
Abstract
Ischemic stroke is a common neurological disease. Currently, there are no Food and Drug Administration-approved drugs that can maximize the improvement in ischemic stroke-induced nerve damage. Hence, treating ischemic stroke remains a clinical challenge. Ferroptosis has been increasingly studied in recent years, and it is closely related to the pathophysiological process of ischemic stroke. Iron overload, reactive oxygen species accumulation, lipid peroxidation, and glutamate accumulation associated with ferroptosis are all present in ischemic stroke. This article focuses on describing the relationship between ferroptosis and ischemic stroke and summarizes the relevant substances that ameliorate ischemic stroke-induced neurological damage by inhibiting ferroptosis. Finally, the problems in the treatment of ischemic stroke targeting ferroptosis are discussed, hoping to provide a new direction for its treatment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Strokes include ischemic stroke (IS) and hemorrhagic strokes. Ischemic strokes account for approximately 85% of all strokes worldwide [1], and its incidence is increasing yearly [2]. The etiology of IS varied [3], with depression [4], cognitive impairment [5], and many other complications. The pathogenesis of IS is complex and unclear [6], and the possible mechanisms include overproduction of reactive oxygen species(ROS), blood-brain barrier (BBB) disruption [7], oxidative Stress [8], secondary neuroinflammation [9], excitatory amino acid toxicity [10], and iron overloading. Studies have found that drugs to improve cerebral circulation, thrombolytic drugs [11], and neuroprotective agents [12]are effective in alleviating neurological impairment after cerebral ischemia, but there are still many problems with clinical application. For example, the thrombolytic drug tissue-type plasminogen activator (tPA), which has been approved by the U.S. Food and Drug Administration (FDA), has a certain time window (TTW) in clinical application [13]. Therefore, it is necessary to further explore the mechanism of IS and develop more drugs for treatment.
In recent years, studies have identified an iron-dependent form of regulated necrosis (RCD) [14] known as ferroptosis. Ferroptosis occurs mainly because of the excessive accumulation of ROS in cells in an iron-dependent form, and it is obviously different from other forms of cell death such as apoptosis, necrosis, autophagy, and pyrodeath with respect to morphology and function [15]. Previous studies on ferroptosis have focused on cancer [16], but in recent years, it has been found that ferroptosis is closely associated with a variety of neurological diseases including Parkinson’s [17] and stroke [18]. At the same time, a growing body of research suggests that IS is strongly associated with ferroptosis. There is a large body of literature on the pathologic changes of IS and the involvement of ferroptosis in the mechanistic process of IS; the use of relevant ferroptosis inhibitors in IS has also been reported. Thus, inhibition of ferroptosis is likely to ameliorate the neurologic impairment caused by IS. However, because of the complex and unclear mechanisms of IS pathology, few IS-related ferroptosis inhibitors have been applied to the clinic. This review systematically introduces the current pathomechanism of IS and the basic process of ferroptosis, explores the pathway of ferroptosis-mediated IS from several aspects, summarizes the relevant substances that improve IS by inhibiting ferroptosis (Table 1), and discusses the future direction of ferroptosis in IS research, with the hope of providing new ideas for the treatment of IS.
Ischemic Stroke
Stroke is the second-leading cause of death and the sixth-leading cause of disability worldwide, with a high proportion of IS. Interruption of cerebral blood flow due to thrombosis or embolism is the underlying pathology for the development of IS [19,20], which causes irreversible neuronal damage. The pathogenesis of IS is complex and unknown. We list below several of its common pathologies:
Blood-Brain Barrier Disruption and Iron Overload
The BBB comprises brain microvascular endothelial cells (BMECs) and their tight junctions (TJ), intact basement membranes, extracellular matrix, pericytes, and plasma membranes surrounded by astrocyte footplates, with the brain microvascular endothelial cells being the main component [21]. The BBB plays a crucial role in maintaining brain tissue homeostasis and limiting the entry of harmful extracellular substances from the bloodstream into the brain [22].
Hypoxia and glucose in the brain after IS causes microglia to release large amounts of matrix metalloproteinase (MMP), which degrades the extracellular matrix of the central nervous system (CNS) and alters the expression and distribution of tight junction (TJ) proteins [23], while MMP-9 promotes BBB disassembly [24,25]. After the destruction of the BBB, increased permeability of brain cell membranes and leakage of brain autoantigens induces secondary autoimmune responses to neuronal antigens and prolonged inflammation and a massive influx of extracellular iron into the brain tissue, resulting in iron accumulation [26], which further exacerbates metabolic disorders in brain tissue and triggers brain edema, iron metabolism disorders, immune infiltration, and other lesions [27]. It was found that serum ferritin levels were significantly higher in patients with cerebral ischemia. Accumulation of iron in the brain tissue and simultaneous generation of large amounts of lipid ROS further induce ferroptosis and promote cerebral infarction, resulting in severe neurological impairment. Therefore, inhibiting ferroptosis may be an effective therapeutic target for IS. Moreover, IS-induced disruption of the BBB has a limiting effect on the therapeutic window of thrombolytic drugs [28]. Therefore, repairing the BBB may also hold new promise for the treatment of IS.
Oxidative Stress
Oxidative stress is cellular damage caused by overproduction of ROS and reactive nitrogen species (RNS) and the imbalance between cellular oxidative and antioxidant systems. Oxidative stress damages organs, tissues, and their functions [29,30], which further leads to damage to DNA, lipids, and proteins and likely also causes severe neurological dysfunction. In cerebral ischemia and hypoxia, a large number of ROS free radicals are generated, which induce the expression of pro-inflammatory mediators aggravating brain tissue injury [31]. Meanwhile, a large accumulation of ROS would further induce ferroptosis and aggravate neurological damage. Therefore, ROS plays a key role in the pathomechanism of IS, and reducing ROS will further alleviate the neurological impairment post-IS, providing an effective therapeutic idea for IS.
Secondary Neuroinflammation
Immune cells in the brain parenchyma include innate immune cells and peripheral immune cells. The former plays an important role in the regulation of neuroinflammation [32], while the latter is the key to inducing neuroinflammation. Studies have shown that neuroinflammation plays a role in IS injury and can attenuate brain damage by removing senescent cells and debris.
After cerebral ischemia, microglia and astrocytes proliferate [33] and secrete pro-inflammatory factors such as interleukin 1β (IL-1β), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6) [9]. At the same time, the excess toxic ROS produced activate MMP, which further exacerbate inflammatory responses and infiltrate leukocytes [34], hence aggravating ischemic injury of the brain tissues. However, the exact mechanism of activation of microglial cells and astrocytes is currently unknown. A growing body of research suggests that neuroinflammation is closely related to IS, which is promising for IS treatment.
Excitatory Amino Acid Toxicity
Excitatory amino acids are closely related to the impairment of neurological function after cerebral ischemia. Glutamate (Glu) is involved in maintaining neurological function and is the most abundant neurotransmitter in the brain [35,36]. The phenomenon of neuronal death caused by the sustained release of excitatory amino acids is also known as “excitatory amino acid toxicity”.
