
Abstract
The composition of a metabolically active prokaryotic community thriving in hydrothermal mud fluids of the deep-sea hypersaline anoxic Western Urania Basin was characterized using rRNA-based phylogenetic analysis of a clone library. The physiologically active prokaryotic assemblage in this extreme environment showed a great genetic diversity. Most members of the microbial community appeared to be affiliated to yet uncultured organisms from similar ecosystems, i.e., deep-sea hypersaline basins and hydrothermal vents. The bacterial clone library was dominated by phylotypes affiliated with the epsilon-Proteobacteria subdivision recognized as an ecologically significant group of bacteria inhabiting deep-sea hydrothermal environments. Almost 18% of all bacterial clones were related to delta-Proteobacteria, suggesting that sulfate reduction is one of the dominant metabolic processes occurring in warm mud fluids. The remaining bacterial phylotypes were related to alpha- and beta-Proteobacteria, Actinobacteria, Bacteroides, Deinococcus-Thermus, KB1 and OP-11 candidate divisions. Moreover, a novel monophyletic clade, deeply branched with unaffiliated 16S rDNA clones was also retrieved from deep-sea sediments and halocline of Urania Basin. Archaeal diversity was much lower and detected phylotypes included organisms affiliated exclusively with the Euryarchaeota. More than 96% of the archaeal clones belonged to the MSBL-1 candidate order recently found in hypersaline anoxic environments, such as endoevaporitic microbial mats, Mediterranean deep-sea mud volcanoes and anoxic basins. Two phylotypes, represented by single clones were related to uncultured groups DHVE-1 and ANME-1. Thus, the hydrothermal mud of hypersaline Urania Basin seems to contain new microbial diversity. The prokaryotic community was significantly different from that occurring in the upper layers of the Urania Basin since 60% of all bacterial and 40% of all archaeal phylotypes were obtained only from mud fluids. The uniqueness of the composition of the active prokaryotic community could be explained by the complex environmental conditions at the site. The interaction of oxygenated warm mud fluids with the cold hypersaline brine of the Urania Basin seems to simultaneously select for various metabolic processes, such as aerobic and anaerobic heterotrophy, sulfide- and methane-dependent chemotrophy along with anaerobic oxidation of methane, sulfate- and metal-reduction.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The world’s deepest and most hypersaline anoxic lakes, occurring on the seafloor of the eastern Mediterranean Sea, represent one of the most hostile environments on Earth, and may serve as analogues for similar potentially life-containing environments of Mars and Europa. Four deep-sea hypersaline anoxic basins (DHABs), L’Atalante, Bannock, Discovery and Urania have recently been discovered on the seafloor of the Mediterranean Sea (De Lange and Ten Haven 1983; Scientific Staff of Cruise Bannock 1984-12 1985; MEDRIFF Consortium 1995; Wallmann et al. 1997). The surface of these brine lakes lies >3.300 m beneath the surface of the Mediterranean Sea and the salinity of the brines is 5 to 10 times higher than that of seawater. The brines of DHABs of the Eastern Mediterranean were formed several thousand years ago through the dissolution of buried Messinian evaporitic deposits, followed by brine accumulation and entrapment in sea floor depressions. As a result, very stable brines sharply stratified from the overlying water column were formed (Thomson et al. 1995; Wallmann et al. 2002). According to calculations, the hydrostatic pressure at the seafloor under the brines corresponds to that of the putative ocean on Europa at the middle of its predicted depth. The main ions composing the brine solution are Na, K, Ca, Mg, Cl and SO4. These facts make DHABs good analogues for deep saline cold waters on Europa (Marion et al. 2003).
Some major discoveries have been made in recent years on very peculiar forms of microbial life adapted to such environments. According to the recent findings, in spite of being so harsh, DHABs contain much more diverse prokaryotic assemblages (including many novel prokaryotic candidate divisions) than other more shallow anoxic marine hypersaline basins (van der Wielen et al. 2005; Daffonchio et al. 2006) indicating that a large fraction of the microorganisms and their role in biogeochemical processes in the DHAB lakes are yet unknown. A first attempt to functionally analyse the metagenome from Urania Basin seawater-brine interface has yielded a number of microbial enzymes which are adapted to operate in the halocline and which exhibit unusual structures, as well as biochemical parameters and substrate specificities (Ferrer et al. 2005).
