
Abstract
Purpose
Single nucleotide polymorphisms (SNPs) of the ATP-binding cassette (ABC) transporter ABCG2 gene have been suggested to be a significant factor in patients’ responses to medication and/or the risk of diseases. We aimed to evaluate the impact of the major non-synonymous SNP Q141K on lysosomal and proteasomal degradations.
Methods
ABCG2 WT and the Q141K variant were expressed in Flp-In-293 cells by using the Flp recombinase system. Their expression levels and cellular localization was measured by immunoblotting and immunofluorescence microscopy, respectively.
Results
The protein level of the Q141K variant expressed in Flp-In-293 cells was about half that of ABCG2 WT, while their mRNA levels were equal. The protein expression level of the Q141K variant increased about two-fold when Flp-In-293 cells were treated with MG132. In contrast, the protein level of ABCG2 WT was little affected by the same treatment. After treatment with bafilomycin A1, the protein levels of ABCG2 WT and Q141K increased 5- and 2-fold in Flp-In-293 cells, respectively.
Conclusions
The results strongly suggest that the major non-synonymous SNP Q141K affects the stability of the ABCG2 protein in the endoplasmic reticulum and enhances its susceptibility to ubiquitin-mediated proteasomal degradation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Pharmacogenomics dealing with heredity and response to drugs is the part of science that attempts to explain patient to patient variability of drug responses and seeks for the genetic basis of such variations or differences (1). As a means to implementing personalized medicine, it is critically important to understand the molecular mechanisms underlying inter-individual differences in the drug response, namely, pharmacological effects vs. side effects. The genetic polymorphisms of drug transporters are currently being studied in several laboratories worldwide. It is expected that efforts to discover and characterize drug transporter gene polymorphisms may lead to diagnostic tests for discriminating between different gene alleles and better strategies for the molecular design of new drugs.
Clinical relevance is implicated between the genetic polymorphisms of the ATP-binding cassette (ABC) transporter ABCG2 and individual differences in drug response (2–7). Sequencing of the ABCG2 gene from human samples has revealed over 80 different, naturally occurring sequence variations (5–16). The most extensively studied of those SNPs with potential clinical relevance is 421 C>A, which results in a glutamine to lysine substitution (Q141K) in the ABCG2 protein. The Q141K SNP was identified with varying frequencies in different ethnic groups and was found to be the most prevalent in Japanese and Chinese populations (approximately 30% in allele frequency) (2). Q141K has been associated with lower levels of protein expression and impaired transport in vitro; however, some controversies exist in the publications characterizing this SNP (12–18). The polymorphism has been studied in vivo; patients carrying the SNP were found to have elevated plasma levels of gefitinib and diflomotecan as well as increased bioavailability of oral topotecan (19–21). Furthermore, the Q141K SNP was reportedly associated with a higher incidence of diarrhea in non-small cell lung cancer patients treated with gefitinib (22).
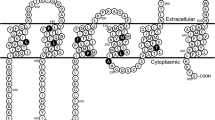
Human ABCG2 (BCRP/MXR1/ABCP) (23–25) is a so-called “half ABC transporter” bearing a single ATP-binding fold at the NH2-terminus and six transmembrane domains. Human ABCG2 exists in the plasma membrane as a homodimer bound through disulfide-bonded cysteine residues (26–30). The expression level of ABCG2 is regulated by both synthesis and degradation of the protein. Recently, our group has demonstrated that the formation of an intramolecular disulfide bond between Cys592 and Cys608 (Fig. 1) and the N-glycosylation at Asn596 are critical check points for the stability and degradation of the de novo synthesized ABCG2 protein (31). Furthermore, certain SNPs, such as F208S and S441N, were found to greatly affect the stability of ABCG2 in the endoplasmic reticulum (ER) and to enhance the protein degradation rate via ubiquitination and proteasomal proteolysis (32). In this context, it has been hypothesized that the reduced expression levels of the Q141K variant may be due to the ubiquitin-mediated proteasomal degradation.

Schematic illustration of the protein structure of human ABCG2. The sites of three non-synonymous SNPs, Q141K, F208S and S441N, are indicated. N-linked glycosylation occurs on Asn596 (N596). Cys603 is required for homodimer formation, whereas Cys592 and Cys608 are required for the formation of an intramolecular disulfide bond. A, B, and C indicate the motifs of Walker A (amino acids #80–86), Walker B (amino acids #205–210), and signature C (amino acids #186–200).
The ER system is the site where newly synthesized secretory and membrane proteins are folded and assembled under a stringent quality control system (33,34). Efficient quality control systems have evolved to prevent incompletely folded molecules from moving along the secretory pathway (35–38). Accumulation of misfolded proteins in the ER would detrimentally affect cellular functions. It is likely that misfolded ABCG2 proteins are removed from the ER by retrotranslocation to the cytosol compartment and degradation by the ubiquitin–proteasome system (39). This process is known as endoplasmic reticulum associated degradation (ERAD). In the present study, we examined the potential link between the nonsynonymous SNP and the stability of ABCG2 protein. We here provide evidence that the major nonsynonymous SNP variant Q141K undergoes both ubiquitin-mediated protein degradation in proteasomes and lysosomal proteolysis and that the protein level of the Q141K variant is thereby significantly reduced.
MATERIALS AND METHODS
Chemicals and Biological Reagents
The following reagents and drugs were purchased from the commercial sources indicated in parentheses: l-glutamine (Wako Pure Chemical Industries, Ltd., Osaka, Japan); Tris and high-glucose Dulbecco’s modified Eagle’s medium (D-MEM) (Nacalai Tesque, Inc., Kyoto, Japan); fetal calf serum (FCS) (ICN Biomedicals, Inc., Aurora, OH, USA); antibiotic–antimycotic cocktail solution, and hygromycin B (Invitrogen, Carlsbad, CA, USA); MG132 (Sigma-Aldrich Co., St. Louis, MO, USA); N-glycosidase F (PNGase F) (New England Biolabs, Inc., Ipswich, MA, USA); the protease inhibitor cocktail Complete™ (Roche Ltd., Mannheim, Germany). All other chemicals used were of analytical grade.