After cerebral ischemia, nerve cells supply insufficient blood and oxygen, causing membrane potential depolarization. On the one hand, presynaptic membrane Ca2+ channels open and inhibit Glu reuptake by neurons, and Glu accumulates in large amounts in the interstitial space of the cell, which is toxic to neurons [37]. On the other hand, activation of the glutamate receptor N-methyl-D-aspartate receptor (NMDA) triggers excitatory amino acid toxicity in the brain [10,38], causes postischemic neuronal damage and oxidative stress, resulting in neuronal injury and even death. Studies have reported that the transcription of the Glu transporter protein Excitatory amino acid transporter 1 (EAAT1) gene is restrained during cerebral ischemia [39]. Meanwhile, the ability of astrocytes to take up Glu is diminished after brain tissue injury [40], but the exact mechanism is still unclear. Therefore, researchers believe that improving amino acid accumulation can alleviate neurological impairment after cerebral ischemia.
Mechanisms of Ferroptosis and Their Relation to Ischemic Stroke
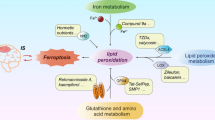
Studies have found multiple forms of cell death in IS, and ferroptosis is one of them [18,41]. Ferroptosis occurs mainly due to the inactivation of intracellular phospholipid peroxidase glutathione peroxidase 4 (GPX4), resulting in the inability of toxic lipid hydroperoxides (L-OOH) to be converted to non-toxic lipid alcohols (L-OH), and the consequent increase in the level of poisonous lipid ROS [42]. Three main metabolic pathways are involved: iron metabolism, lipid metabolism, and amino acid metabolism (Fig. 1). Furthermore, ferroptosis is subject to other avenues and mechanisms within the cell. An increasing number of studies have shown that serum iron levels are significantly higher in patients with IS, and brain tissue also exhibits significant iron deposition in animal models of IS [43,44]. It is suggested that there is a potential link between ferroptosis and IS and that intervening with the metabolic pathway of ferroptosis may provide new options for IS treatment.

Major metabolic mechanisms of ferroptosis and their mediation of IS pathways
Serum Transferrin (TF); The membrane protein Transferrin Receptor1 (TFR1); Six-Transmembrane Epithelial Antigen of Prostate 3 (STEAP3); Ceruloplasmin (CP); Divalent Metal Transporter 1 (DMT1); Recombinant Solute Carrier Family 7, Member 11 (SLC7A11); Recombinant Solute Carrier Family 3, Member 2 (SLC3A2); Polyunsaturated fatty acids (PUFAs); Phosphatidyl Ethanolamine-Polyunsaturated Fatty Acids (PE-PUFA); Lyso-Phosphatidylcholine Acyltransferase-3 (LPCAT3); Glutathione (GSH); Glutathione Peroxidase 4 (GPX4); oxidized glutathione, GSSG; glutamic acid (Glu); Cystine; Cys (Cys); Lipoxygenase (LOX); Arachidonic Acid (AA); Adrenaline (AdA); Lipid hydroperoxide (PL-PUFA-OOH); Lipid alcohol (PL-PUFA-OH); Acyl-CoA Synthetase Long-Chain Family Member 4 (ACSL4); Membrane iron transport protein 1 (Ferroportin1, FPN1); NMDA (NMDAR); Cytoplasmic phospholipase A2α (cPLA2α)
Relationship between iron Metabolism and Ischemic Stroke
Iron is necessary for lipid ROS accumulation and ferroptosis mechanisms; thus, any abnormal process of iron storage, release, or uptake may affect ferroptosis. Under normal conditions, free Fe3+ from the blood enters the cell via serum transferrin (TF) and the membrane protein transferrin receptor 1 (TFR1) and gets deposited in the nuclear endosomes, where it is reduced to Fe2+ by the enzyme Six-Transmembrane Epithelial Antigen of Prostate 3 (STEAP3) and then transported to the cell under the action of Divalent Metal Transporter 1 (DMT1). This later participates in the synthesis of metalloproteins in the cytoplasm or is stored directly in the cell [45]. When Fe2+ is in excess, the Fenton reaction of excess Fe2+ with hydrogen peroxide generates toxic lipid ROS and activates lipoxygenases (especially 15-LOX). The synthesized lipoxygenase (LOX) further participates in lipid peroxidation [46], inducing ferroptosis. Ferroportin 1 (FPN-1) is the only protein that transfers intracellular iron to the extracellular compartment [45], and ceruloplasmin (CP) converts Fe2+ to Fe3+ and translocates it to TF proteins [47] (Fig. 1). In some pathological states, when iron exceeds the iron-binding capacity of TF, non-transferrin-bound iron (NTBI) is produced in the cells, causing iron deposition in the cells [48,49].
The BBB permeability significantly changes after cerebral ischemia, and iron metabolism in the brain tissue is abnormal. The imbalance of iron intake and excretion induces ferroptosis. Intracellular iron uptake increases, acidic conditions generate separation of iron from TF, and the expression of ferritin, TFR1, and DMT1 is up-regulated in the brain. Moreover, iron removal from the cell is blocked, FPN-1 downward adjustment, and Tau expression is inhibited [18]. Large amounts of iron accumulate in the brain parenchyma, inducing the onset of the Fenton reaction and producing excessive lipid ROS in brain cells, exacerbating brain tissue damage. HIF-1α, BACH1, NCOA4, and ferredoxin are potent ferroptosis inhibitors [50] that modulate iron metabolism disorders after cerebral ischemia. For example, cerebral hypoxia promotes the binding of hypoxia-inducible factor-1 (HIF-1) to hypoxia-responsive progenitor (HRE), which upregulates DMT1, TF, and TFR1 expression [51]. However, inactivation of the HIF-1α subunit increases brain damage after IS [52], and DFO was also found to promote HIF-1α expression and inhibit cerebral ischemia [6]. Therefore, inhibiting iron overload might provide a new therapeutic strategy to alleviate the neurological impairment caused by IS.
Environmental enrichment, an effective intervention for IS, was found to down-regulate TFR1, DMT1, and MDA and up-regulate FPN1 and GPX4 expression after ischemia/reperfusion (I/R) injury, which significantly treated neuronal ferroptosis and alleviated neurological damage after IS and promoted the recovery of motor function in rats [53]. A study showed that inflammatory stimuli increased hepcidin expression and degraded FPN-1 [54]. It was found that the natural complex herbal Naotaifang extract (NTE) significantly down-regulated the expression of TFR1 and DMT1, reducing ROS, MDA, and iron accumulation in MCAO rats [55]. Moreover, in recent years, the flavonoid compound Cathamin yellow was found to inhibit TFR1 expression and down-regulate Fe2+ and ROS in a rat model of MCAO, effectively reducing the area of cerebral infarction as well as cerebral edema after IS [56,57]. At the same time, injection of desferrioxamine (DFO) [58] reduced the area of cerebral infarction after cerebral ischemia; whereas, liposome-coated DFO nanoparticles [59] delayed the recovery of neurological function after stroke. Moreover, the natural small molecule hinokitiol also can chelate iron and inhibit ferroptosis by affecting Fe2+ and up-regulating antioxidant factors, thereby ameliorating nerve damage after IS and showing stronger iron chelating ability than DFO [60,61]. In addition, iron chelators such as thymosin β4 [62], 1,10-fenpyrroline [63,64], 2,2’-bipyridine [64], and purine derivatives [55] have been found to play a role in other diseases by affecting free Fe2+ and reducing ROS levels. Therefore these iron chelators are also expected to offer hope for future IS treatments. Overall, modulation of iron metabolism is an effective way to ameliorate ferroptosis-mediated neurologic injury after IS.