The aim of the present study was to characterize for the first time metabolically active prokaryotic assemblage thriving in a hydrothermal (45 °C) mud vent habitat underneath the deep-sea brine lake Urania (Eastern Mediterranean).
Experimental Section
Sampling of hydrothermal mud fluids of deep-sea hypersaline anoxic lake Urania
To explore the microbial diversity of the Urania West Basin mud fluids, high-precision sampling was carried out during the BIODEEP-II cruise of RV Urania in September 2003 at location 35°13′51″N, 21°28′24″E; 3,524, 3,542, 3,623 and 3,727 m depth. Immediately after sampling, the Niskin bottles were retrieved, and their contents carefully sub-sampled. Five hundred milliliters of mud samples from the depth of 3,727 m were taken from the top and bottom of the bottles and were immediately analyzed for salinity. Values of 97‰ were observed in both cases, indicating that little or no mixing with overlying hypersaline brine had occurred.
RNA extraction from Urania mud fluids and synthesis of cDNA by reverse transcription reaction
Extraction of total RNA was performed on-board of research vessel ‘Urania’ using a QIAGEN® RNA/DNA mini extraction kit (QIAGEN, Valencia, CA) according to the manufacturer’s protocol. Total RNA was precipitated with isopropanol, washed with 70% ethanol and after air-drying was resuspended in 50 μl of diethylpyrocarbonate (DEPC)-treated sterile water and subsequently treated with ‘RNA-only’ DNase I, as described elsewhere (Mills et al. 2004). The quality of the RNA samples was examined by agarose electrophoresis and concentrations were determined using spectrophotometry (Biophotometer, Eppendorf). The crDNA was synthesized by reverse transcription using the 16S universal reverse primer Uni_1492R (5′-TACGYTACCTTGTTACGACTT-3′; Lane 1991) and SuperScript II RNase H-free reverse transcriptase (Life Technologies) according to the manufacturer’s protocol. In brief, total RNA was initially denatured by heating at 70 °C for 10 min. The reverse transcription reaction mix containing 5 μM of a 16S rRNA reverse primer 1492R (specific to the majority of prokaryotic organisms, including both Bacteria and Archaea domains; Yakimov et al. 2001), was added to 50 to 100 ng of denatured RNA, and 200 μM of deoxynucleoside triphosphate mix. The mixture was incubated for 5 min at 65 °C and 2 min at 4 °C, followed by the addition of 1X first-strand buffer (50 mM Tris–HCl [pH 8.3], 75 mM KCl, 3 mM MgCl2) and 75-U of RNase inhibitor and heating at 37 °C for 2 min. A 200 U aliquot of SuperScript II RNaseH-free reverse transcriptase (Life Technologies) was added prior to a 50 min incubation at 42 °C that resulted in the transcription of RNA into complementary 16S ribosomal DNA (crDNA). The reaction was stopped by heating at 80 °C for 5 min, and the crDNA was used as the template for a further PCR amplification.
Possible DNA contamination of RNA templates was routinely monitored by PCR amplification of RNA aliquots that were not reverse transcribed. No contaminating DNA was detected in any of these reactions.
PCR-amplification, cloning and sequencing
Primers used for standard crDNA PCR amplification included the above reverse primer (Uni_1492R) and 16S rDNA forward domain-specific Bacteria, Bac27_F (5′-AGAGTTTGATCCTGGCTCAG-3′; Lane 1991), and Archaea, Arc20_F (5′-TTCCGGTTGATCCYGCCRG-3′; Hallam et al. 2003), primers. The PCR mix contained 10 to 50 ng of crDNA, 1X Qiagen reaction buffer, 1X solution Q (Qiagen), 1 pM of each primer, 200 μM dNTPs (Gibco), and 2.5 U of Qiagen Taq polymerase. The PCR amplification involved a 3 min activation of the polymerase at 95 °C prior to 30 cycles each consisting of 1 min at 94 °C, 1 min at 50 °C and 2 min at 72 °C after which a 10 min extension at 72 °C was performed. Obtained PCR products were purified with QIAQuick PCR purification columns (Qiagen) and the amplicons were analyzed on 1.0% agarose gels run in Tris-borate-EDTA buffer stained with ethidium bromide and UV illuminated. Purified amplicons were cloned into the pGEM T-easy Vector II according to the manufacturer’s instructions (Promega). Inserts were subsequently PCR-amplified from lysed colonies with primers specific for the vector, M13F (5′-GTAAAACGACGGCCAG-3′) and M13R (5′-CAGGAAACAGCTATGAC-3′). Sequencing was performed with an ABI PRISM 3100-Avant Genetic Analyzer (PE Applied Biosystems, Foster City, CA) using the ABI PRISM BigDye® Terminator v 3.1 cycle sequencing kit (PE Applied Biosystems).
