4), its solubility is expected to increase with decreasing pH (increased fentanyl ionization). Based on the solubility data, the maximum final concentration in the receiving media (HBSS buffered to pH < 5.5) was calculated to maintain sink conditions during the skin permeation study. A pH of 5.5 was chosen due to its close proximity to the pH of the skin.
In Vitro Drug Release and Permeation Using EVA Membrane
In vitro drug release and permeation profiles (using EVA-9 membrane) of different ages of D-FTS are represented in Figs. 3 and 4, respectively. The release data obtained were treated according to zero-order (cumulative amount of drug release versus time), first-order (log cumulative percentage of drug remaining versus time), and Higuchi (cumulative percentage of release versus square root of time) models. The linear region was selected based on the largest R 2 value and the region (time points) is kept constant for each study (dissolution, EVA permeation, skin permeation). The R2 value in all cases was greater than 0.995. Higuchi equation gave the best fit; therefore, the release rate constant was calculated from the linear portion of the Higuchi plots for dissolution and permeation data. The fitted data are presented in Table III.

Fentanyl release from D-FTS as a function of patch age. a Cumulative percent drug released plotted against time (filled symbols indicate data points used in linear regression); b cumulative drug released per unit area plotted against square root of time (filled symbols indicate data points used in linear regression; c drug release rate (0–5 h); d drug release rate (5–72 h).

Fentanyl permeation through EVA-9 membrane as a function of patch age. a Cumulative percent drug permeated plotted against time (filled symbols indicate data points used in linear regression; b cumulative drug permeated per unit area plotted against square root of time (filled symbols indicate data points used in linear regression; c drug permeation rate (0–5 h); d drug permeation rate (0–72 h).
The release rates for every time interval were calculated and the average release rates for select intervals are shown in Table IV.
In Vitro Permeation Using Heat Separated Epidermis
In vitro skin permeation profiles as a function of patch age are represented in Fig. 5. As is the case for drug release and permeation through EVA-9 membrane, permeation through skin also followed Higuchi model and hence the permeation rate constant was calculated from the slope of the Higuchi plots (A representative profile is indicated in Fig. 5b). The calculated permeation rate constant values for the 22 months old patches (28.6 ± 6.0 mcg/cm2/h1/2) and 5 months old patches (29.6 ± 3.4 mcg/cm2/h1/2) were not significantly different. The permeation rates were calculated between every consecutive sampling time points and plotted versus time (Fig. 5). The average permeation rate for 22 and 5 month old patches over the 72 h duration of patch application, about 20 ± 1 and 24 ± 6 mcg/h respectively, are not significantly different.

Fentanyl skin permeation as a function of patch age. a Cumulative percent drug permeated plotted against time (filled symbols indicate data points used in linear regression; b cumulative drug permeated per unit area plotted against square root of time (filled symbols indicate data points used in linear regression; c drug permeation rate (0–72 h).
To understand the inter-individual skin variability, replicate studies were done using samples of skin from the same person for the three different lots (22, 8 and 5 months). These data are illustrated in Fig. 6 and Table V. These data indicate that skin variability is higher than the variability of the patch as a function of its age and higher than variability observed using EVA membrane. Indeed, the permeation profiles indicate that all the three different lots (different patch ages) when studied on skin from the same individual were found to have no difference in their permeation properties. However, the values obtained for the same lot (patch age) when studied using skin samples from two different individuals, were different indicating that the variability of the skin is higher than the variability of the product.

Variability in drug permeation profiles of two lots of patches between skin samples from two subjects.
DISCUSSION
The commonly used Franz diffusion cell could not be used for the current research due to the reservoir design and large size of the D-FTS (patch can not be cut without losing structural integrity). Hence, to facilitate measurement of fentanyl permeability, a novel sample preparation technique was developed using the USP dissolution apparatus 5.
An ideal TDDS has a drug delivery rate controlled primarily by drug release from the system as opposed to being controlled by the rate of skin permeability. In this way, variability in drug delivery due to skin inconsistency is minimized. A reservoir system design has inherent secondary control on drug delivery due to its rate controlling membrane. For D-FTS, variation in the fentanyl skin permeation rate is reduced by 50% due to the presence of the rate controlling membrane (6). The drug release profiles indicate a controlled release of fentanyl for 72 h with a rate (excluding the initial burst due to the drug in the EAL) similar to that of the drug delivery rate through the cadaver skin (Table IV). Other product variables such as the amount of drug in the EAL must also be evaluated and its effect on drug delivery should be understood. According to the manufacturer of Duragesic®, the fentanyl base is incorporated only into the drug reservoir during manufacture. After manufacture, fentanyl from the reservoir migrates through the rate-controlling membrane until the fentanyl concentration in the adhesive/EVA (EAL) reaches equilibrium with that in the reservoir (6). The amount of fentanyl in the adhesive is now referred to as the loading dose (6). Since any changes in the drug concentration within the EAL may have a significant effect on drug delivery, patches having different ages (time since manufacturer) are evaluated for drug content in the EAL, drug release and skin permeation.