Preparation of Plasmids Carrying ABCG2 Q141K Variant cDNA
Wild-type (WT) ABCG2 cDNA inserted into the pcDNA5/FRT plasmid was used as the template, and the nonsynonymous SNP Q141K variant was generated by using the QuikChange® Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). Polymerase chain reaction (PCR) was carried out in an iCycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA) by using PfuTurbo® DNA polymerase, the ABCG2-pcDNA5/FRT plasmid, and specific primers. The PCR was initiated by incubation at 95°C for 30 s and then followed by 12 cycles of reactions at 95°C for 30 s, at Tm for 1 min, and at 68°C for 14 min. After the PCR, the reaction mixture was incubated with Dpn I endonuclease at 37°C for 1 h to digest the original template plasmid. The variant cDNA generated in the pcDNA5/FRT plasmid was subjected to nucleotide sequence analysis (Hitachi, Ltd., Tokyo, Japan).
Expression of ABCG2 WT and Q141K Variant in Flp-In-293 Cells
Flp-In-293 cells were transfected with the ABCG2-pcDNA5/FRT plasmid, the Flp recombinase expression plasmid pOG44, and LipofectAmine™-2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions (see Fig. 2A). Single colonies resistant to hygromycin B (Invitrogen, Carlsbad, CA, USA) were picked and subcultured. Selection of positive colonies was performed by immunoblotting, as described below. The resulting cells are described as Flp-In-293/ABCG2 cells throughout this manuscript. Mock cells (Flp-In-293/Mock) were prepared by transfecting Flp-In-293 cells with pcDNA5/FRT and pOG44 plasmids in the same manner as described above.

Flp-mediated integration of the ABCG2 cDNA into FRT-tagged genomic DNA (A), the mRNA and protein levels of ABCG2 WT and Q141K (B) and the relationship between the relative intensity of ABCG2 immunoreactivity and the amount of cellular proteins (C). A Genomic DNA of Flp-In-293 cells contains a single integrated Flp recombination target (FRT) site. The cells were co-transfected with the pcDNA5/FRT vector carrying the ABCG2 cDNA and the Flp recombinase expression plasmid pOG44. Flp recombinase mediates insertion of the expression construct with the ABCG2 cDNA into the genome at the integrated FRT site through site-specific DNA recombination. B The mRNA level was analyzed by RT-PCR with total RNA extracted from Flp-In-293 cells expressing ABCG2 WT or Q141K. For comparison of the protein levels, the cell lysate of each cell population was treated with PNGase F and then analyzed by immunoblotting with the ABCG2-specific monoclonal antibody (BXP-21) or the GAPDH-specific antibody. The level of the Q141K variant protein was normalized to the WT level. C Relationship between the ABCG2 protein level and the intensity of chemiluminescence. Cell lysate samples (2.5, 5, 10 and 20 μg protein) of Flp-In-293/ABCG2 WT cells were treated with PNGase F and then analyzed by immunoblotting. The signal intensity of ABCG2 was plotted as a function of the logarithmic value of protein.
Flp-In-293 cells expressing ABCG2 WT or Q141K, named Flp-In-293/ABCG2 (WT) or Flp-In-293/ABCG2 (Q141K), were maintained in high-glucose Dulbecco’s modified Eagle’s medium (D-MEM) supplemented with 10% (v/v) heat-inactivated fetal calf serum (ICN Biomedicals, Inc., Aurora, OH, USA), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 250 ng/ml amphotericin B, and 100 μg/ml hygromycin B at 37°C in a humidified atmosphere of 5% (v/v) CO2 in air. The number of viable cells was determined from counts made in a hemocytometer with Trypan Blue dye exclusion.
Detection of mRNA by RT-PCR
Total RNA was extracted from cultured cells with NucleoSpin® RNA II (MACHEREY-NAGEL GmbH & Co. KG, Duren, Germany) according to the manufacturer’s protocol. cDNA was prepared from the extracted RNA in a reverse transcriptase reaction with the High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA) and random hexamers according to the manufacturer’s instructions. The mRNA level of ABCG2 was determined by PCR in an iCycler™ thermal cycler (BIO-RAD, Hercules, CA, USA) with the following specific primer sets: ABCG2 (5′-GATCTCTCACCCTGGGGCTTGTGGA, 5′-TGTGCAACAGTGTGATGGCAAGGGA). The PCR reaction consisted of hot-start incubation at 94°C for 2 min and 30 cycles of 94°C for 30 s, 59°C for 30 s, and 72°C for 30 s. The PCR products were then separated by 1.2% (w/v) agarose gel electrophoresis and detected with ethidium bromide under UV light.
Immunoblotting to Detect ABCG2 Proteins
The ABCG2 protein expressed in Flp-In-293 cells was detected by immunoblotting with BXP-21 (ALEXIS Co., Lausen, Switzerland), a specific antibody to human ABCG2. Cells were rinsed with ice-cold PBS and subsequently treated with lysis buffer containing 50 mM Tris/HCl (pH 7.4), 1% (w/v) Triton X-100, 1 mM DTT, and a protease inhibitor cocktail (Roche Ltd., Mannheim, Germany). The samples were homogenized by passage through a 27-guage needle and then centrifuged at 800×g for 10 min at 4°C. For glycosidase treatments, 20 μg of protein of the cell lysate sample was incubated with 20 U of PNGase F at 37°C for 10 min.
Equal amounts of the resulting cell lysate (10 μg of protein) were subjected to SDS-PAGE in the presence of mercaptoethanol (see “RESULTS AND DISCUSSION” for more details). Briefly, proteins were separated by electrophoresis on 7.5% (w/v) polyacrylamide gels and then electroblotted onto Hybond-ECL nitrocellulose membranes (Amersham, Buckinghamshire, UK). Immunoblotting was performed by using BXP-21 (1:1,000 dilution) as the first antibody and anti-mouse IgG-horseradish peroxidase (HRP)-conjugate (1:3,000 dilution; Cell Signaling Technology, Beverly, MA, USA) as the secondary antibody. HRP-dependent luminescence was developed by using Western Lighting Chemiluminescent Reagent Plus (PerkinElmer Life Sciences, Boston, MA, USA) and detected in a Lumino Imaging Analyzer FAS-1000 (TOYOBO, Osaka, Japan).