Phylogenetic analysis
Sequence analysis was preformed as previously described (Yakimov et al. 2004). From the total of 200 clones obtained and sequenced, 96 bacterial and 54 archaeal 16S crDNA sequences with a length 400–1400 bp were analyzed. The sequences of individual inserts were initially aligned with the program bl2seq (Altschul et al. 1997; Tatusova and Madden, 1999) available at the National Center for Biotechnology Information website. To determine the presence of any hybrid sequences the sequencing data were checked using the CHECK_CHIMERA programme, the individual sequences were subsequently aligned using the RDP Phylip Interface package (Maidak et al. 1997; Felsenstein 2001). Nucleotide sequences were manually aligned to sequence data from the RDP database considering their secondary structure using the Se–Al sequence alignment editor version 1.0 alpha 1 (Rambaut 1996). Single phylogenetic groups or phylotypes were defined at the cutoff of 99% of sequence identity The phylogenetic tree was constructed with MacVector software (Accelrys, San Diego, CA) version 7.2.2 (Rajagopal 2000) using Neighbor Joining method and Jukes–Cantor distance matrix; 100 bootstrap re-samplings were performed to estimate the robustness of the tree.
Nucleotide sequence accession numbers
The sequences determined in this study have been deposited at GenBank/EMBL/DDBJ databases under accession numbers DQ536433 and AM268241-268272.
Results and Discussion
Sampling site characterization
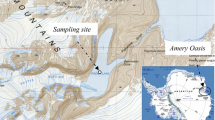
The Urania Basin is one of the few known depressions in the eastern Mediterranean Sea filled with anoxic and highly saline brine (up to 270 g/l) with the highest concentration of sulfide ever encountered in the marine environment (12–20 mM). The basin is located about 200 km west of Crete on the Mediterranean Ridge (Figure 1a), at the boundary of the accretionary complex and the so-called Inner Plateau of the ridge (Lallemant et al. 1994). Urania Basin, as all Mediterranean DHABs, is separated from the water column by a 2.0 m-thick halocline with a steep salt gradient (3.9% to 27% [w/w] salinity) between anoxic hypersaline brine and overlying seawater (Figure 1b). The uniqueness of the horseshoe-like Urania Basin is due to the presence in its deepest western part of an active mud volcano with a thermal anomaly of up to 45 °C (Figure 1; Corselli et al. 1996). The formation of Mediterranean mud volcanoes has been considered to result from the destruction of subsurface gas hydrate accumulations, the piercing of the surface by shale diapirs, and the rise of fluidized mud along faults (Milkov 2000; Charlou et al. 2003). The chemical analysis indicated that warm and methane-rich (800 μM) Urania mud fluids come from a deeper and hotter sedimentary reservoir below the Messinian evaporates (Charlou et al. 2003). In spite of the fact that the Urania mud fluids are hotter and less saline than overlying brine (10% salinity, 45 °C and 27% salinity, 16 °C, respectively), the mixing of these two compartments does not occur due to the higher clay content and higher density of the former, and stable formation of a thin interface was detected (Corselli et al. 1998; Charlou et al. 2003). The occasional upward expulsion of brine basin sediments due to extreme gas pressure in the mud fluids has also been documented (Hübner et al. 2003).