Another important characteristic of the reservoir system that usually does not receive adequate attention is the lateral migration of the drug into the peripheral EAL from the inner EAL which, in turn, is in equilibrium with the drug reservoir. Because of this complex design, the drug content in these two regions was analyzed separately. The concentrations (the term concentration is used to represent drug content per unit area) of the drug in the different EAL regions of the D-FTS (Table I) are calculated based on their respective areas and reported in mcg/cm2. Because the drug concentration in the inner EAL appears to remain constant with time, it was predicted that the equilibrium concentration of the drug in the EAL is about 25 ± 1 mcg/cm2. However, the drug concentration in the peripheral EAL (heat sealed area) increased from 23 to 40 mcg/cm2 indicating that there is a continuous migration of the drug towards the periphery with time and consequent migration of the drug from the reservoir gel to the inner EAL to maintain equilibration in the inner EAL. The lateral distance for migration of the drug (heat seal width) was calculated to be about 6.3 mm (for 25, 50, 75 and 100 mcg/h strengths while it is 5 mm for 12.5 mcg/h strength) while the vertical migration of the drug from the reservoir into the inner EAL is only about 0.05 mm (more than 100 fold decrease in the path length) which explains the quick equilibration of the inner EAL while there was a continuous migration into the peripheral adhesive with time until the end of the study time of 22 months. Assuming that the peripheral EAL is equilibrated at 22 months, the excess drug per square cm in the peripheral adhesive is about 51%, which can be explained by the fact that the backing membrane of the Duragesic membrane has an inner coating of EVA which is about 75% in thickness when compared to the EVA membrane and hence can hold an additional 75% drug. In addition, the presence of only 51% drug (compared to 75% excess EVA) indicates that the peripheral adhesive is not yet completely saturated. This is confirmed from the fact that the lateral migration path length into the heat sealed peripheral EAL is about 125 times that of the vertical path length into the inner EAL and hence it would be expected that it would take more than 2 years (about 125 weeks) for it to be equilibrated even if the equilibration time of the inner EAL was considered to be as low as 1 week.
It should also be realized that this phenomenon will be more important for the 25 mcg/h strength when compared to the higher doses (50, 75 and 100 mcg/h) which have the same heat seal width but increasing inner EAL area (20, 30 and 40 cm2). The heat seal width of the 12.5 mcg/h dose (which has an inner EAL area of 5 cm2) has been reduced to 5 mm in order to minimize the peripheral drug migration.
As indicated earlier, fentanyl release from the patch and the permeation through either EVA membrane or skin follows Higuchi model. Modeling the release characteristics of reservoir devices in which the transport of the drug occurs primarily by a solution-diffusion mechanism typically involves solution of Fick’s second law (unsteady-state conditions; concentration dependent flux) for the relevant boundary conditions. If the device contains dissolved active agent, the rate of release decreases exponentially with time as the concentration (activity) of the agent (i.e., the driving force for release) within the device decreases (i.e., first order release). However, if the active agent is in a saturated suspension (as in case of D-FTS), then the driving force for release is expected to remain constant (zero order) until the device is no longer saturated (8). For D-FTS, the release rate is complicated by concurrent release of ethanol (penetration enhancer) from the reservoir and hence, the ethanol concentration decreases with time, which is expected to decrease the driving force for drug permeation within the skin or the EVA membrane resulting in a deviation from zero-order kinetics. A similar release profile from fentanyl reservoir systems (Higuchi model) was reported by Mehdizadeh et al. (9).
The initial drug release (0–0.5 h) and EVA permeation (0–2 h) rates were higher in 22 month old patches than in 5 month old patches but the release rate constant and EVA permeation rate constant values (Table III) showed the opposite trend, with the 22 month old patches having a lower permeation rate constant than the 5 month old patches. The higher initial drug release rate and EVA permeation rate in older patches (22 months) may be attributed to the increased drug content in the EAL; however, the lower steady-state release and permeation rate constants and the lower average drug release rate (0–72 h; Table IV) of old patches when compared to the newer patches may be attributed to loss of drug into the peripheral EAL with time, specifically into the EVA lining of the backing which does not contain permeation enhancer and/or to the decreasing alcohol content of the system with patch age. The alcohol content in 22 month old patches was statistically lower (p < 0.05) than that found in the 5 month old patches though the difference was small.
Neither of the above effects was significant when examining skin permeation though a slight decrease in the permeation rate constant was observed in the older patches which can be again attributed to the same reasons as discussed for the EVA permeation study. The effect of excess drug migration into the peripheral EAL, although significant with regard to the initial release rate/EVA permeation rate, is not apparent in the skin permeation study since the excess drug was in the peripheral region which does not have a supply of ethanol from the gel reservoir (skin permeation enhancer) and represents only 14% of the total drug available. Another possible reason for the differential effects is that fentanyl absorbed into the skin in the initial time period accumulates and forms a depot in the skin, a phenomenon that does not occur for EVA. However, the observed lower steady-state permeation rate constant (though not significant at p < 0.05) observed in older patches may be indeed due to the lower alcohol content in the older patches. In other words, the observed differences in skin permeation are sufficiently small that they are obscured by the high skin variability. In a study to assess skin variability (performed in duplicate), it was found that the skin sample variability was higher than the variability from the patch age (Fig. 6, Table V). Patches of different ages when compared using the skin from the same donor (samples from adjacent areas) showed no significant difference in their permeation rate constants. In addition, permeation rate constants obtained from patches from the same lot (same age) but two different donors showed a significant difference; however, since the study was performed only in duplicate, no firm conclusions about the role of skin variability can be made.