Based on the amino acid sequence (NM_004827) of human ABCG2, the molecular weight of non-glycosylated ABCG2 WT was calculated to be 72,314 by using the ExPASY Compute pI/Mw tool (http://us.expasy.org/tools/pi_tool.html). This molecular weight was referred as the non-glycosylated nascent peptide (monomer) of ABCG2.
Immunofluorescence Analysis for Expression Levels and Distribution of Intracellular ABCG2
ABCG2-expressing Flp-In-293 cells (2 × 104 cells) were seeded onto 96-well SensoPlates™ (Greiner Bo-one Co., Ltd., Tokyo, Japan) in which the bottom of each well had been coated with mouse collagen type IV (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) at a density of 1.6 μg/well. In the plate wells, cells were pre-cultured for 24 h under the above-mentioned culture conditions and then incubated in the presence or absence of 2 μM MG132 for 24 h. Thereafter, cells were fixed with 4% paraformaldehyde in PBS at room temperature for 20 min and then cell membranes were permeabilized by incubation with 0.02% Triton X-100 in PBS at room temperature for 5 min. To block free aldehyde groups in the formaldehyde, cells were treated with glycine (10 mg/ml) in PBS at room temperature for 10 min followed by a further incubation with 0.5% (w/v) BSA in PBS at room temperature for 1 h. To detect the ABCG2 protein, cells were treated with ABCG2-specific monoclonal antibody BXP-21 antibody (1:1,000 dilution) as the first antibody and subsequently with Alexa Fluor 488-conjugated anti-mouse IgG antibody (1:1,000 dilution; Invitrogen, Carlsbad, CA, USA).
To selectively detect the ABCG2 protein localized on the plasma membrane of cells, we used ABCG2-specific monoclonal antibody 5D3 (1:1,000 dilution; R&D Systems, Inc., Minneapolis, MN) as the first antibody, since it reacts with an epitope in the extracellular loop of ABCG2 (40). During the incubation with the 5D3 antibody, we avoided treatments with 0.02% Triton X-100 in order to keep the plasma membrane impermeable to the antibody. In the same preparations, nuclear DNA was stained with Hoechst 33342 (1 μg/ml; Invitrogen, Carlsbad, CA, USA) in PBS containing 0.5% (w/v) BSA.
Immunofluorescence of the Flp-In-293 cells was detected with a newly developed system of confocal fluorescence microscopy (Yokogawa Electric Corp., Kanazawa, Japan). The fluorescence of Alexa Fluor 488 and Hoechst 33342 were observed with excitation laser light at 488 and 405 nm, respectively. To quantitatively analyze the distribution of the ABCG2 protein localized on the plasma membrane or in the cytosol compartment, the immunofluorescence images captured by the confocal fluorescence microscopy system were processed by means of computer software, where a single immunofluorescent cluster with a diameter of 0.5 to 10 μm was counted as one ABCG2-positive dot. Such digital counting of ABCG2-positive dots was performed over 400 cells for each immunofluorescence preparation. The resulting data were accumulated and then statistically analyzed.
Statistical Analysis
Statistical analyses were performed by using Microsoft Excel 2003 software (Microsoft Co., Redmond, WA, USA). The statistical significance of differences was determined according to the Student’s t test. P values <0.01 were considered statistically significant.
RESULTS AND DISCUSSION
Protein Expression Levels of ABCG2 WT and Q141K in Flp-In-293 Cells
We previously found that protein expression levels varied among those SNP variants (18). It was speculated that such reduced protein levels are associated with instability and/or degradation of those variant proteins. The Q141K variant is reportedly associated with lower levels of protein expression; however, little is known about the underlying molecular mechanism. In the present study, ABCG2 WT and the Q141K variant were individually expressed in Flp-In-293 cells by using the Flp recombinase system, where one single copy of the cDNA encoding either the WT or the variant was integrated into the genomic DNA at the designated FRT site prepared in chromosome 12 (Fig. 2A).
As shown in Fig. 2B, mRNA levels of ABCG2 WT as well as Q141K were evenly represented in Flp-In-293 cells, where the mRNA levels of ABCG2 and GAPDH were measured by RT-PCR. Both WT and Q141K proteins were found as glycosylated homodimers. To quantify the levels of ABCG2 WT and Q141K proteins, we treated the samples with PNGase F and mercaptoethanol (ME) to remove the glycomoieties and to break the cysteinyl disulfide bond forming the homodimers (Fig. 2B). Since there was a linear relationship between the signal intensity of immunoblotting and the logarithmic value of the amount of ABCG2 protein applied to the electrophoresis gels (Fig. 2C), the expression level of ABCG2 in cell lysate samples could be quantitatively estimated based on the linear relationship (41). As demonstrated in Fig. 2B, the protein level of the Q141K variant was about 45% of the ABCG2 WT protein level.
Effect of MG132 and Bafilomycin A1 on the Protein Expression Levels of ABCG2 WT and Q141K Variant
We investigated the effect of MG132, an inhibitor of proteasomal proteolysis, on the protein expression levels of those SNP variants. Flp-In-293 cells expressing WT or Q141K were incubated in the presence of MG132 at a concentration of 2.0 μM for 24 h, and then cell lysate samples were immediately prepared. This incubation condition (2.0 μM MG132 for 24 h) was selected as the optimal one based on our previous studies (Fig. 3 in Ref. (32)). Protein expression levels of the WT and the Q141K variant were determined by immunoblotting after PNGase F treatment in the same way as described above. As shown in Fig. 3A, the protein level of the Q141K variant was approximately two-fold enhanced by treatment with the proteasome inhibitor MG132. In contrast, the protein level of ABCG2 WT was little affected by MG132 treatment (Fig. 3A). Similar results were obtained with clasto-lactacystin beta-lactone (data not shown). These results suggest that the Q141K variant undergoes proteasomal proteolysis but the WT does not.

