
Abstract
In this work, 3-nitroanilinium nitrate (3NAN) has been synthesized and crystallized successfully by solution growth combined with solvent evaporation technique. 3NAN molecular structure has been optimized with Density Functional Theory (DFT) using B3LYP function and Hartree–Fock method with a 6–311 + + G(d,p) basis set. The geometrical parameters of the title molecules have been analyzed. The computed vibrational spectra were compared with experimental result which show appreciable agreement. Thermal stability of the crystal was analyzed with TGA/DTA and the melting points of the salt identified at 209 ºC. HOMO–LUMO energy calculations have shown the charge transfer within the molecule. The possible pharmaceutical/biological activity of the salts confirmed by the Frontier Molecular Orbital (FMO) analysis have lower band gap value. The antimicrobial activity of grown crystals has been tested against certain potentially threatening microbes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Generally nitroaniline is called as push–pull molecule. Because of the intramolecular charge transfer (ICT) from the –NH2 (electron-donor group) to –NO2 (electron-acceptor group) through the phenyl ring. 3-Nitroaniline and 4-nitroaniline are used as reference compounds for optical nonlinearity (Krishnakumar et al. 2008; Bishop et al. 1998; Reis et al. 2000) calculated by computational and experimental method. 3-Nitroaniline and its derivatives are biologically important compounds, such compounds are produced significant antihyperglycemic as well as hypoglycemic effects in alloxan-induced and normal diabetic rabbits (Jain et al. 2005). Also m-Nitroaniline plays an essential role in several biological processes and has considerable chemical and pharmacological importance. Since a long time, schiff based compounds are generally considered due to their importance in several electro-optical and other biological applications (Siva et al. 2019).
In this context, a new family of organic–inorganic crystals with the biological importance, with chemical, physical and thermal properties through inorganic sub networks is proposed. Based on the above specifics, 3-nitroaniline was reacted with nitric acid and the crystals of 3-nitroanilinium nitrate were grown. In this paper, the synthesis, crystal growth, structural, spectroscopic, thermal analyses and biological activity of 3NAN is reported.
2 Experimental
2.1 Material preparation
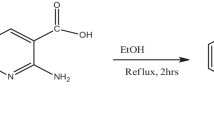
3-nitroanilinium nitrate is synthesized from the AR grade 3-nitroaniline and nitric acid obtained from Himedia fine chemicals taken in 1:1 ratio using acetone, as solvent. This solution was stirred well for about 1 h to get clear solution filtered and kept in a clean beaker for evaporation. The title crystal was synthesized according to the scheme as shown in Fig. 1. The reaction was allowed in room temperature. After many attempts, the compound 3-nitroaniline nitrate has not yielded good quality single crystal; which is not suitable for single crystal X-ray diffraction. Hence, the powder XRD analysis of 3NAN was taken to prove the new phase of the complex.

Reaction scheme of 3-Nitroanilinium nitrate
2.2 Characterization techniques
Powder XRD study of 3NAN was carried out by Bruker X-ray diffractometer of D8 advanced ECO XRD system with SSD160 1D Detector. The diffraction was collected in the range of 2θ, 0° to 50°. The preliminary data of the polycrystalline sample were calculated by indexing in the first 2θ strong intensity peaks using DASH 3.3.2 program (David et al. 2006). FT-IR spectrum of grown crystal was recorded using Nexus 670 FTIR spectrometer and FT-Raman spectrum were recorded using BRUKER RFS 27 FT-Raman Spectrometer. Both the spectra were recorded in the range of 400 to 4000 cm−1 with Nd:YAG laser at 1064 nm as source. DTA/TGA of grown crystals were carried out by SII (SEIKO) Japan Analytical Instrument, Model No: TG/DTA-6200 in N2 atmosphere with heating rate 20 °C/min.
2.3 Computational profile
The 3NAN molecular parameters in the ground state were computed by performing both HF and B3LYP with basis set 6–311 + G(d,p). Computational calculations were performed using GAUSSIAN 09 W (Frisch et al. 2010) program package on i5 processor/3.20 GHz personal computer without any constraint on the geometry (Schlegel 1982). The geometry of the title compound was optimized by the computational method. The HF and B3LYP method (Hohenberg and Kohn 1964) acknowledged as a practical approach for the computation of molecular structure and vibrational frequencies with the Becke’s three parameter exact exchange-functional (B3)(Becke 1993) combined with gradient-corrected correlational functional of Lee, Yang and Parr (LYP) (Lee et al. 1988). Using principle of statistical mechanics, thermodynamic properties of 3NAN have been calculated from normal mode of vibrational frequencies which provide the details about title compound in gas phase. GAUSSVIEW program (Dennington et al. 2013) with symmetry considerations were used to assign vibrational frequencies. The HOMO–LUMO energies were calculated and computed by the HF and B3LYP method.
3 Results and discussion
3.1 Structural and molecular geometry analyses
The possible unit cell parameters for 3NAN is given in Table 1. By comparing the powder XRD pattern of grown crystal 3NAN compared with its pure 3-nitroaniline and shown in Fig. 2. With the help of XRD pattern and the unit cell values are confirmed the 3NAN salt formation. Some of the new intensity peaks are appear in 3NAN crystals and some of the peaks are disappear compared to 3-nitroaniline. The possible unit cell parameters were identified and 3NAN crystal belongs to orthorhombic system was notable using DASH 3.3.2 program.