Relationship between Lipid Metabolism and Ischemic Stroke
Lipid peroxidation is a central process in ferroptosis [65]. Acyl-Coenzyme A Synthetase Long-Chain Family Member 4 (ACSL4) promotes the binding of long-chain polyunsaturated fatty acids (PUFAs) to CoA, preferentially recognizes activated PUFA family arachidonic acid (AA)/adrenaline (AdA) [66], and acylates it to form AdA-CoA and AA-CoA derivatives in the oxidation center, which are then catalytically acylated and converted to AdA-PE and AA-PE in conjunction with lysophosphatidylcholine acyltransferase-3 (LPCAT3). Lipid peroxides are then produced by enzymatic and non-enzymatic reactions in the presence of lipoxygenase-15 (15-LOX) and Fe2+, respectively [67], thereby promoting lipid peroxidation and leading to ferroptosis (Fig. 1). The generation of lipid peroxides is accompanied by a chain reaction, which may result in more severe cellular damage [68]. Inhibition of lipid peroxidation has emerged as an important target for inhibiting ferroptosis at present.
Studies have shown that ROS generated by lipid peroxidation is the primary cause of brain damage and a key trigger of ferroptosis. Unlike other organs, the brain is rich in oxidation-reducing metal ions and unsaturated lipids and is prone to oxidative stress [69]. The pathways associated with lipid metabolism after cerebral ischemia are as follows:
-
(1)
LOX is a key oxygenase that induces ferroptosis. It has been found that LOX inhibitors can ameliorate IS-induced neurologic impairment. For example, injection of ALOX12/15 inhibitor in IS rats was effective in reducing the area of cerebral infarction and prolonging the time window in rats [70,71], and it reduced the risk of hemorrhage after thrombolysis in combination with tPA [51]. Hence, inhibition of LOX may be an effective target for treating IS [69,72]. (2) Cerebral ischemia and hypoxia lead to inadequate energy supply to the brain and massive ion leakage, resulting in membrane depolarization and release of neurotransmitters such as Glu. Glu accumulates in the middle cerebral artery occlusion (MCAO) rat model with high iron content. Excessive Glu release activates phospholipases and accelerates lipid peroxidation [73], while NMDA promotes iron accumulation, and DFO can protect against this damage. Thus, Glu can induce iron metabolism disorder and increased neural function impairment in brain tissue [74]. (3) After cerebral ischemia, Ca2+ inflow and activation of Ca2+ dependent cytoplasmic enzyme cPLA2α [18], promoting lipid peroxidation. Calcium antagonists have been shown to significantly reduce the size of cerebral infarcts and exert neuroprotective effects in rats. (4) Studies have shown that ACSL4 overexpression further aggravates brain tissue injury, and pro-inflammatory cytokine expression is reduced after down-regulation of ACLS4, suggesting that down-regulation of ACSL4 expression attenuates ischemic brain injury [75]. (5) It was found that both thrombin inhibitors and the potent bioactive ingredient astragaloside IV (As-IV) had inhibitory effects on ACSL4 [76,77]. (6) In addition, Danhong injection (DHI) was found to alleviate lipid peroxidation and inhibit ferroptosis by modulating the SATB1/SLC7A11/HO-1 signaling pathway and was neuroprotective in both an in vivo pMCAO model and an in vitro oxygen glucose deprivation (OGD) model [78]. (7) It has been shown that the lipid antioxidants ferritin-1 (Fer-1) [74] and liproxstatin-1 [79] are effective in alleviating neurological damage after cerebral ischemia. Free radical trapping antioxidants (RTAs) ameliorate post-ischemic neurological damage by trapping lipid peroxyl radicals such as α-tocopherol (α-TOH) [80] and diarylamine compounds [81] (phenoxazine, phenothiazine). It remains to be determined whether there are other RTAs that likely play a role in IS. In addition, in recent years, a novel small molecule compound, olanzapine, has been discovered, which is similar to Fer-1 and also exerts ferroptosis inhibition through antioxidationand improves the easy metabolism of Fer-1, and also brings new hope for future IS treatment [82].
Relationship between Amino Acid Metabolism and Ischemic Stroke
It is known that System Xc- and GPX4 are important pathways for blocking ferroptosis [83,84]. The System Xc-GSH-GPX4 axis is a critical step in the death of iron. Its carrier is a dimer comprising light chain solute carrier family 7, Member 11 (SLC7A11) and heavy chain solute carrier family 3, and Member 2 (SLC3A2) linked by disulfide bonds, which has high transport activity and specificity [85]. System Xc- transports intracellular Glu and takes up extracellular cystine in a 1:1 ratio. The rapid reduction of cystine to cysteine (Cys) is involved in the synthesis of GSH. This GSH is synthesized in the cytoplasm from cysteine, glycine (Gly), and Glu. GPX4 converts cellular reduced GSH to oxidized glutathione (GSSG). Meanwhile, GPX4 converts toxic lipid peroxides in cells to nontoxic lipid alcohol (PL-PUFA-OH) in the presence of GSH, maintaining redox homeostasis [86,87] (Fig. 1). It has been shown that the neurotransmitter dopamine [88] achieves the effect of limiting lipid peroxidation and inhibiting ferroptosis by enhancing GPX4 stability.
Glu is the principal excitatory neurotransmitter in the CNS. It is an essential factor in maintaining neural function. Studies have shown that after cerebral ischemia, brain metabolism is disturbed, Glu accumulates, and excess Glu inhibits the Xc−system, which in turn triggers ferroptosis [89,90]. Elevated lipid peroxidation levels and decreased GSH levels were detected in the MCAO model, thereby interfering with the synthesis and expression of GSH. GPX4 could effectively inhibit ferroptosis and alleviate neurological damage caused by IS. Meanwhile, exogenous GSH administration has a mitigating effect on post-IS neurological damage and promotes the neuroprotective effect of intra-striatal dopamine in post I/R rats [56,91].
Studies have shown that trace elements have a role in IS, such as: Se [92] which induces GPX4 transcriptional activation, plays an antioxidant role and alleviates neurological damage induced by cerebral ischemia. However, the specific mechanism is unknown. Zn2+ [93] is involved in postischemic BBB injury, and the over-accumulation of intracellular Zn2+ induces neuronal damage after ischemia. The free radical scavenger edaravone [94]is the only ferroptosis inhibitor currently used to treat cerebral ischemia by alleviating ferroptosis in cerebral ischemia caused by cystine deficiency. Furthermore, the monoterpene phenol carvacrol was found to up-regulate GPX4 levels and reduce peroxides in brain tissue, effectively improving memory and reducing hippocampal neuronal damage after IS in gerbils [95,96,97]. Meanwhile, in recent years, glycyrrhizin (GL), a natural glycosylated triterpenoid, has been found to inhibit ferroptosis and inflammatory factors by modulating the GPX4 pathway, which in turn ameliorates neuronal damage after neonatal hypoxic-ischemic brain damage (HIBD) [98]. In addition, natural products such as galangin [99], Angong Niuhuang Pills (AGNHW) [100], Kaempferol [101], and icaritin II [102,103] have also been found to inhibit ferroptosis and exhibit neuroprotective effects in IS by modulating the GPX4 pathway and thereby inhibiting ferroptosis. Thus, it is hoped that intervening in the Xc−system could result in an improvement of neurological impairment due to cerebral ischemia.