Effect of MG132 (A) or bafilomycin A1 (B) on the protein levels of ABCG2 WT and Q141K variant. A Flp-In-293 cells expressing ABCG2 WT or Q141K were incubated with 2 μM MG132 for 0 and 24 h. ABCG2 protein levels were analyzed by immunoblotting after PNGase F treatment. B Flp-In-293 cells expressing ABCG2 WT or Q141K were incubated with 10 nM bafilomycin A1 for 0, 12, and 24 h. ABCG2 protein levels were analyzed as described above. Data are expressed as mean values ± SD in triplicate experiments. Statistical significance (*P < 0.01) was evaluated by Student’s t test.
Interestingly, the protein level of ABCG2 WT increased more than 5-fold when cells were treated with bafilomycin A1, which inhibits lysosomal degradation (Fig. 3B). The Q141K variant protein level was only two-fold enhanced by the same treatment (Fig. 3B). It is suggested that the WT is degraded substantially in lysosomes and that the Q141K variant protein undergoes degradation in both proteasomes and lysosomes.
Effect of MG132 on the Cellular Localization of ABCG2 WT and Q141K Variant
Fig. 4A depicts the immunofluorescence images of Flp-In-293 cells expressing ABCG2 WT or Q141K that were incubated with or without 2 μM MG132 for 24 h. The ABCG2 protein was probed with either BXP-21 or 5D3 antibody. By using the BXP-21 antibody, which recognizes an epitope (amino acids 271–396) in the intracellular loop of ABCG2, we detected the localization of ABCG2 both at the plasma membrane and within intracellular compartments. In contrast, with the 5D3 antibody, which recognizes an epitope in the extracellular loop of the ABCG2 protein (40), we could observe that ABCG2 was localized solely at the plasma membrane.


Immunocytochemical staining of Flp-In-293 cells expressing ABCG2 WT or Q141K proteins (A) and statistical data to analyze the effects of MG132 on the cellular localization of ABCG2 WT or Q141K proteins (B, C). A Cells were incubated with or without 2 μM MG132 for 24 h. ABCG2 proteins were immunologically detected with an ABCG2-specific monoclonal antibody (either BXP-21 or 5D3) and Alexa Fluor 488 (green). Cellular nuclei were stained by Hoechst 33342 (blue), as described in “MATERIALS AND METHODS”. The horizontal white bar corresponds to 20 μm. B Based on the data shown in C, the average intensity per cell has been calculated. Data are expressed as mean values ± SD (*P < 0.005). C Based on the immunofluorescence microscopic images, the intensity of ABCG2-positive dots was measured for more than 300 cells for each preparation. The number of cells is plotted as a function of total intensity per cell.
As demonstrated in Fig. 4A, the ABCG2 protein was labeled with green fluorescence dye (Alexa Fluor 488), whereas DNA in the nuclei was stained with Hoechst 33342 (blue fluorescence). In the case of ABCG2 WT, strong green fluorescence was observed at the plasma membrane and within intracellular compartments in Flp-In-293 cells expressing ABCG2 WT. The localization and intensity of the WT was not greatly affected by the MG132 treatment.
In contrast with the WT, immunofluorescence of the Q141K variant was relatively weak at the plasma membrane as well as within intracellular compartments. After MG132 treatment, however, the localization of the ABCG2 variant protein in intracellular compartments was enhanced. The 5D3 antibody-immunofluorescence image demonstrated even more clearly that MG132 treatment enhanced the localization of the Q141K variant protein at the plasma membrane (Fig. 4A).
Fig. 4B and C depict the statistical data on the cellular localization of ABCG2 WT and Q141K. The data were acquired from a large number of cells, as described below. In each cell, immunofluorescent clusters with a diameter of 0.5 to 10 μm were counted as ABCG2-positive dots. The intensity of each ABCG2-positive dot cluster was counted over 300 cells for each immunofluorescence preparation. We analyzed the digital data to exhibit as histograms (Fig. 4C), in which the number of cells is represented as a function of the intensity of each cluster of ABCG2-positive dot per each cell. Marked differences were observed after the MG132 treatment in terms of the cellular localization and/or amount of the Q141K variant protein. It is noteworthy that the protein level of the Q141K variant increased in the plasma membrane after the MG132 treatment (Fig. 4B).
Effect of MG132 on the Drug Resistance of Flp-In-293 Cells Expressing ABCG2 Q141K Variant
Under the standard incubation conditions, Flp-In-293 cells expressing the Q141K variant exhibited a cellular resistance to SN-38, but their resistance profile was relatively weaker than that of cells expressing the WT (Fig. 5). This may be due to the lower expression level of the Q141K variant protein as compared with the ABCG2 WT in Flp-In-293 cells.

Drug resistance profiles of Flp-In-293 cells expressing ABCG2 WT or Q141K (A) and the effect of MG132 on the SN-38 resistance of Flp-In-293 cells expressing Q141K (B). A Cells were incubated with SN-38 at different concentrations for 72 h in the absence of MG132. Cell viability was determined by MTT assay. Data are expressed as mean values ± SD in multi-replicated experiments (n = 4). *P < 0.01 as compared with Flp-In-293 cells expressing ABCG2 WT. B Cells were incubated with SN-38 at different concentrations for 24 h in the presence or absence of 2 μM MG132. Cell viability was determined as described above. Data are expressed as mean values ± SD in multi-replicated experiments (n = 8). *P < 0.01 as compared with Flp-In-293 cells expressing ABCG2 Q141K treated with MG132.
It was of great interest to know how the inhibition of proteasomal protein degradation by MG132 affects the drug resistance of cells expressing the Q141K variant protein. As demonstrated in Fig. 5B, the MG132 treatment enhanced cellular resistance to SN-38 in Flp-In-293 cells expressing the Q141K variant, whereas little was changed in cells expressing ABCG2 WT, even after the MG132 treatment (data not shown). Thus, it is suggested that the Q141K variant sorted to the plasma membrane domain after the MG132 treatment was functionally active to extrude SN-38 out of the cells.