Location of deep anoxic hypersaline basins in the Eastern Mediterranean Sea. The map was constructed with ocean data view software http://awi-bremerhaven.de/GEO/ODV). (a) The detailed map of Urania Basin profiles demonstrating the position of mud volcano is shown in the insert map (bottom right). (b) Salinity and temperature profiling of seawater column, brine and mud fluids of Urania Basin. (c) The sampling site (35°13′51″N; 21°28′24″E; 3,524, 3,542, 3,623 and 3,727 m depth) is shown by red arrows.
The microbial community of Urania West Basin mud fluids
To survey the composition of the metabolically active fraction of the microbial community of the Urania West Basin mud environment, a total of 96 bacterial and 54 archaeal riboclones were recovered in the corresponding clone libraries, sequenced and characterized. No chimeric sequences were detected among the clones. Sequences that were >99% similar were considered to have the same phylotype, or operational taxonomic units (OTUs). Figure 2 shows the taxonomic affiliation of a total of 17 bacterial OTUs. In terms of absolute numbers of clones and the diversity of their sequences, the epsilon-and the delta-Proteobacteria were the most prominent groups of organisms represented in clone library. These two groups accounted together for more than 67% of the clones and 41% of the phylotypes detected in hydrothermal mud fluids (Figures 2 and 4). According to the recent classification (Corre et al. 2001; Takai et al. 2003), the most abundant phylotype UMP-54B (32% of all clones) and phylotypes UMP-50B and UMP-57B (12.5% of all clones) were affiliated with the Group B and Group F of the epsilon-Proteobacteria, respectively. Known members within these phylogenetic subgroups are mesophiles retrieved from deep-sea hydrothermal vents and sediments (Reysenbach et al. 2000; Teske et al. 2002; Nakagawa et al. 2005a), which is consistent with the temperatures measured in situ in Urania mud fluds. Remarkably, the OTU UMP-52B accounting for 5% of the bacterial clones could not be categorized by Corre’s grouping and obviously represents a new group. Previous reports have suggested the epsilon-Proteobacteria to be microaerobic sulfur oxidizers (Wirsen et al. 1993; Taylor et al. 1999). However, more recent studies have emphasized not only the prevalence of epsilon-Proteobacteria in deep-sea hydrothermal environments, but also their metabolic versatility and therefore importance in fluxes of other elements (Alain et al. 2002; Inagaki et al. 2004; Takai et al. 2005; Nakagawa et al. 2005b). For instance, the majority of isolates belonging to Group B and F of the epsilon-Proteobacteria were found to utilize a wide spectrum of substrates including H2, S0, S2O3 2−, O2 and NO3 −. Hydrogen and nitrate were used as electron donor and acceptor respectively (Nakagawa et al. 2005a). The phylotypes UMP-24B, UMP-41B and UMP-77B of delta-Proteobacteria were members of predominantly sulfate-reducing group of Desulfobacterales and showed high sequence similarities to Desulfospira joergensenii and to environmental clone HMMVBeg-47, retrieved from microbial methanotrophic communities at the Haakon Mosby mud volcano (Barents Sea). They constituted more than 17% of the clone library, suggesting that reduction of sulfate to sulfide is one of the major metabolic pathways occurring in Urania West Basin mud fluids. A number of alpha-and beta-Proteobacteria was also detected in the mud fluids. These organisms were closely related to facultatively methylotrophic, methane-utilizing and chemolithoautotrophic, ammonium-oxidizing bacteria, respectively (Figure 2).

Phylogenetic tree of 16S rRNA sequences of the most abundant bacterial phylotypes recovered in the Western Urania Basin mud fluids. The tree was constructed using sequences of comparable region of the 16S rRNA gene sequences available in public databases. Neighbor-joining analysis using 100 bootstrap replicates was used to infer tree topology. The bar represents 5% sequence divergence. Abbreviation used: PB, Proteobacteria; KB1, Kebrit Basin candidate division 1; OP-11, Obsidian Pool candidate division 11. The Urania Lake clones retrieved from seawater-interface, water body and thermal mud fluids are shown by filled circles, filled squares and asterisks, respectively.