Powder XRD patterns of 3NAN and 3NA
In 3NAN, asymmetric part of unit cell contains 3-nitroaniline (cation) charge is neutralized with nitrate (anion). The optimized molecular structure of 3-nitroanilinium nitrate diagram and atom numbering scheme are shown in the Fig. 3 and geometrical parameters are given in Table 2. In phenyl ring, optimized C–C bond length is calculated by HF method around 1.381–1.390 Å and by B3LYP method around 1.389–1.400 Å. The N–O bond lengths are calculated around 1.167–1.321 Å by HF method and 1.199–1.382 Å by B3LYP method in nitrate anion.

Optimized molecular structure of 3NAN by a HF and b B3LYP method
3.2 Mulliken charge analysis
The mulliken atomic charges of 3NAN given in Table 3 and the histogram is shown in Fig. 4. By quantum chemical methods, Mulliken atomic charge distribution was calculated, which has a vital role to the molecular system because atomic charges effect dipole moment, polarizability and electronic structure (Siva et al. 2017b). The hydrogen atom (H11) (0.497e in HF and 0.451e in B3LYP) in HNO3 group of 3NAN was more electropositive, this hydrogen atom was attached with electronegative atoms (N18, O19, O20, O21). Also, nitrogen atom has more electro negativity (N1) (-0.535e in HF and -0.474e in B3LYP), this nitrogen atom enclosed by H12 and H13 (electropositive atom) and C2 (carbon atom).

Histogram of atomic charges on 3NAN by HF and B3LYP methods
3.3 Vibrational analysis
The 3NAN consists of 21 atoms, with 57 modes of vibrations. The comparison of experimental and computed IR and Raman spectra of 3NAN are given in Fig. 5 and frequency values are given in Table 4.