Nonsynonymous Polymorphisms of ABCG2 and Proteasomal Degradation
Hitherto a total of 17 nonsynonymous polymorphisms have been reported for the ABCG2 gene (2). In the previous study, we functionally validated those SNP variants (18). Based on our functional validation, the above-mentioned non-synonymous polymorphisms as well as two acquired mutants (R482G and R482T) of ABCG2 were classified into four groups (18). The nonsynonymous SNP variants of Q126stop, F208S, S248P, E334stop, S441N, and F489L were defective in the active transport of methotrexate and hematoporphyrin (42). Furthermore, the F208S, S248P, F431L, S441N, and F489L variants exhibited greatly altered protein expression levels and drug resistance profiles (18). In particular, expression levels of the F208S and S441N variant proteins were markedly low. We recently found that such reduced protein levels were associated with the instability and ubiquitin-mediated proteasomal degradation of those variant proteins (32).
In our functional validation, the Q141K variant and the WT showed very similar substrate specificities and drug resistance profiles when they were expressed in Sf9 insect cells (42) and Flp-In-293 cells (18). The level of the Q141K variant protein was significantly lower than that of the WT, however when it was expressed in Flp-In-293 cells (Fig. 2B) and in other mammalian cell lines (12–18). Based on those recent findings, we have examined the contribution of proteasomal degradation to the lower-level expression of the Q141K variant by testing the effect of MG132. In fact, MG132 enhanced both the protein level of the Q141K variant (Figs. 3A and 4A) and the drug resistance profile of the Q141K-expressing Flp-In-293 cells (Fig. 5B). It is strongly suggested that the lower protein expression level of the Q141K variant is due to both ubiquitin-mediated proteasomal degradation and the lysosomal proteolysis.
Protein Quality Control in ER
The ER is the site for synthesis and maturation of proteins destined for the plasma membrane, for the secretory and endocytic organelles, and for secretion (34). While the native conformation of a protein lies encoded in its primary amino acid sequence, the ER greatly enhances protein folding efficacy (43). Accumulating evidence suggests that mammalian secretory and membrane proteins are synthesized and translocated into the ER. Misfolded proteins are considered to be removed from the ER by retrotranslocation to the cytosol compartment and then degraded by the ubiquitin–proteasome system. This process is known as endoplasmic reticulum-associated degradation (ERAD) (37). We recently provided evidence that the disulfide bond formation and N-glycosylation of ABCG2 are critical check points in protein folding in the ER (31) (Nakagawa et al., unpublished data), while N-linked glycosylation per se is NOT essential for trafficking of ABCG2 to the plasma membrane, protein expression, or transport activity (44,45). On the other hand, the substitution of Gln141 to Lys141 owing to the 421C>A SNP may destabilize the cytosolic loop of the ABCG2 protein and thereby enhance ubiquitin-mediated proteasomal degradation (Fig. 6).

Schematic illustration of plausible pathways for protein folding and degradation of ABCG2 WT and Q141K variant protein. BiP(Grp78) is one of the abundant ER chaperones and is closely related to cytosolic Hsp70. Because of its location, BiP can immediately interact with the unfolded nascent peptide chain and hence contribute to the translocation of nascent chains into the ER (49). BiP reportedly has an ATPase domain and a peptide-binding domain that coordinate cycles of ATP hydrolysis and ADP exchange, which stimulate the binding and release of the unfolded protein, respectively (50). During de novo synthesis in the ER, cysteine disulfide bonds are formed and oligosaccharides are added to asparagine (N-glycosylation) or serine residues (O-glycosylation) of glycoproteins. The ER has a unique oxidizing potential that supports disulfide bond formation during protein folding (51). In addition to the well-known oxidoreductases, such as PDI and ERp57, many novel oxidoreductases have been identified over the past few years whose functions and substrates are unknown (52,53). Some of them are likely to be involved in the formation of disulfide bonds at Cys592, Cys603, and Cys608 in the ABCG2 protein. Furthermore, N-linked glycans are added en block to proteins as “core oligosaccharides” (Glc3Man9GlcNAc2). In the ABCG2 protein, Asn596 is an N-glycosylation site (44,45). Calnexin (CNX) is located near the translocon and can interact with nascent peptide chains of N-glycosylated proteins. N-linked glycans are subjected to extensive modification as glycoproteins mature and move through the ER via the Golgi apparatus to their final destination, for example the plasma membrane. After remaining in the plasma membrane domain for a certain period, ABCG2 WT is degraded by the endosome–lysosome pathway. In contrast, Q141K undergoes both lysosomal proteolysis and ubiquitination-mediated proteasomal degradation. The proteasomal degradation is inhibited by MG132.
At present, however, it remains unclear how misfolded membrane proteins are selected and destroyed during ERAD. Chaperones are considered to solubilize aggregation-prone motifs. In the case of the yeast ABC transporter Ste6p, a 12-transmembrane protein, it has recently been shown that Hsp70/40s act before ubiquitination and facilitate Ste6p association with an E3 ubiquitin ligase (46). Furthermore, polyubiquitination was a prerequisite for retrotranslocation, which required the Cdc48 complex and ATP (46). Wang et al. (47) and Younger et al. (48) recently provided new insights into the complexity of protein networks that govern the fate of the cystic fibrosis transmembrane conductance regulator (CFTR), an apical membrane ABC transporter. In this context, it is increasingly important to identify and to characterize multiple chaperone proteins that control the folding and degradation of ABC transporter proteins.
Conclusion
The clinical impact of the Q141K SNP has been most extensively studied. The Q141K SNP was identified with varying frequencies in different ethnic groups and was found to be the most prevalent in Japanese and Chinese populations (2). Q141K is reportedly associated with lower levels of protein expression and impaired transport (12–18), whereas some controversies exist in the publications characterizing this SNP (15,16). Zamber et al. (15) and Urquhart et al. (16) reported that the levels of ABCG2 in the intestine were not significantly affected by the Q141K SNP. It is important to note, however, that ABCG2 protein levels in the intestinal biopsy samples were measured by immunoblotting and compared between homozygous WT/WT and heterozygous WT/Q141K human subjects in those reports (15,16). No data was demonstrated for the ABCG2 protein level in homozygous Q141K/Q141K subjects. Since ABCG2 forms homo- and hetero-dimers linked through a cysteinyl disulfide bond, it is plausible that the heterodimer consisting of the WT and the Q141K variant is preferentially preserved in the ER as is the WT homodimer. The present in vitro study provides clear evidence that this major non-synonymous SNP Q141K affects the protein stability of ABCG2 in the ER and enhances its susceptibility to proteasomal degradation, thus affecting the plasma membrane localization of this nonsynonymous SNP. Further studies will be needed to verify the impact of the homozygous Q141K SNP on ABCG2 protein degradation in the intestine in vivo.