Two phylotypes UMP-17B and UMP-47B were related to phylum Bacteroidetes. Similar sequences were retrieved from deep-sea hydrothermal vent environment of the Mid-Atlantic Ridge. The majority of cultured members of Bacteroidetes are heterotrophic organisms, utilizing a variety of sugars and biopolymers as sources of carbon and energy both under aerobic and anaerobic conditions. Such metabolic versatility would be benefitial at the Urania brine–mud interface where organic polymers are likely to accumulate. The phylotype UMP-11B belonged to class Actinobacteria and was closely affiliated to 16S rDNA sequences, recovered from deep-sea hydrocarbon seep and brine–seawater interface in the Kebrit Deep Basin (Figure 2). The Deinoccocus-Thermus-related OTU UMP-71B constituted 8% of clone library and exhibited >98% sequence similarity to Thermus scotoductus SA-01, a thermophilic metal reducer, carrying out the oxidation of organic compounds coupled with iron or manganese reduction (Kieft et al. 1999). This finding indirectly supports the possibility that metal reduction may be one of the metabolic processes occurring in the Urania West Basin mud fluids.
Three phylotypes were affiliated with the members of candidate division KB1, found exclusively in anaerobic hypersaline sediments (Eder et al. 2001; Mouné et al. 2003; van der Wielen et al. 2005; Daffonchio et al. 2006). Similar to the KB1 bacteria, the habitat characteristics of members of the OP-11 candidate division (three clones in the clone library) suggest an anaerobic phenotype. Other components of the bacterial community included three similar clones with uncertain affiliation. No cultured representatives of these three groups of phylotypes are known at present and thus their physiology and ecological importance remain unknown.
A total of 54 archaeal sequences were retrieved from Urania mud fluid sample. They were distributed among five phylotypes, all affiliated with three uncultured groups of Euryarchaeota (Figures 3 and 4). Only UMP-32 was attributed to the group of organisms with known metabolic features. The presence of organisms related to ANME-1 in mud fluids indicates that anaerobic oxidation of methane is possibly one of the metabolic pathways occurring in this environment. Another phylotype, UMP-82A, is a member of deep-sea hydrothermal vent group I (DHVE-I) of euryarchaeotes (Takai and Horikoshi 1999). Three remaining phylotypes yielded more than 96% of all archaeal clones and were affiliated to MSBL1 candidate division. The members of this division were found only in anoxic hypersaline ecosystems, like Mediterranean DHABs, mud volcanoes and endoevaporitic microbial mats (Sørensen et al. 2005; van der Wielen et al. 2005; Daffonchio et al. 2006).

Phylogenetic tree of 16S rRNA sequences of the most abundant archaeal phylotypes recovered in the Western Urania Basin mud fluids. Neighbor-joining analysis using 100 bootstrap replicates was used to infer tree topology. The phylogenetic groups and clones retrieved from overlaying seawater and the Urania Lake seawater-interface, water body and thermal mud fluids are shown by open circles, filled circles, filled squares and asterisks, respectively. The bar represents 5% sequence divergence. Abbreviation used: ANME-1, Anoxic Methane-oxidizing Euryarchaeota Group 1; DHVE-1, Deep-sea Hydrothermal Vent Euryarchaeota Group 1; MEG, Miscellaneous Euryarchaeotic Group; MSBL1, Mediterranean Sea Brine Lake candidate division 1 of Euryarchaeota; SAGMEG, South-African Gold Mine Euryarchaeota Group.

Overview on prokaryotic diversity and relative abundance of phylogenetic groups recovered from Archaea and Eubacteria clone libraries of Urania Basin warm mud fluids.
In conclusion, the thermal mud fluids, located at the bottom of Urania West Basin, harbor an active and highly diverse prokaryotic community. Our culture-independent analysis demonstrated that these warm mud fluids provide a suitable habitat for prokaryotic organisms belonging to at least 13 high taxonomic lineages (Figure 4). Similarly to other marine hydrothermal systems (Nakagawa et al. 2005a; Kormas et al. 2006), the members of epsilon-Proteobacteria dominate within the bacterial community of Urania mud fluids, but overall the diversity is high. The difference between deep-sea hydrothermal fields and the Urania mud fluids was more evident by the presence of groups KB-1, OP-11 and dominant archaeal order MSBL-1, previously found only in anoxic hypersaline ecosystems. Comparing the mud prokaryotic community with those of overlaying compartments of the Urania Basin and seawater, we found that 60% of all bacterial and 40% of all archaeal phylotypes were mud-specific. This uniqueness of the active prokaryotic community of the deep-sea hydrothermal mud fluids of the Urania Basin could be explained by the complex physical–chemical conditions, which exist at the studied site. Data on microbial community structure were compared to the chemical and physical characteristics of this habitat to infer community function. The interaction of slightly oxygenated and methane-saturated warm mud fluids with the cold sulfide-rich hypersaline brine of Urania Basin appears to simultaneously support various microbial lifestyles, such as aerobic and anaerobic heterotrophy, sulfide-and methane-dependent chemotrophy, chemolithotrophy along with anaerobic oxidation of methane, and sulfate reduction.