Experimental and computed IR & Raman spectra of 3NAN
3.3.1 NH2 vibrations
The NH2 group vibrations are expressed as antisymmetric stretching, symmetric stretching, scissoring, wagging, rocking and twisting vibrations are spread over a region in the spectrum (Siva et al. 2017a). In theoretical spectrum the peak at 3826 cm−1 in HF and 3596 cm−1 in B3LYP method are assigned to NH2 antisymmetric stretching. NH2 symmetric stretching wavenumbers are calculated at 3742 and 3510 cm−1 in HF and B3LYP respectively. NH2 scissoring are calculated at 1808 and 1654 cm−1 in HF and B3LYP respectively. NH2 twisting is calculated at 1628 and 1494 cm−1 in HF and B3LYP respectively. NH2 wagging along with scissoring NO2 vibrations are observed as weak peak at 1036 in Raman and theoretically scaled by HF and B3LYP at 1037 and 942 cm−1 respectively. The rocking NH2 vibrations are calculated at mode 34 corresponding experimental spectrum identified as a weak peak in Raman at 1198 cm−1 and a medium peak at 1174 cm−1 in IR.
3.3.2 NO2 vibrations
In nitrobenzene and substituted nitrobenzene, the band around 1500–1570 cm−1 and 1300–1370 cm−1 are represents the asymmetric and symmetric stretching vibrations of NO2 group, respectively (Prabavathi et al. 2013). The NO2 asymmetric stretching vibrations are theoretically computed by HF and B3LYP at 1772 and 1591 cm−1 respectively. The NO2 symmetric stretching vibration theoretically scaled by HF and B3LYP method at 1367 and 1325 cm−1, respectively. The scissoring NO2 vibrations are observed as weak peak at 1036 cm−1 computed at mode 30 and mode 4 represents NO2 twisting vibrations in theoretical calculation.
3.3.3 C–C and C–N vibrations
The title compounds have C–C and C–N stretching vibrations is usually occur in the region 1300–1600 cm−1. In theoretical spectrum the peak at 1837 cm−1 by HF and 1665 cm−1 in B3LYP method are assigned to C–C stretching vibrations. Similarly, C–N stretching vibrations are assigned to 1444 cm−1 in HF and 1333 cm−1 in B3LYP.
3.3.4 C-H vibrations
In 3NAN, the C–H stretching vibrations confirm aromatic structure. These stretching vibrations are occurs in the characteristic region around 3100 cm−1B (Bhuvaneswari et al. 2018; Thangarasu et al. 2018). The C–H stretching vibrations peaks are assigned in the theoretical spectrum in the region 3657–3176 cm−1. The in-plane bending vibrations are observed with O–H stretching in theoretical spectrum at 1782 cm−1 in HF and 1632 cm−1 in B3LYP.
3.3.5 Vibrations of nitrate anion
The title compound, \({\mathrm{NO}}_{3}^{-}\) asymmetric stretching vibrations are observed as a weak peak at 2003 cm−1 in IR and in theoretical spectrum at 1932 and 1735 cm−1 in the HF and B3LYP respectively. \({\mathrm{NO}}_{3}^{-}\) symmetric stretching vibrations are observed at 1607 and 1372 cm−1 in the HF and B3LYP respectively. The O–H stretching is observed as a weak peak in IR at 3073 cm−1, it is scaled at 3326 cm−1 in HF and 3016 cm−1 in B3LYP. The O–H in-plane banding vibrations are calculated 1611 and 1482 cm−1 in the HF and B3LYP respectively. The O–H out-plane banding vibrations observed as a weak peak at 1082 cm−1 in IR and it is well matches with experimental spectra.
3.4 Frontier moleculer orbital analysis
In the organic molecules, optical and electric properties are proposed by using frontier molecular orbital analysis (Jean and Volatron et al. 2005; Drozd and Dudzic et al. 2013; Mary Novena et al. 2017). The molecular orbital have two important interactions; such as HOMO means the ability to donate an electron and LUMO represents accept an electron. Also, these orbitals are named as frontier orbitals. Frontier electron density (Palafox et al. 2000) is used to explain the several types of reaction in conjugated system and most reactive position in π-electron systems. Also, FMO plays an essential part in abiding chemical stability of the compound (Fleming et al. 1976). The calculated HOMO and LUMO energies and some important parameters of the 3NAN were calculated by HF and DFT/B3LYP method as shown in Table 5. The stimulated energy level diagrams of 3NAN molecule was shown in Fig. 6. A huge amount of charge transfer occurred in 3NAN is indicated by small energy gap, which is connected with a high chemical reactivity. The small value of energy gap and high value of charge transfer leads the molecules to biologically active.