CONCLUSIONS
A novel sample preparation technique was developed and successfully used to study the skin permeation of reservoir type systems. EVA-9 membrane is suggested as being well suited to study TDDS lot-to-lot variability since the membrane itself exhibits good reproducibility and is able to mimic the permeation characteristics (but not the depot effect) of the skin.
The current study demonstrated that drug molecules continuously diffuse into the peripheral EAL of the reservoir type transdermal system. This phenomenon may be predominantly due to the drug migration into the EVA lining of the backing membrane in the heat seal area (peripheral EAL). Patch age had a significant effect on drug content in the peripheral EAL and drug release rates from the system; however, the increased drug content in the peripheral EAL with patch age did not have a significant effect on the skin permeation rate. Indeed, there was a decrease in the total alcohol content of the older patches when compared to the newer patches which resulted in a small decrease (not statistically significant) in the permeation rate constant of the system with increasing patch age. With increasing patch age, there are two competing variables with respect to skin permeation, increased drug in peripheral EAL, which should yield higher permeation (given that the absence of alcohol in the peripheral region does not affect the drug permeation rate significantly from this region); and decreased total alcohol content of the patch, which should reduce permeation. It appears that, for the reservoir TDDS studied here, these opposing effects have canceled each other. Though a significant in vitro effect was not observed in this particular case (Duragesic FTS), it is important to monitor and determine the amount of the drug in the different EAL segments of all reservoir-based systems in order to ensure quality, safety and efficacy of such systems. In addition, if the patient dose is increased by applying multiple patches, then this affect will be exacerbated since multiple patches have more peripheral heat seal area when compared to unit patch of same strength. Because skin permeation is drug, dose, dosage form design and formulation dependent, it is important to incorporate a risk assessment plan for transdermal drug products, especially for reservoir-based systems in order to ensure quality, safety and efficacy of such systems, many of which are designed to deliver potent or narrow therapeutic index drugs.
References
http://www.fda.gov/bbs/topics/NEWS/2005/NEW01206.html. FDA Issues Public Health Advisory on the Fentanyl Patch. In 2005.
R. B. R. Muijsers, and A. J. Wagstaff. Transdermal Fentanyl an updated review of its pharmacological properties and therapeutic efficacy in chronic cancer pain control. Drugs. 61:2289–2307 (2001). doi:10.2165/00003495-200161150-00014.
L. P. Janssen Pharmaceutica Products. Duragesic (fentanyl transdermal system) Full Prescribing Information. In 2007.
A. Aguiar, R. A. A. Nascimento, L. P. Ferreti, and A. R. Gonçalves. Determination of organic acids and ethanol in commercial vinegars. Braz. J. Food Technol. 5:52–56 (2005).
T. W. Moore. Dissolution testing: A fast, efficient procedure for degassing dissolution media. Dissolution Technol. 3:3–5 (1996).
S. K. Gupta, M. Southam, R. Gale, and S. S. Hwang. System functionality and physicochemical model of fentanyl transdermal system. Journal of Pain and Symptom Management. 7:S17–S26 (1992). doi:10.1016/0885-3924(92)90049-N.
T. Higuchi. Rate of release of medicaments from ointment bases containing drug in suspension. J. Pharm. Sci. 50:874–875 (1961). doi:10.1002/jps.2600501018.
T. DeV Naylor. Polymer properties. In G. J. C. AllenBevington (ed.), Comprehensive Polymer Science, Pergamen, Oxford, 1988.
A. Mehdizadeh, T. Toliate, M. R. Rouini, S. Abashzadeh, and F. Dorkoosh. Design and in vitro evaluation of new drug-in-adhesive formulations of fentanyl transdermal patches. Acta Pharm. 54(4):301–317 (2004).
Acknowledgements
The authors would like to acknowledge John Black and Dr. Michael Trehy at the Division of Pharmaceutical Analysis, FDA for their help with the studies associated with this manuscript. The authors would also like to thank the scientists and regulatory and scientific personnel of the manufacturer of Duragesic fentanyl transdermal systems and Dr. Ravi Harapanhalli for their valuable comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Prodduturi, S., Smith, G.J., Wokovich, A.M. et al. Reservoir Based Fentanyl Transdermal Drug Delivery Systems: Effect of Patch Age on Drug Release and Skin Permeation. Pharm Res 26, 1344–1352 (2009). https://doi.org/10.1007/s11095-009-9843-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-009-9843-0