This work is the first demonstration of an active microbial community thriving in deep and hydrothermal mud fluids, covered by the high-density, cold and hypersaline water masses of Urania Basin. The data obtained through this study represent an additional input to our knowledge about microbial life in extreme ecosystems and may form the basis for future research in this field and in astrobiology.
References
Alain K, Querellou J, Lesongeur F, Pignet P, Crassous P, Raguénès G (2002) Caminibacter hydrogeniphilus gen. nov., sp. nov., a novel thermophilic, hydrogen-oxidizing bacterium isolated from an east pacific rise hydrothermal vent. Int J Syst Evol Microbiol 52:1317–1323
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Charlou JL, Donval JP, Zitter T, Roy N, Jean-Baptiste P, Foucher JP, Woodside J, MEDINAUT Scientific Party (2003) Evidence of methane venting and geochemistry of brines on mud volcanoes of the Eastern Mediterranean Sea. Deep-Sea Res 50:941–958
Corre E, Reysenbach A-L, Prieur D (2001) Epsilon-proteobacterial diversity from a deep-sea hydrothermal vent on the mid-atlantic ridge. FEMS Microbiol Lett 205:329–335
Corselli C, Basso D, De Lange GJ, Thomson J (1996) Mediterranean ridge accretionary complex yields rich surprises. EOS 77:227
Corselli C, Della Vedova B, Camerlenghi A, De Lange GJ, Westbrook GK (1998). Emission of warm fluids and high temperature in the Urania basin: observations from 1993 to 1998, International Workshop ‘Extreme Marine Environments’ GEOMAR Kiel, 19–22 November 1998, Volume of Abstracts: pp. 9–10
Daffonchio D, Borin S, Brusa T, Brusetti L, van der Wielen PWJJ, Bolhuis H, D’Auria G, Yakimov MM, Giuliano L, Marty D, Tamburini C, McGenity TJ, Hallsworth JE, Sass AM, Timmis KN, Tselepides A, De Lange GJ, Andreas Hübner A, Thomson J, Varnavas SP, Gasparoni F, Gerber HW, Malinverno E, Corselli C, Biodeep Scientific Party (2006) Stratified prokaryote network in the oxic–anoxic transition of a deep sea halocline. Nature 440:203–207
De Lange GJ, Ten Haven HL (1983) Recent sapropel formation in the Eastern Mediterranean. Nature 305:797–798
Eder W, Jahnke LL, Schmidt M, Huber R (2001) Microbial diversity of the brine–seawater interface of the Kebrit Deep, Red Sea, studied via 16S rRNA gene sequences and cultivation methods. Appl Environ Microbiol 67:3077–3085
Felsenstein J (2001) PHYLIP phylogenetic inference package, version 3.6 ed. Department of Genetics, University of Washington. Seattle, Washington
Ferrer M, Golyshina OV, Chernikova TN, Khachane AN, Martins Dos Santos VA, Yakimov MM, Timmis KN, Golyshin PN (2005) Microbial enzymes mined from the Urania deep-sea hypersaline anoxic basin. Chem Biol 12:895–904
Hallam SJ, Girguis PR, Preston CM, Richardson PM, DeLong EF (2003) Identification of methyl coenzyme m reductase A (mcrA) genes associated with methane-oxidizing archaea. Appl Environ Microbiol 69:5483–5491
Hübner A, De Lange GJ, Dittmer J, Halbach P (2003) Geochemistry of an exotic sediment layer above sapropel S-1: mud expulsion from the Urania Basin, Eastern Mediterranean? Mar Geol 197:49–61
Inagaki F, Takai K, Nealson KH, Horikoshi K (2004) Sulfurovum lithotrophicum gen. nov., sp. nov., a novel sulfur-oxidizing chemolithoautotroph within the epsilon-Proteobacteria isolated from the Okinawa trough hydrothermal sediments. Int J Syst Evol Microbiol 54:1477–1482