Abbreviations
- ABC:
-
ATP-binding cassette
- ABCP:
-
Placenta-specific ABC transporter
- BCRP:
-
Breast cancer resistance protein
- BMA:
-
Bafilomycin A1
- BSA:
-
Bovine serum albumin
- D-MEM:
-
High-glucose Dulbecco’s modified Eagle’s medium
- DTT:
-
Dithiothreitol
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- FCS:
-
Fetal calf serum
- FISH:
-
Fluorescence in situ hybridization
- FRT:
-
Flp recombination target
- HRP:
-
Horseradish peroxidase
- MTT:
-
3-(4,5-dimethyl-2-thiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
- MXR:
-
Mitoxantrone resistance-associated protein
- PBS:
-
Phosphate-buffered saline without both Ca2+ and Mg2+
- RT-PCR:
-
Reverse transcriptase-polymerase chain reaction
- SNP:
-
Single nucleotide polymorphism
- TBS:
-
Tris-buffered saline
- TTBS:
-
TBS with 0.05% (v/v) Tween 20
- WT:
-
Wild type
References
E. Kalow, U. Meyer, and R. F. Tyndale. Pharmacogenomics. Marcel Dekker, New York, 2001.
T. Ishikawa, A. Tamura, H. Saito, K. Wakabayashi, and H. Nakagawa. Pharmacogenomics of the human ABC transporter ABCG2: from functional evaluation to drug molecular design. Naturwissenschaften. 92:451–463 (2005).
F. A. de Jong, M. J. de Jonge, J. Verweij, and R. H. Mathijssen. Role of pharmacogenetics in irinotecan therapy. Cancer Lett. 234:90–106 (2006) doi:10.1016/j.canlet.2005.04.040.
K. Yanase, S. Tsukahara, J. Mitsuhashi, and Y. Sugimoto. Functional SNPs of the breast cancer resistance protein—therapeutic effects and inhibitor development. Cancer Lett. 234:73–80 (2006) doi:10.1016/j.canlet.2005.04.039.
G. Backstrom, J. Taipalensuu, H. Melhus, H. Brandstrom, A. C. Svensson, P. Artursson, and A. Kindmark. Genetic variation in the ATP-binding cassette transporter gene ABCG2 (BCRP) in a Swedish population. Eur. J. Pharm. Sci. 18:359–64 (2003) doi:10.1016/S0928-0987(03)00038-1.
T. M. Bosch, L. M. Kjellberg, A. Bouwers, B. P. Koeleman, J. H. Schellens, J. H. Beijnen, P. H. Smits, and I. Meijerman. Detection of single nucleotide polymorphisms in the ABCG2 gene in a Dutch population. Am. J. Pharmacogenomics. 5:123–31 (2005) doi:10.2165/00129785-200505020-00005.
F. A. de Jong, S. Marsh, R. H. Mathijssen, C. King, J. Verweij, A. Sparreboom, and H. L. McLeod. ABCG2 pharmacogenetics: ethnic differences in allele frequency and assessment of influence on irinotecan disposition. Clin. Cancer Res. 10:5889–94 (2004) doi:10.1158/1078-0432.CCR-04-0144.
Y. Honjo, K. Morisaki, L. M. Huff, R. W. Robey, J. Hung, M. Dean, and S. E. Bates. Single-nucleotide polymorphism (SNP) analysis in the ABC half-transporter ABCG2 (MXR/BCRP/ABCP1). Cancer Biol. Ther. 1:696–702 (2002).
A. Iida, S. Saito, A. Sekine, C. Mishima, Y. Kitamura, K. Kondo, S. Harigae, S. Osawa, and Y. Nakamura. Catalog of 605 single-nucleotide polymorphisms (SNPs) among 13 genes encoding human ATP-binding cassette transporters: ABCA4, ABCA7, ABCA8, ABCD1, ABCD3, ABCD4, ABCE1, ABCF1, ABCG1, ABCG2, ABCG4, ABCG5, and ABCG8. J. Hum. Genet. 47:285–310 (2002) doi:10.1007/s100380200041.
D. Kobayashi, I. Ieiri, T. Hirota, H. Takane, S. Maegawa, J. Kigawa, H. Suzuki, E. Nanba, M. Oshimura, N. Terakawa, K. Otsubo, K. Mine, and Y. Sugiyama. Functional assessment of ABCG2 (BCRP) gene polymorphisms to protein expression in human placenta. Drug Metab. Dispos. 33:94–101 (2005) doi:10.1124/dmd.104.001628.
M. Itoda, Y. Saito, K. Shirao, H. Minami, A. Ohtsu, T. Yoshida, N. Saijo, H. Suzuki, Y. Sugiyama, S. Ozawa, and J. Sawada. Eight novel single nucleotide polymorphisms in ABCG2/BCRP in Japanese cancer patients administered irinotacan. Drug Metab. Pharmacokinet. 18:212–7 (2003) doi:10.2133/dmpk.18.212.
Y. Imai, M. Nakane, K. Kage, S. Tsukahara, E. Ishikawa, T. Tsuruo, Y. Miki, and Y. Sugimoto. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol. Cancer Ther. 1:611–6 (2002).
S. Mizuarai, N. Aozasa, and H. Kotani. Single nucleotide polymorphisms result in impaired membrane localization and reduced ATPase activity in multidrug transporter ABCG2. Int. J. Cancer. 109:238–46 (2004) doi:10.1002/ijc.11669.