Relationship between Oxidative Stress and Ischemic Stroke
As is well known, oxidative cell damage is associated with the pathogenesis of many diseases. Studies have found that oxidative stress, which is caused by intracellular pro-oxidant accumulation and antioxidant deficiency, may be one of the important mechanisms that induce brain tissue damage after IS. Ferroptosis as a distinct oxidative stress-induced cell death pathway is mainly characterized by glutathione depletion and lipid peroxidation. (1) GPX4 converts GSH to GSSG, constituting a GSH/GSSG antioxidant system to eliminate oxides. (2) GPX4 converts L-OOH to L-OH in cells, which in turn reduces ROS [104] (Fig. 1).
NADPH oxidase 2 (NOX2) has been found to be a key enzyme for ROS production in cerebral ischemia-induced oxidative stress injury [105]. MiR-532-3p has been found to attenuate postischemic reperfusion injury by directly inhibiting NOX2 expression and activity [106]. Superoxide dismutase (SODs, a free radical scavenger in the body) can inhibit free radical damage and protect cells from oxidative stress [107]. It has been reported that the flavonoid compound astragalin effectively improves the neurological impairment caused by cerebral ischemia by regulating the SODs and MDA contents in the brain of model rats, thereby reducing lipid peroxidation, significantly reducing the area of cerebral infarction, and alleviating cerebral edema [108]. Meanwhile, poly(lactic acid)-glycolic acid copolymerization-nanoparticles (SOD-PLGA-NPs) showed enhanced free radical-scavenging ability and significant reduction of the cerebral infarct area in MCAO mice, which ultimately ameliorated neurological impairment after IS [109]. However, the mechanism of ROS and RNS accumulation during cerebral ischemia is still unclear. In addition, it has been found that electroshock (EA) [110] can alleviate cerebral ischemic injury and reduce the area of cerebral infarction in the MCAO model by up-regulating FSP1, GSH, and GPX4 in the brain tissues, and by down-regulating TFR1 in the Baihui, bilateral Sanyinjiao, and bilateral Neiguan acupoints [111,112]. Several studies have found that oxidative stress has been involved in the pathology of various diseases, including atherosclerosis, hypertension, and neurodegenerative diseases [29].
Relationship between Nuclear Factor Erythroid 2-Related Factor 2 and Ischemic Stroke
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a salient anti-oxidative stress transcription factor that improves cellular oxidative tolerance and plays an important role in inhibiting ferroptosis [113]. Recombinant Kelch-like ECH-Associated Protein 1 (Keap1) is a regulator of Nrf2. The downstream target proteins and enzymes associated with it are as follows:
Known regulators of ferroptosis: light and heavy chains of ferritin (FTL, FTH1) and FPN1, which is responsible for extracellular efflux from cells [114]. (2) Peroxisome proliferator-activated receptor γ (PPARγ): It is a ligand-activated receptor that synergizes with the Nrf2 pathway to inhibit ferroptosis [113]. (3) Enzymes used for GSH regulation: the glutathione synthesis gene γ-glutamylCys synthetase (GCS), the Glu-Cys ligase catalytic subunit (GCLC) [115], the subunit of the cystathionine/Glu transporter protein xCT, and the light-chain SLC7A11 [116]. (4) Enzymes related to NADPH regeneration: glucose-6-phosphate dehydrogenase (G6PD) and phosphogluconate dehydrogenase (PGD), and malic enzyme (ME) [117] are essential for GPX4 activity. Transcriptional regulators related to Nrf2 and many of its target genes have been shown to play important roles in the regulation of ferroptosis [115] and are the current new hotspots for ferroptosis therapy.
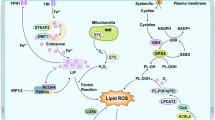
In summary, Nrf2 is a key transcription factor involved in ferroptosis and iron homeostasis in the brain. Several studies have shown that it is possible to affect Nrf2 to regulate ferroptosis and thereby treat neurodegenerative diseases, delay the progression of diabetic nephropathy (DN), and inhibit the death of vascular endothelial cells (VEC) in atherogenesis. Studies have shown that cerebral ischemia produces excess ROS that activate Nrf2. The Keap1/Nrf2 signaling pathway is the most important antioxidant defense pathway identified to date [118] (Fig. 2). Chrysanthemum extract (CME) modulates the Keap1/Nrf2 pathway to significantly inhibit oxidative stress and alleviate neurological damage after ischemia/reperfusion (CR/IR) in vivo and in vitro [119]. Elevated levels of Nrf2 were detected in the brains of experimental rats 2 h after MCAO-reperfusion (MCAO/R), which increased GSH synthesis and GPX4 transcription. In turn, Nrf2 binds with antioxidant response elements (AREs) in the promoter region to drive the expression of antioxidant genes for antioxidant effects. Under the influence of ROS, the Nrf2/ARE complex dissociates and Nrf2 translocates, triggering the voice of the ARE sequence genes in the promoter (Fig. 2) to produce endogenous antioxidants [120]. Nrf2 can form an antioxidant pathway with heme oxygenase (HO-1) (Fig. 2) to regulate cellular inflammation and oxidative stress [121]. Geraniin activates the HO-1-Nrf2 pathway, inhibits ROS production and anti-inflammatory factor expression, and significantly reduces neurological damage after cerebral ischemia [122]. A large number of studies have also shown that procyanidins (PACs) can ameliorate cerebral ischemia-reperfusion injury (CIRI) by activating the Nrf2-HO-1 pathway and thereby bringing a new direction for CIRI treatment [123].

Mechanisms of ferroptosis regulators Nrf2 and p53 involved in IS
Nuclear factor E2-related factor 2 (Nrf2); p53 (tumor suppressor); Recombinant Kelch-like ECH Associated Protein 1 (Keap1); Antioxidant response element (ARE); Heme oxygenase-1 (HO-1); Recombinant Solute Carrier Family 7, Member 11 (SLC7A11)
Silent information regulator factor 2-related enzyme 1 (SIRT1) [31] was found to upregulate Nrf2 and GPX4 in a hypoxic-ischemic brain injury (HIBI) model, and significantly improved cognitive deficits and exerted neuroprotective effects in rats [124]. Meanwhile, in 2017, Guo et al. found that the dithionite compound 3 H-1,2-Dithiole-3-thione (D3T) activated Nrf2 in the MCAO model and reduced the area of cerebral infarcts and ameliorated neurological impairments in rats after IS [125]. In addition, many inhibitors ameliorate ferroptosis after IS by acting on Nrf2, but the exact mechanism remains unclear. In addition, natural compounds such as 15,16-dihydrotanshinone I (DHT) [126], forsythia fruit extract forsythia glycoside A (FA) [127], and β-caryophyllene (BCP) [128] have been found to ameliorate ferroptosis after IS by acting on Nrf2. Meanwhile, exogenous melatonin administered to rats also showed significant ferroptosis inhibition and neuroprotective effects in rats [129]. In addition to this, there are several ferroptosis inhibitors that can play a role in other diseases by modulating the Nrf2 signaling pathway. For example, Withaferin A (WFA) inhibits neurological impairment after cerebral hemorrhage by activating the Nrf2/HO−1 pathway [130], Ajuga nipponensis (ADC) can also play a role in other classes of neurological disorders mediated by ferroptosis by scavenging free radicals and activating the Nrf2-AREs signaling pathway [131]. Thus these ferroptosis inhibitors also hold great promise for future use in the treatment of ferroptosis-mediated IS. In general, Nrf2 is critical for neurological damage after cerebral ischemia, and Nrf2 has the potential to become a hotspot for the treatment of IS.