Molecular orbital energy level diagram of 3NAN by a HF and b B3LYP levels
For 3NAN, 356 energy levels are observed in the energy range −20.668 au to 51.757 au and −19.214 au to 49.990 au in HF and B3LYP method respectively. The calculated HOMO and LUMO energy values of 3NAN molecule in HF and B3LYP are −0.357/0.022 au and −0.275/−0.116 au repectively. The energy gap value between the frontier orbital was 0.379 a.u in HF and 0.159 a.u in B3LYP.
3.5 Thermal analysis
Thermal behaviour was analyzed by using simultaneous DTA/TGA analysis in the temperature range 30–650 °C in air atmosphere. The TGA/DTA curves of 3NAN are given in the Fig. 7. The TGA/DTA curve represent that the decomposition in the range of 190–650 °C. The three main stages of decomposition in 3NAN occurred within this temperature range as given Fig. 8. In TGA/DTA curve, weight loss 11% noted in temperature range 150–190 °C due to the elimination of water group from 3NAN, it indicates first stage of decomposition. The second stage of major weight loss 41% occurs at 190–220 °C temperature range, which is due to the liberation of NO and CO from the earlier intermediate compound. The next decomposition stage is owing to the removal of NO and C2H4 in the temperature range 220–420 °C with weight loss of 29%. An endothermic peak observed at 209 °C is may be the melting point of the compound. The soft exothermic peaks at 485 °C are due to the decomposition of the organic compound.

DTA/TGA curve of 3NAN

Decomposition steps of 3NAN
3.6 Antimicrobial activity analysis
To analyze the antimicrobial activity of 3NAN, the crystal was tested against Staphylococcus Aureus (G+ve) and Klebsiella Pneumoniae (G–ve) by disc diffusion method with 50 and 100 mg mL–1 concentrations. Similarly, the title compound antifungal activity was tested against Candida Albicans (C. albicans). The zone of inhibition were measured in diameter are shown in Table 6. The biological activity of 3-nitroanilinium nitrate complex results indicate, it is a moderately active material against all the organisms (Karthiga Devi et al. 2017; Chitradevi et al. 2015) (Figs. 9, 10, 11).