C. Kondo, H. Suzuki, M. Itoda, S. Ozawa, J. Sawada, D. Kobayashi, I. Ieiri, K. Mine, K. Ohtsubo, and Y. Sugiyama. Functional analysis of SNPs variants of BCRP/ABCG2. Pharm. Res. 21:1895–903 (2004) doi:10.1023/B:PHAM.0000045245.21637.d4.
C. P. Zamber, J. K. Lamba, K. Yasuda, J. Farnum, K. Thummel, J. D. Schuetz, and E. G. Schuetz. Natural allelic variants of breast cancer resistance protein (BCRP) and their relationship to BCRP expression in human intestine. Pharmacogenetics. 13:19–28 (2003) doi:10.1097/00008571-200301000-00004.
B. L. Urquhart, J. A. Ware, R. G. Tirona, R. H. Ho, B. F. Leake, U. I. Schwarz, H. Zaher, J. Palandra, J. C. Gregor, G. K. Dresser, and R. B. Kim. Breast cancer resistance protein (ABCG2) and drug disposition: intestinal expression, polymorphisms and sulfasalazine as an in vivo probe. Pharmacogenet. Genomics. 18:439–48 (2008) doi:10.1097/FPC.0b013e3282f974dc.
K. Morisaki, R. W. Robey, C. Ozvegy-Laczka, Y. Honjo, O. Polgar, K. Steadman, B. Sarkadi, and S. E. Bates. Single nucleotide polymorphisms modify the transporter activity of ABCG2. Cancer Chemother. Pharmacol. 56:161–72 (2005) doi:10.1007/s00280-004-0931-x.
A. Tamura, K. Wakabayashi, Y. Onishi, M. Takeda, Y. Ikegami, S. Sawada, M. Tsuji, Y. Matsuda, and T. Ishikawa. Re-evaluation and functional classification of non-synonymous single nucleotide polymorphisms of the human ATP-binding cassette transporter ABCG2. Cancer Sci. 98:231–9 (2007) doi:10.1111/j.1349-7006.2006.00371.x.
J. Li, G. Cusatis, J. Brahmer, A. Sparreboom, R. W. Robey, S. E. Bates, M. Hidalgo, and S. D. Baker. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol. Ther. 6:432–8 (2007).
A. Sparreboom, H. Gelderblom, S. Marsh, R. Ahluwalia, R. Obach, P. Principe, C. Twelves, J. Verweij, and H. L. McLeod. Diflomotecan pharmacokinetics in relation to ABCG2 421C>A genotype. Clin. Pharmacol. Ther. 76:38–44 (2004) doi:10.1016/j.clpt.2004.03.003.
A. Sparreboom, W. J. Loos, H. Burger, T. M. Sissung, J. Verweij, W. D. Figg, K. Nooter, and H. Gelderblom. Effect of ABCG2 genotype on the oral bioavailability of topotecan. Cancer Biol. Ther. 4:650–8 (2005) doi:10.1158/1535-7163.MCT-04-0238.
G. Cusatis, V. Gregorc, J. Li, A. Spreafico, R. G. Ingersoll, J. Verweij, V. Ludovini, E. Villa, M. Hidalgo, A. Sparreboom, and S. D. Baker. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J. Natl. Cancer Inst. 98:1739–42 (2006).
L. A. Doyle, W. Yang, L. V. Abruzzo, T. Krogmann, Y. Gao, A. K. Rishi, and D. D. Ross. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 95:15665–70 (1998) doi:10.1073/pnas.95.26.15665.
R. Allikmets, L. M. Schriml, A. Hutchinson, V. Romano-Spica, and M. Dean. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 58:5337–9 (1998).
K. Miyake, L. Mickley, T. Litman, Z. Zhan, R. Robey, B. Cristensen, M. Brangi, L. Greenberger, M. Dean, T. Fojo, and S. E. Bates. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. 59:8–13 (1999).
K. Kage, S. Tsukahara, T. Sugiyama, S. Asada, E. Ishikawa, T. Tsuruo, and Y. Sugimoto. Dominant-negative inhibition of breast cancer resistance protein as drug efflux pump through the inhibition of S–S dependent homodimerization. Int. J. Cancer. 97:626–30 (2002) doi:10.1002/ijc.10100.
H. Mitomo, R. Kato, A. Ito, S. Kasamatsu, Y. Ikegami, I. Kii, A. Kudo, E. Kobatake, Y. Sumino, and T. Ishikawa. A functional study on polymorphism of the ATP-binding cassette transporter ABCG2: critical role of arginine-482 in methotrexate transport. Biochem. J. 373:767–74 (2003) doi:10.1042/BJ20030150.
K. Kage, T. Fujita, and Y. Sugimoto. Role of Cys-603 in dimer/oligomer formation of the breast cancer resistance protein BCRP/ABCG2. Cancer Sci. 96:866–72 (2005) doi:10.1111/j.1349-7006.2005.00126.x.
U. Henriksen, J. U. Fog, T. Litman, and U. Gether. Identification of intra- and intermolecular disulfide bridges in the multidrug resistance transporter ABCG2. J. Biol. Chem. 280:36926–34 (2005) doi:10.1074/jbc.M502937200.
K. Wakabayashi, H. Nakagawa, T. Adachi, I. Kii, E. Kobatake, A. Kudo, and T. Ishikawa. Identification of cysteine residues critically involved in homodimer formation and protein expression of human ATP-binding cassette transporter ABCG2: a new approach using the flp recombinase system. J. Exp. Ther. Oncol. 5:205–22 (2006).
K. Wakabayashi, H. Nakagawa, A. Tamura, S. Koshiba, K. Hoshijima, M. Komada, and T. Ishikawa. Intramolecular disulfide bond is a critical check point determining degradative fates of ATP-binding cassette (ABC) transporter ABCG2 protein. J. Biol. Chem. 282:27841–6 (2007) doi:10.1074/jbc.C700133200.
H. Nakagawa, A. Tamura, K. Wakabayashi, K. Hoshijima, M. Komada, T. Yoshida, S. Kometani, T. Matsubara, K. Mikuriya, and T. Ishikawa. Ubiquitin-mediated proteasomal degradation of non-synonymous SNP variants of human ABC transporter ABCG2. Biochem. J. 411:623–31 (2008) doi:10.1042/BJ20071229.