Relationship between p53 and Ischemic Stroke
p53 is an important tumor suppressor gene that regulates ferroptosis [132]. It is divided into canonical and non-canonical pathways [133]. (1) Canonical pathway (GPX4 is a central factor): p53 promotes Fe3+ production and ROS and lipid peroxidation, while inhibiting GSH production directly inhibits GPX4 activity, which in turn promotes ferroptosis. (2) Non-canonical pathway: The p53/SLC7A11/ALOX12 and p53/SLC7A11/ALOXE3 axes cooperate and are independently involved in the regulation of ferroptosis, while not being dependent on GPX4 [133]. Under specific conditions, p53 is involved in the dual regulation of inhibition and promotion of ferroptosis [134]; furthermore, the regulation of ferroptosis is complex.
A growing body of research suggests that p53 plays an important role in central nervous system tumors, cardiovascular disease, IS, and neurodegenerative diseases [132,135]. It was shown that activation of p53 by cerebral ischemia, down-regulation of ferritin levels, up-regulation of SLC7A11 and GPX4, and inhibition of p53 activation reduced early brain damage associated with ferroptosis (Fig. 2). The small molecule protein-protein interaction (PPI) inhibitor K-181 has been reported to inhibit p53 transcription and alleviate postischemic nerve injury [136], and novel long-stranded noncoding RNAs [137] may have the same effect. In addition, the herbal Pueraria Mirifica extract Pueraria Mirifica was also found to be effective in alleviating brain tissue damage after IS by down-regulating p53 and it can also be used in conjunction with conventional therapies in IS treatment [112,138]. Therefore, the regulation of ferroptosis by inhibiting p53 activation is expected to be a new strategy for the treatment of IS.
Endoplasmic Reticulum Stress and Ischemic Stroke
The endoplasmic reticulum (ER) has since long played an important role in protein synthesis, folding, and structural maturation. A correlation was also found between ferroptosis and oxidative stress in the ER, usually expressed in Nrf2-HO-1, Xc-, and GPX4 [102]. When ER proteins are overloaded because of altered genetic or external conditions, including Ca2+ imbalance and altered ER lumen conditions, the cell will activate the unfolded protein response (UPR), thereby initiating the endoplasmic reticulum stress response to balance its protein folding capacity. However, once its regulatory capacity is exceeded, the endoplasmic reticulum stress will lead to cell death [139]. UPR activation is regulated by three type I transmembrane proteins upstream of it, including inositol-essential protein 1 (IRE1), activating transcription factor-6 (ATF6), and protein kinase RNA-like ER kinase (PERK) [102].
Several inhibitors have been found to inhibit the endoplasmic reticulum stress response by acting on ferroptosis, which in turn exhibits neuroprotective effects in IS models. In 2023, Ileriturk et al. found that carvacrol down-regulated GRP78, ATF-6, PERK, IRE1α, and CHOP gene expression and corrected λ-cyfluthrin-induced misfolding rates [96], and up-regulated GPX4 and inhibited ferroptosis in gerbil brain I/R injury thus exhibiting neuroprotective effects [97]. It was seen that baicalein could exhibit a significant inhibitory effect on ferroptosis in rat cerebral I/R injury by up-regulating GPX4 and ACSL4 in a transient MCAO (tMCAO) model [140]. Baicalein could also partially reduce ROS by suppressing endoplasmic reticulum stress [141]. In addition, there are natural agents such as BCP [142] and carvacrol [96,102] that are associated with endoplasmic reticulum stress. Thus, inhibition of endoplasmic reticulum stress may also be an effective way to inhibit ferroptosis and provide a new direction for neurologic recovery after IS.
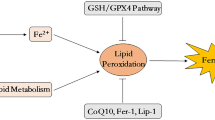
The above metabolic pathways regulate ferroptosis, which plays a role in alleviating the neurological damage after IS and is also a key target for researchers to develop novel ferroptosis inhibitors and provide a new direction for the treatment of IS. It is hoped that the inhibition of ferroptosis can provide new therapeutic options and effective drugs to improve the neurological impairments caused by IS. In addition to the above mechanisms, many other pathways mediating ferroptosis have been reported (Fig. 3). Therefore, interfering with these pathways may provide ideas for alleviation of neurological impairment after IS.

Other pathways mediate ferroptosis
Ferroptosis Inhibitory Protein 1 (FSP1); Coenzyme Q10 (CoQ10); Panthenol (CoQ10H2); Nicotinamide Adenine Dinucleotide Phosphate (NADPH); Dihydroorotate dehydrogenase (DHODH); GTP Cyclohydrolase-1 (GCH1); Tetrahydrobiopterin (BH4); GTP Cyclohydrolase-1-Tetrahydrobiopterin (GCH1-BH4); Mevalonate (MVA); Cys-tRNA Synthetase (CARS); Heat Shock Protein Beta-1 (HSPB1); Heat shock factor (HSF1)
Other Ferroptosis Pathways
The Nicotinamide Adenine Dinucleotide Phosphate-Ferroptosis Inhibitory Protein 1-Coenzyme Q10 Pathway
Ferroptosis Inhibitory Protein 1(FSP1) [143], a member of the type II NADH quinone oxidoreductase (NDH-2) family of enzymes, is a potent ferroptosis inhibitor [18,143,144]. At the same time, studies have shown that vitamin K (VK) is converted to the corresponding hydroquinone (VKH2) catalyzed by FSP1, which inhibits lipid peroxidation and thus ferroptosis [145,146]. Genetic screens revealed that FSP1 catalyzes H2O2 production and converts FAD to 6-hydroxy-FAD in the presence of O2 and NADPH thereby directly inhibiting ferroptosis [143,147]. Moreover, inhibitor diphenyl butene (DPT) has recently been found to inhibit ferroptosis through upregulation of FSP1, and its derivative 3f can alleviate neurological impairments after IS to a certain extent in the MCAO model [148]. In addition, it was found that Nrf2 and p53 can all act as FSP1 transcription factors to regulate FSP1 levels [149].
Coenzyme Q10 (CoQ10) is a lipophilic free radical trapping antioxidant that mediates the inhibition of ferroptosis by FSP1 [74]. FSP1 inhibits lipid peroxidation by reducing CoQ10 to ubiquinol (CoQ10H2) in the presence of NADPH, which traps and reduces lipid peroxides [146,147,150] Dihydroorotate dehydrogenase (DHODH), a ferroptosis inhibitor present in the mitochondria, independently reduces ubiquinone to ubiquinol to protect cells from lipid peroxidation [151] (Fig. 3). Administration of CoQ10H2 to neonatal rats after IS increases motor function and decreases infarct size [152]. The NADPH-FSP1-CoQ10 pathway cooperates with GSH and GPX4 to inhibit lipid peroxidation and ferroptosis. Therefore, interfering with FSP1 expression may become one of the hotspots for future ferroptosis research. Researchers hope that FSP1 has the potential to become an effective target for the treatment of IS in the future.