The antibacterial activity of 3NAN against Staphylococcus Aureus

The antibacterial activity of 3NAN against Klebsiella Pneumoniae

The antifungal activity of 3NAN against Candida Albicans
4 Conclusions
3-nitroanilinium nitrate was synthesized and characterized by various characterization techniques. The detailed interpretation of the vibrational spectra between calculated (IR, Raman) and computed spectra, experimental results of 3NAN molecule show the good reliability with computed results. The higher stability of the title molecule is confirmed from the computed HOMO–LUMO energy gap. TGA and DTA exposed about the material, thermal analysis confirmed the absence of phase transition before the material reach the melting point. Antimicrobial activity of the title compound was investigated.
References
Becke, A.D.: Density‐functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5653 (1993)
Bhuvaneswari, N., Priyadharsini, N., Sivakumar, S., Venkatachalam, K., Siva, V.: Growth, vibrational, optical, thermal, magnetic and dielectric behavior of organo-metallic tetramethyl ammonium cadmium chloride crystal. J. Therm. Anal. Calorim. 136, 411–417 (2018)
Bishop, D.M., Champagne, B., Kirtman, B.: Molecular vibration and nonlinear optics. J. Chem. Phys. 109, 9987–9994 (1998)
Chitradevi, A., Suresh Kumar, S., Athimoolam, S., Asath Bahadur, S., Sridhar, B.: Single crystal XRD, vibrational spectra, quantum chemical and thermal studies on a new semi-organic crystal: 4-Aminium antipyrine chloride. J. Mol. Struct. 1099, 58–67 (2015). https://doi.org/10.1016/j.molstruc.2015.06.022
David, W.I.F., Shankland, K., Vandestreek, J., Pidcock, E., Motherwell, W.D.S., Cole, J.C.: DASH: a program for crystal structure determination from powder diffraction data. J. Appl. Cryst. 39, 910–915 (2006)
Dennington, R., Keith, T., Millam, J., Gauss View Version 5.0.8 Semichem Inc., Shawnee Mission KS, (2013)
Drozd, M., Dudzic, D.: Guanidinium perchlorate ferroelectric crystal. Study of vibrational properties based on experimental measurements and theoretical calculations. Spectrochim. Acta Part A 115, 345–356 (2013). https://doi.org/10.1016/j.saa.2013.06.023
Fleming, I.: Frontier Orbitals and Organic Chemical Reactions. John Wiley & Sons, New York (1976)
Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A., Jr., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J.B., Ortiz, J.V., Cioslowski, J., Fox, D.J.: Gaussian 09, Revision B.01. Gaussian Inc., Wallingford (2010)
Hohenberg, P., Kohn, W.: Inhomogeneous electron gas. Phys. Rev. B 136, 864–871 (1964)
Jain, A.K., Mehta, S.C., Shrivastava, N.M.: Hypoglycemic and antihyperglycemic effects of newly synthesized sulfonyloxy derivatives of azalactone in normal and alloxan diabetic rabbits. Indi. J. Pharmaco. 37, 395–396 (2005)
Jean, Y., Volatron, F.: An Introduction to Molecular Orbitals. Oxford University Press, Oxford (2005)
Karthiga Devi, P., Venkatachalam, K.: Growth, spectroscopic, mechanical, thermal, antimicrobial and DFT studies of piperidinium hydrogen oxalate. J. Mater Sci: Mater Electron. 28, 8061–8073 (2017). https://doi.org/10.1007/s10854-017-6512-7
Krishnakumar, V., Nagalakshmi, R.: Terahertz generation in 3-Nitroaniline single crystals. Cryst. Growth Des. 8, 3882–3884 (2008)
Lee, C., Yang, W., Parr, R.G.: Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 37, 785–789 (1988)
Mary Novena, L., Suresh Kumar, S., Athimoolam, S., Saminathan, K., Sridhar, B.: Single crystal, vibrational and computational studies of Theophylline (a bronchodilator drug) and its chloride salt. J. Mol. Struct. 1133, 294–306 (2017). https://doi.org/10.1016/j.molstruc.2016.11.087
Palafox, M.A.: Scaling factors for the prediction of vibrational spectra. I. Benzene molecule. Int. J. Quant. Chem. 77, 661–684 (2000)
Prabavathi, N., Nilufer, A., Krishnakumar, V.: Spectroscopic (FT-IR, FT-Raman, UV and NMR) investigation, conformational stability, NLO Properties, HOMO-LUMO and NBO Analysis of Hydroxyquinoline Derivatives by Density Functional Theory Calculations. Spectrochim. Acta A 114, 449–474 (2013). https://doi.org/10.1016/j.saa.2013.05.011
Reis, H., Papadopoulos, M.G., Calaminici, P., Jug, K., Köster, A.M.: Calculation of macroscopic linear and nonlinear optical susceptibilities for the naphthalene, anthracene and meta-nitroaniline crystals. Chem. Phys. 261, 359 (2000)
Schlegel, H.B.: Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 3(2), 214–218 (1982)
Siva, V., Shameem, S., Suresh Kumar, S., Raja, M., Athimoolam, S., Asath Bahadur, S.: Structural, spectral, Quantum chemical and thermal studies on a new NLO crystal: Guanidinium Cinnamate. J. Mat. Sci: Mat. Electr. 28, 12484–12496 (2017a). https://doi.org/10.1007/s10854-017-7070-8
Siva, V., Suresh Kumar, S., Suresh, M., Raja, M., Athimoolam, S., Asath Bahadur, S.: N-H∙∙∙O hydrogen bonded novel nonlinear optical semiorganic crystal (4-methoxyanilinium trifluoroacetate) studied through theoretical and experimental methods. J. Mol. Struct. 1133, 163–171 (2017b). https://doi.org/10.1016/j.molstruc.2016.11.088
Siva, V., Asath Bahadur, S., Shameem, A., Murugan, A., Athimoolam, S., Suresh, M.: Supramolecular architecture, hydrogen bonding assembly and Hirshfeld surface analysis of charge transfer adipate salt of 4-methoxyaniline. Chem. Data Collect. 24, 100281 (2019)
Thangarasu, S., Siva, V., Athimoolam, S., Asath Bahadur, S.: Molecular structure, spectroscopic and quantum chemical studies on benzoic acid and succinic acid co-crystals of 2-Aminopyrimidine. J. Theor. Comput. Chem. 17(4), 1850021 (2018)
Acknowledgements
The authors are grateful to the management of Kalasalingam Academy of Research and Education, Krishnankoil, India for their support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Thangarasu, S., Siva, V., Asath Bahadur, S. et al. Structural, vibrational, quantum chemical calculations, thermal and antimicrobial studies on nitrate salt of 3-nitroaniline. Opt Quant Electron 53, 570 (2021). https://doi.org/10.1007/s11082-021-03146-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-021-03146-w