A. Helenius, and M. Aebi. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73:1019–49 (2004) doi:10.1146/annurev.biochem.73.011303.073752.
L. Ellgaard, M. Molinari, and A. Helenius. Setting the standards: quality control in the secretory pathway. Science. 286:1882–8 (1999) doi:10.1126/science.286.5446.1882.
K. Mori. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 101:451–4 (2000) doi:10.1016/S0092-8674(00)80855-7.
L. Ellgaard, and A. Helenius. ER quality control: towards an understanding at the molecular level. Curr. Opin. Cell Biol. 13:431–7 (2001) doi:10.1016/S0955-0674(00)00233-7.
R. Y. Hampton. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 14:476–82 (2002) doi:10.1016/S0955-0674(02)00358-7.
B. Kleizen, and I. Braakman. Protein folding and quality control in the endoplasmic reticulum. Curr. Opin. Cell Biol. 16:343–9 (2004) doi:10.1016/j.ceb.2004.06.012.
K. Wakabayashi, A. Tamura, H. Saito, Y. Onishi, and T. Ishikawa. Human ABC transporter ABCG2 in xenobiotic protection and redox biology. Drug Metab. Rev. 38:371–91 (2006) doi:10.1080/03602530600727947.
S. Zhou, J. D. Schuetz, K. D. Bunting, A. M. Colapietro, J. Sampath, J. J. Morris, I. Lagutina, G. C. Grosveld, M. Osawa, H. Nakauchi, and B. P. Sorrentino. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 7:1028–34 (2001) doi:10.1038/nm0901-1028.
T. Ishikawa, A. Sakurai, Y. Kanamori, M. Nagakura, H. Hirano, Y. Takarada, K. Yamada, K. Fukushima, and M. Kitajima. High-speed screening of human ATP-binding cassette transporter function and genetic polymorphisms: new strategies in pharmacogenomics. Methods Enzymol. 400:485–510 (2005) doi:10.1016/S0076-6879(05)00027-3.
A. Tamura, M. Watanabe, H. Saito, H. Nakagawa, T. Kamachi, I. Okura, and T. Ishikawa. Functional validation of the genetic polymorphisms of human ATP-binding cassette (ABC) transporter ABCG2: identification of alleles that are defective in porphyrin transport. Mol. Pharmacol. 70:287–96 (2006).
C. B. Anfinsen. Principles that govern the folding of protein chains. Science. 181:223–30 (1973) doi:10.1126/science.181.4096.223.
K. Mohrmann, M. A. van Eijndhoven, A. H. Schinkel, and J. H. Schellens. Absence of N-linked glycosylation does not affect plasma membrane localization of breast cancer resistance protein (BCRP/ABCG2). Cancer Chemother. Pharmacol. 56:344–50 (2005) doi:10.1007/s00280-005-1004-5.
N. K. Diop, and C. A. Hrycyna. N-Linked glycosylation of the human ABC transporter ABCG2 on asparagine 596 is not essential for expression, transport activity, or trafficking to the plasma membrane. Biochemistry. 44:5420–9 (2005) doi:10.1021/bi0479858.
K. Nakatsukasa, G. Huyer, S. Michaelis, and J. L. Brodsky. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell. 132:101–12 (2008) doi:10.1016/j.cell.2007.11.023.
X. Wang, J. Venable, P. LaPointe, D. M. Hutt, A. V. Koulov, J. Coppinger, C. Gurkan, W. Kellner, J. Matteson, H. Plutner, J. R. Riordan, J. W. Kelly, J. R. Yates 3rd, and W. E. Balch. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 127:803–15 (2006) doi:10.1016/j.cell.2006.09.043.
J. M. Younger, L. Chen, H. Y. Ren, M. F. Rosser, E. L. Turnbull, C. Y. Fan, C. Patterson, and D. M. Cyr. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 126:571–82 (2006) doi:10.1016/j.cell.2006.06.041.
B. D. Hamman, L. M. Hendershot, and A. E. Johnson. BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell. 92:747–58 (1998) doi:10.1016/S0092-8674(00)81403-8.
M. J. Gething. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 10:465–72 (1999) doi:10.1006/scdb.1999.0318.
B. P. Tu, and J. S. Weissman. Oxidative protein folding in eukaryotes: mechanisms and consequences. J. Cell Biol. 164:341–6 (2004) doi:10.1083/jcb.200311055.
E. Sargsyan, M. Baryshev, L. Szekely, A. Sharipo, and S. Mkrtchian. Identification of ERp29, an endoplasmic reticulum lumenal protein, as a new member of the thyroglobulin folding complex. J. Biol. Chem. 277:17009–15 (2002) doi:10.1074/jbc.M200539200.
B. Knoblach, B. O. Keller, J. Groenendyk, S. Aldred, J. Zheng, B. D. Lemire, L. Li, and M. Michalak. ERp19 and ERp46, new members of the thioredoxin family of endoplasmic reticulum proteins. Mol. Cell Proteomics. 2:1104–19 (2003) doi:10.1074/mcp.M300053-MCP200.
Acknowledgments
We thank Drs. Kenta Mikuriya, Takayoshi Matsubara, and Satoshi Kometani (Yokogawa Electric Corporation) for generous support in fluorescence-microscopic observations. This study was supported by the NEDO International Joint Research Grant program “International standardization of functional analysis technology for genetic polymorphisms of drug transporters” as well as Grant-in-Aid for Scientific Research (A) (No. 18201041), Grant-in-Aid for Exploratory Research (No. 19659136) from the Japanese Society for the Promotion of Science (JSPS), and Grant-in-Aid for Young Scientists (B) (No. 19791361) from the Ministry of Education, Culture, Sports, Science and Technology. In addition, research in the Osawa laboratory at the University of Michigan Medical School was supported by an NIH grant GM077430.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Furukawa, T., Wakabayashi, K., Tamura, A. et al. Major SNP (Q141K) Variant of Human ABC Transporter ABCG2 Undergoes Lysosomal and Proteasomal Degradations. Pharm Res 26, 469–479 (2009). https://doi.org/10.1007/s11095-008-9752-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-008-9752-7