GTP Cyclohydrolase-1-tetrahydrobiopterin (GCH1-BH4) Pathway
GCH1 is another ferroptosis regulator that produces BH4. BH4 acts as a potent endogenous free radical-trapping antioxidant, reduces lipid peroxidation protecting cells from ferroptosis, and is also involved in the formation of CoQ10H2 [153] (Fig. 3). At the same time, GCH1 overexpression can eliminate lipid peroxidation [154] and can selectively enhance BH4 synthesis and reduce ROS production [155]. The GCH1-BH4 pathway is an endogenous antioxidant pathway. In recent years, the histone methylation transferase inhibitor BRD4770 was found to downregulate the GCH1-BH4 pathway and thus inhibit ferroptosis, which was comparable to Fer−1 inhibition at optimal concentrations [156]. And BRD4770 plays an inhibitory role in aortic dislocation (AD) caused by smooth muscle cell (SMC) loss [155]. GCH1 inhibitors have been reported to play an effective role in the treatment of colorectal cancer [157]. Researchers have explored the role of this pathway in neurodegenerative diseases. It is hoped that the GCH1-BH4 pathway may provide new therapeutic strategies for neurodegenerative diseases.
Heat Shock Factor 1-heat Shock Protein beta-1(HSF1-HSPB1) Pathway
HSF1 is an important transcription factor for cellular adaptation to stimuli that promote cancer development and plays an important role in HSPB1 transcription [158]. HSPB1 is a member of the small heat shock proteins family and is upregulated in breast cancer [159]. Meanwhile, HSPB1 was found to up-regulate G6PD, GPX4, SLC7A11, and TFR1 in an OGD-mediated neonatal hypoxic-ischemic brain damage (HIBD) model, and significantly reduced the area of cerebral infarction in rats [160]. In addition, combining the induction of ferroptosis with anticancer agents is a potential therapeutic strategy to overcome drug resistance in breast cancer [159]. Knockdown of HSF1 and HSPB1 increases the concentration of iron and ROS in cells, which in turn enhances elastin-induced ferroptosis and ultimately inhibits tumor growth, while HSPB1 overexpression inhibits ferroptosis [161]. HSF1 contributes to nuclear translocation and stabilization of p53 [162]. The HSF1-HSPB1 pathway can negatively regulate elastin-induced ferroptosis in human cervical cancer cells [163] and prostate cancer cells [164] (Fig. 3). However, this pathway has hardly been reported in neurological diseases. Hence, it is important to further explore the application of this pathway in neurological diseases to add a new treatment direction.
Sulfur Transfer Pathway
Deletion of Cys-tRNA Synthetase (CARS) (Fig. 3) increased intracellular Cys, GSH, and GSSG, while inhibiting ROS production, which in turn inhibited Xc−system-mediated ferroptosis, but had no effect on GPX4-induced ferroptosis [165]. Overall, the sulfur transfer pathway regulates ferroptosis to some extent. The sulfur transfer pathway plays a central role in the maintenance of redox homeostasis and oxidative stress response and has been implicated in a many diseases such as vascular dysfunction and Huntington’s disease (HD) and in aging. It is therefore speculated that the transsulfur pathway might also play a role in neurological disorders by regulating ferroptosis.
The Mevalonate Pathway
The mevalonate (MVA) pathway is an important route for regulating ferroptosis. The MVA pathway regulates selenoCys tRNA transporter activity involved in the synthesis of the selenoprotein GPX4, and isopentenyl pyrophosphate (IPP) promotes selenoCys tRNA maturation and GPX4 synthesis. SelenoCys tRNA maturation and GPX4 synthesis are impeded by MVA pathway inhibitors (e.g., statins) [166]. IPP is the precursor of CoQ10, and the MVA pathway synthesizes coenzyme CoQ10 in the presence of acetyl-coenzyme A (Acetyl-CoA) [167] (Fig. 3). Thus, the MVA pathway is involved in the Xc−GSH-GPX4 axis and the NADPH-FSP1-CoQ10 pathway. However, further studies on the relationship between MVA and ferroptosis are needed. MVA is expected to be a new target for the treatment of ferroptosis, and MVA has rarely been reported in IS. Hence, it also provides a new idea for the study of IS.
Clinical Translational Applications of Ferroptosis Inhibitors
Inhibitors of Ferroptosis that have Entered the Clinic for the Treatment of Ischemic Stroke
Ferroptosis as a new mode of cell death plays an important role in IS, and three ferroptosis inhibitors—edaravone dexborneol, thrombin inhibitor and Dl-3-n-butylphthalide (NBP), have already entered the clinical translational stage of IS treatment.
Edaravone dexborneol has synergistic effects in neuroprotection. In animal experiments, it was found to ameliorate neuronal damage and improve memory in mice after CIRI mainly through activating the Nrf2-HO-1 pathway and inhibiting NOX2 [170]. Moreover, it is currently in phase III clinical trials as a ferroptosis inhibitor, and administration within 48 h after acute IS (AIS) has revealed an increase in the number of patients with better neurological recovery on day 90, while the trials have demonstrated an improvement in both drug efficacy and drug safety profile [171]. Meanwhile the dual-target drug edaravone-dexamethasone has been approved for marketing in China in 2020. Edaravone, a neuroprotectant and free radical scavenger, has been approved for the treatment of AIS in Japan in 2021 [172]. It is also an inhibitor of ferroptosis that is currently marketed and widely used in the clinical treatment of IS. Edaravone significantly reduced the area of cerebral infarction after IS by upregulating Nrf2, FPN, and GPX4 and activating the Nrf2-FPN pathway [173]. Additionally, edaravone is approved by the FDA for the treatment of amyotrophic lateral sclerosis (ALS) [174]. Dexborneol is a natural terpene bicyclic organic compound with a protective effect on the brain. Studies have shown that edaravone dexborneol is significantly better than edaravone alone in alleviating neurological impairment after cerebral ischemia [171]. Therefore, the development of multi-targeted drugs provides a more effective clinical treatment for AIS patients, which is currently an important direction for optimizing neuroprotective agents and is expected to become a new trend in future drug development.
Thrombin has been reported to be one of the main targets of IS. It has also been shown that thrombin promotes ferroptosis and further promotes neuronal cell death by promoting ACSL4, thus inhibiting thrombin is expected to be one of the effective measures to inhibit ferroptosis. The thrombin inhibitor dabigatran is currently in a phase III (NCT03961334) clinical trial for IS [77]. The thrombin inhibitor argatroban has also been used in the acute phase of ischemic cerebral infarction, and studies have found that the combination of argatroban with aspirin has been shown to increase neuroprotection and significantly reduce the adverse prognosis [175].