Kieft TL, Fredrickson JK, Onstott TC, Gorby YA, Kostandarithes HM, Bailey TJ, Kennedy DW, Li SW, Plymale AE, Spadoni CM, Gray MS (1999) Dissimilatory reduction of Fe (III) and other electron acceptors by a Thermus isolate. Appl Environ Microbiol 65:1214–1221
Kormas KA, Tivey MK, Von Damm K, Teske A (2006) Bacterial and archaeal phylotypes associated with distinct mineralogical layers of a white smoker spire from a deep-sea hydrothermal vent site. Environ Microbiol 8:909–920
Lallemant S, Truffert C, Jolivet N, Henry P, Chamot-Rooke N, De Voogd B (1994) Spatial transition from compression to extension in the Western Mediterranean ridge accretionary complex. Tectonophysics 234:33–52
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackerbrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, New York, pp 115–175
Maidak BL, Olsen GJ, Larsen N, Overbeek R, McCaughey MJ, Woese CR (1997) The RDP (Ribosomal Database Project). Nucleic Acids Res 25:109–111
Marion GM, Fritsen CH, Eicken H, Payne MC (2003) The search for life on Europa: limiting environmental factors, potential habitats, and earth analogues. Astrobiology 3:785–811
MEDRIFF Consortium (1995) Three brine lakes discovered in the seafloor of the Eastern Mediterranean. EOS 76:313
Milkov AV (2000) Worldwide distribution of submarine mud volcanoes and associated gas hydrates. Mar Geol 167:29–42
Mills HJ, Martinez RJ, Story S, Sobecky PA (2004) Identification of members of the metabolically active microbial populations associated with Beggiatoa species mat communities from Gulf of Mexico cold-seep sediments. Appl Environ Microbiol 70:5447–5458
Mouné S, Caumette P, Matheron R, Willison JC (2003) Molecular sequence analysis of prokaryotic diversity in the anoxic sediments underlying cyanobacterial mats of two hypersaline ponds in Mediterranean salterns. FEMS Microbiol Ecol 44:117–130
Nakagawa S, Takai K, Inagaki F, Hirayama H, Nunoura T, Horikoshi K, Sako Y (2005a) Distribution, phylogenetic diversity and physiological characteristics of epsilon-Proteobacteria in a deep-sea hydrothermal field. Environ Microbiol 7:1619–1632
Nakagawa S, Takai K, Inagaki F, Horikoshi K, Sako Y (2005b) Nitratiruptor tergarcus gen. nov., sp. nov. and Nitratifractor salsuginis gen. nov., sp. nov., nitrate-reducing chemolithoautotrophs of the epsilon-Proteobacteria isolated from a deep-sea hydrothermal system in the mid-Okinawa trough. Int J Syst Evol Microbiol 55:925–933
Rajagopal I (2000) SOFTWARE: genomics made easy. Science 290:474
Rambaut A (1996) Se–Al (Sequence Alignment Editor) version 1.0 alpha 1. WWW Page http://evolve.zoo.ox.ac.uk/Se–Al/Se–Al.html
Reysenbach AL, Longnecker K, Kirshtein J (2000) Novel bacterial and archaeal lineages from an in situ growth chamber deployed at a mid-Atlantic ridge hydrothermal vent. Appl Environ Microbiol 66:3798–3806
Scientific Staff of Cruise Bannock 1984-12 (1985) Gypsum precipitation from cold brines in an anoxic basin in the Eastern Mediterranean. Nature 314:152–154
Sørensen KB, Canfield DE, Teske AP, Oren A (2005) Community composition of a hypersaline endoevaporitic microbial mat. Appl Environ Microbiol 71:7352–7365
Takai K, Horikoshi K (1999) Genetic diversity of archaea in deep-sea hydrothermal vent environments. Genetics 152:1285–1297
Takai K, Inagaki F, Nakagawa S, Hirayama H, Nunoura T, Sako Y (2003) Isolation and phylogenetic diversity of members of previously uncultivated epsilon-Proteobacteria in deep-sea hydrothermal fields. FEMS Microbiol Lett 218:167–174