NBP is a neuroprotective agent isolated from Apium graveolens Linn, which was approved by the China Food and Drug Administration (CFDA) for the treatment of IS as early as 2002 [176] and has completed FDA-approved Phase II clinical trials [177]. It exerts neuroprotective effects in IS mainly by lowering ROS, inhibiting inflammatory responses, reducing oxidative stress, and thus attenuating ferroptosis [177,178]. Meanwhile, NBP was found to significantly improve postoperative cognitive impairment in IS patients in a clinical study [179]. In addition, upregulation of Keap1 levels in patient’s serum and decreases in Nrf2, ARE, and NQO1 levels were found after administration of the drug in a study of 127 IS patients between January 2019 and January 2021 [180,181,182]. And zhang et al. obtained the same results in a rat MCAO model [179]. Meanwhile, Lu et al. found that NBP treatment upregulated SLC7A11 and GPX4 expression in rat neuronal cells in an MCAO model [178]. Meanwhile, NBP was found to synergistically improve IS neurological function and attenuate cognitive deficits with edaravone dextroamphetamine in the MCAO model [183].
Inhibitors of Ferroptosis that have Entered the Clinic for the Treatment of Other Diseases
In addition to the above inhibitors, other ferroptosis inhibitors have also been introduced for clinical use in recent years, so it can be conjectured whether these ferroptosis inhibitors could play a neuroprotective role in IS patients by inhibiting ferroptosis. For example, in 1991, Crapper McLachlan et al. found that intramuscular injections of DFO significantly improved cognitive performance in AD patients in a phase II clinical trial of AD [184,185]. In 2013, Guo et al. showed therapeutic promise when they found that intranasal injection of DFO reversed memory deficits in the APP/PS1 bi-genic mouse model [186,187]. Meanwhile, deferiprone (DFP), another iron chelator with the same function as DFO, is in a phase II clinical trial for the treatment of Parkinson’s disease (PD), where it significantly reduces brain iron deposition and improves motor function in patients [188]. In addition, the copper compound CuII (atsm) showed significant neuroprotective effects in phase I clinical trials in ALS and PD [189]. CuII (atsm) is currently in phase II clinical trials for ALS, after successful completion of phase I trials [185,190]. In addition, several iron chelators including DXZ [191], CPX [192], and DFX [193] have entered clinical trials for other diseases. It is encouraging to note that the above ferroptosis inhibitors have shown some neuroprotective effects in animal models of IS, but whether they can be successfully translated into clinical drugs needs to be further investigated.
Problems that may be Encountered in the Translational Clinical use of Ferroptosis Inhibitors
The development of drugs for neurological disorders has been a major challenge in the drug development phase for decades. Currently IS drug therapy is mostly symptomatic and no specific drug has been found. Although ferroptosis inhibitors are now available for clinical translation, there are still problems associated with their use. For example, taste disturbances may occur after administration of edaravone dextran in patients with hypertension [194]. In phase II, multicenter, randomized, double-blind, multi-dose, active-controlled clinical trials, patients were still found to experience adverse reactions such as itch, rash, acute liver injury, and renal injury [195]. In addition, there may be sex-related differences in thrombin production, so the use of thrombin inhibitors in male and female patients with AIS requires further attention [196]. In addition, several iron-death neuroprotective agents have still not translated successfully in clinical trials. Meanwhile, the brain is a very challenging organ, and the current very limited knowledge of the brain is also a major challenge for drug development [197]. In addition, the complexity of neurological injury response and the single mechanism of drug research are also important reasons for the low number of effective drugs for IS in recent years. Furthermore, basic research mainly evaluates early outcomes, while clinical trials need to evaluate long-term efficacy. Therefore, the clinical translation of ferroptosis inhibitors still faces great challenges, and more research, including basic experiments in animal models and clinical trials, is needed to understand in greater detail the process of brain cell death in disease, further improve the specificity of drugs targeting ferroptosis, and reduce adverse effects [185].
Discussion and Conclusion
Although improvements have been made over the decades for IS prevention and post-IS neurologic impairment, it is still very challenging to treat IS completely. Acute stress in the brain after cerebral ischemia is complex and unclear, with inflammatory responses and immune processes playing a key role in the development of IS. Cerebral ischemic injury is an inflammatory stimulus-response. After cerebral ischemia, microglia and astrocytes are activated and secrete pro-inflammatory factors, but exactly how they are activated is unclear. Investigations have found that drugs such as anticoagulants, antiplatelet agents, thrombolytics, anti-inflammatory drugs, and neuroprotectants, alone or in combination, are effective in alleviating neurological impairment after cerebral ischemia, but they mostly lack targeting and have been used less frequently in clinical practice. Therefore, exploring new drug targets is crucial to alleviate the neurologic impairment caused by IS.
In recent years, a growing body of literature has reported that ferroptosis is closely related to IS. Ferroptosis has also been found to play an important role in neurological diseases in recent years. Disturbed iron metabolism, lipid peroxidation, and ROS accumulation are key to ferroptosis. Numerous studies have shown that after cerebral ischemia, the iron levels of brain tissue are significantly elevated, accelerating lipid peroxidation, promoting the occurrence of ferroptosis, and exacerbating the damage to neurological function. In this paper, we specifically summarize the pathological mechanisms of IS, describe the mechanistic pathways related to ferroptosis, and introduce the mechanism of ferroptosis-mediated IS from four aspects: iron metabolism, lipid metabolism, amino acid metabolism, and oxidative stress. The effects of two ferroptosis regulators, Nrf2 and p53, and their related pathways on alleviating neurological impairment after cerebral ischemia by regulating ferroptosis have also been introduced. The application of related ferroptosis inhibitors in alleviating the neurological impairment caused by IS is summarized, which brings new hope for the treatment of IS. There are many enzymes related to ferroptosis which play an important role in IS. It has been reported that Ring Finger Protein 146 (RNF146) was overexpressed in the MCAO mouse model, preventing the protein expression of Fe2+ in genes related to ferroptosis, including SLC7A11 and GPX4, and alleviating neurological impairment after cerebral ischemia, while RNF146 inhibition significantly enhanced ACLS4 levels [198]. ACSL4 is the key to ferroptosis enforcement [199] and has been associated with cerebral postischemic reperfusion injury, renal injury, cancer, and other diseases.
There are still several limitations in IS therapy targeting ferroptosis. First, the pathological mechanism of IS is very complex, and the specific mechanism is still unclear. Although there is a certain relationship between ferroptosis and the pathological mechanism of IS, the specific details need to be explored more extensively. Studies have shown that ferroptosis can be accompanied by inflammation, which is closely related to oxidative stress and affects the expression of factors such as Nrf2, GPX4, and GSH [200], but studies targeting inflammation in ferroptosis are still in the early stages. Second, IS is accompanied by multiple forms of cell death modalities, and there are signaling pathways that interact between ferroptosis and other cell death modalities, but their relationship needs to be further investigated. Third, although some of the ferroptosis mechanisms (e.g., GCH1-BH4 pathway, HSF1-HSPB1 pathway, sulfur transfer pathway, MVA pathway) have been reported, their specific mechanisms are still unknown, and there is no research on whether the above mechanisms can play a role in IS, which also provides more targets for researchers to study ferroptosis-mediated IS. Fourth, small-molecule ferroptosis inhibitors suffer from low targeting, low solubility, and low clinical use. Targeted ferroptosis nanomedicines have been reported to ameliorate these problems [201] and have been reported in ferroptosis-mediated diseases such as cancer and tumors [202]. Meanwhile, some studies have shown that the combination of nanoliposomal Fasudil (Fasudil-Lip) and tPA effectively alleviated the neurological impairment caused by cerebral ischemia and prolonged the TTW of tPA [203], which further improved the drug targeting. Progress has also been made in recent years with penetration-targeted nano-delivery systems via the BBB. Therefore, future research should focus on combining small-molecule ferroptosis inhibitors with nanoparticles.