Takai K, Hirayama H, Nakagawa T, Suzuki Y, Nelson KH, Horikoshi K (2005) Lebetimonas acidiphila gen. nov., sp. nov., a novel thermophilic, acidophilic, hydrogen-oxidizing chemolithoautotroph within epsilon-Proteobacteria isolated from a deep-sea hydrothermal fumarole in the mariana arc. Int J Syst Evol Microbiol 55:183–189
Tatusova TA, Madden TL (1999) BLAST 2 SEQUENCES, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett 177:187–188
Taylor CD, Wirsen CO, Gaill F (1999) Rapid microbial production of filamentous sulfur mats at hydrothermal vents. Appl Environ Microbiol 65:2253–2255
Teske A, Hinrichs KU, Edgcomb V, de Vera Gomez A, Kysela D, Sylva SP, Sogin ML, Jannasch HW (2002) Microbial diversity of hydrothermal sediments in the guaymas basin: evidence for anaerobic methanotrophic communities. Appl Environ Microbiol 68:1994–2007
Thomson J, Higgs NC, Wilson TRS, Croudace IW, De Lange GJ, Van Santvoort PJM (1995) Redistribution and geochemical behaviour of redox-sensitive elements around S1, the most recent Eastern Mediterranean sapropel. Geochim Cosmochim Acta 59:3487–3501
van der Wielen PW, Bolhuis H, Borin S, Daffonchio D, Corselli C, Giuliano L, D’Auria G, De Lange GJ, Huebner A, Varnavas SP, Thomson J, Tamburini C, Marty D, McGenity TJ, Timmis KN, BioDeep Scientific Party (2005) The enigma of prokaryotic life in deep hypersaline anoxic basins. Science 307:121–123
Wallmann K, Suess E, Westbrook GH, Winkler G, Cita MB, MEDRIFF Consortium (1997) Salty brines on the Mediterranean Sea floor. Nature 387:31–32
Wallmann K, Aghib FS, Castradori D, Cita MB, Suess E, Greinert J, Rickert D (2002) Sedimentation and formation of secondary minerals in the hypersaline discovery basin, Eastern Mediterranean. Mar Geol 186:9–28
Wirsen CO, Jannasch HW, Molyneaux SJ (1993) Chemosynthetic microbial activity at mid-Atlantic ridge hydrothermal vent sites. J Geophys Res 98:9693–9703
Yakimov MM, Giuliano L, Timmis KN, Golyshin PN (2001) Upstream-independent ribosomal RNA amplification analysis (URA): a new approach to characterizing the diversity of natural microbial communities. Environ Microbiol 3:662–666
Yakimov MM, Gentile G, Bruni V, Cappello S, D’Auria G, Golyshin PN, Giuliano L (2004) Crude oil-induced structural shift of coastal bacterial communities of Rod Bay (Terra Nova Bay, Ross Sea, Antarctica) and characterization of cultured cold-adapted hydrocarbonoclastic bacteria. FEMS Microbiol Ecol 49:419–432
Acknowledgments
This study was supported by the European Commission’s Sustainable Marine Ecosystem program, under BIODEEP (EVK3-2000-00042) and COMMODE (EVK3-2002-00077) Projects. We thank Giuseppe D’Auria for his valuable help at sea and in the laboratory. We thank the crew and the pilot of RV Urania for their expert handling of our samplers and equipment at the Urania Basin and for highly productive cruises. P.N.G. acknowledges the support of DFG, Project Nr Ti86/8. Priority Program ‘Mars and Terrestrial Planets’, and GenoMikPlus Program of the Federal Ministery of Science and Education (BMBF).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yakimov, M.M., Giuliano, L., Cappello, S. et al. Microbial Community of a Hydrothermal Mud Vent Underneath the Deep-Sea Anoxic Brine Lake Urania (Eastern Mediterranean). Orig Life Evol Biosph 37, 177–188 (2007). https://doi.org/10.1007/s11084-006-9021-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11084-006-9021-x