Although there are still many problems with ferroptosis-based regulatory mechanisms for therapeutic application in IS, ferroptosis still has great research prospects and development potential for alleviating neurological impairments after IS.
References
Xu S, Li X, Wang Y (2023) Regulation of the p53–mediated ferroptosis signaling pathway in cerebral ischemia Stroke (review). Experimental and Therapeutic Medicine, 25(3)
Wei Z et al (2022) New insights in ferroptosis: potential therapeutic targets for the treatment of ischemic Stroke. Front Pharmacol, 13
Li Y-X et al (2022) Advances in the research of nano delivery systems in ischemic Stroke. Front Bioeng Biotechnol, 10
Li Y et al (2021) Paradoxical effect of statin medication on depressive disorder in first-ever ischemic Stroke patients: possible antidepressant-like effect prestroke and the opposite in continuous medication poststroke. Int Clin Psychopharmacol 36(3):147–153
Kwon HS et al (2020) Post-stroke cognitive impairment as an Independent predictor of ischemic Stroke recurrence: PICASSO sub-study. J Neurol 267(3):688–693
Liu C et al (2024) Ferroptosis: a potential therapeutic target for Stroke. Neural Regen Res 19(5):988–997
Guo X et al (2023) Ischemia Reperfusion Injury Induced Blood Brain Barrier Dysfunction and the involved molecular mechanism. Neurochem Res 48(8):2320–2334
Orellana-Urzúa S et al (2020) Pathophysiology of ischemic Stroke: role of oxidative stress. Curr Pharm Des 26(34):4246–4260
Jayaraj RL et al (2019) Neuroinflammation: friend and foe for ischemic Stroke. J Neuroinflamm, 16(1)
Amantea D, Bagetta G (2017) Excitatory and inhibitory amino acid neurotransmitters in Stroke: from neurotoxicity to ischemic tolerance. Curr Opin Pharmacol 35:111–119
Lyden S, Wold J (2022) Acute Treatment of ischemic Stroke. Neurol Clin 40(1):17–32
Xu H et al (2021) Neuroprotective Phytochemicals in Experimental Ischemic Stroke: Mechanisms and Potential Clinical Applications Oxid Med Cell Longev, 2021: p. 6687386
Yoon E-J et al (2022) The neuroprotective effects of Exosomes Derived from TSG101-Overexpressing human neural stem cells in a Stroke model. Int J Mol Sci, 23(17)
Chen X et al (2021) Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol 18(5):280–296
Tang D et al (2021) Ferroptosis: molecular mechanisms and health implications. Cell Res 31(2):107–125
Mou Y et al (2019) Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol 12(1):34
Wu L et al (2023) The role of Iron metabolism, lipid metabolism, and Redox Homeostasis in Alzheimer’s Disease: from the perspective of Ferroptosis. Mol Neurobiol 60(5):2832–2850
Xu Y et al (2023) Role of ferroptosis in Stroke. Cell Mol Neurobiol 43(1):205–222
Qin C et al (2022) Signaling pathways involved in ischemic Stroke: molecular mechanisms and therapeutic interventions. Signal Transduct Target Therapy, 7(1)
Nair R, Wagner AN, Buck BH (2023) Advances in the management of acute ischemic Stroke. Curr Opin Neurol 36(2):147–154
Whelan R, Hargaden GC, Knox AJS (2021) Modulating the blood–brain barrier: a Comprehensive Review. Pharmaceutics, 13(11)
Zeng M et al (2021) Danhong injection enhances the therapeutic effect of mannitol on hemispheric ischemic Stroke by ameliorating blood-brain barrier disruption. Biomedicine & Pharmacotherapy, p 142
Zheng X, Ren B, Gao Y (2023) Tight junction proteins related to blood-brain barrier and their regulatory signaling pathways in ischemic Stroke. Biomed Pharmacother 165:115272
Chen X et al (2022) The Molecular mechanisms of Ferroptosis and its role in blood-brain barrier dysfunction. Front Cell Neurosci, 16
Abdullahi W, Tripathi D, Ronaldson PT (2018) Blood-brain barrier dysfunction in ischemic Stroke: targeting tight junctions and transporters for vascular protection. Am J Physiol Cell Physiol 315(3):C343–c356
Tiwari YV et al (2016) Magnetic resonance imaging of blood–brain barrier permeability in ischemic Stroke using diffusion-weighted arterial spin labeling in rats. J Cereb Blood Flow Metabolism 37(8):2706–2715
Zhao Y et al (2022) The role of ferroptosis in blood–brain barrier Injury. Cell Mol Neurobiol 43(1):223–236
Qiu L et al (2022) Mesenchymal stem cell-derived extracellular vesicles attenuate tPA-induced blood-brain barrier disruption in murine ischemic Stroke models. Acta Biomater 154:424–442
Su XT et al (2020) Mechanisms of Acupuncture in the Regulation of Oxidative Stress in Treating Ischemic Stroke Oxid Med Cell Longev, 2020: p. 7875396
Pawluk H et al (2022) Increased oxidative stress markers in Acute ischemic Stroke patients treated with thrombolytics. Int J Mol Sci, 23(24)
Tang H et al (2023) New insights into Sirt1: potential therapeutic targets for the treatment of cerebral ischemic Stroke. Front Cell Neurosci 17:1228761
Lu W, Chen Z, Wen J (2023) The role of RhoA/ROCK pathway in the ischemic stroke-induced neuroinflammation. Biomedicine & Pharmacotherapy, p 165
Jayaraj RL et al (2019) Neuroinflammation: friend and foe for ischemic Stroke. J Neuroinflammation 16(1):142
Cheng W et al (2023) Neuroinflammation and brain–peripheral interaction in ischemic Stroke: a narrative review. Front Immunol, 13
Ge L et al (2022) Research Progress on Neuroprotection of insulin-like growth Factor-1 towards Glutamate-Induced neurotoxicity. Cells, 11(4)
Lewerenz J, Maher P (2015) Chronic glutamate toxicity in neurodegenerative diseases-what is the evidence? Front Neurosci 9:469
Kostandy BB (2012) The role of glutamate in neuronal ischemic injury: the role of spark in Fire. Neurol Sci 33(2):223–237
Ren J-X et al (2021) Crosstalk between Oxidative Stress and Ferroptosis/Oxytosis in Ischemic Stroke: Possible Targets and Molecular Mechanisms Oxidative Medicine and Cellular Longevity, 2021: p. 1–13
Malik AR, Willnow TE (2019) Excitatory amino acid transporters in Physiology and disorders of the Central Nervous System. Int J Mol Sci, 20(22)
Liu Y et al (2021) Exogenous Adenosine antagonizes excitatory amino acid toxicity in primary astrocytes. Cell Mol Neurobiol 41(4):687–704
Adedoyin O et al (2018) Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am J Physiology-Renal Physiol 314(5):F702–F714
Yang F et al (2023) Ferroptosis heterogeneity in triple-negative Breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab 35(1):84–100e8
Petrova J et al (2015) Ischemic Stroke, inflammation, iron overload – connection to a hepcidin. Int J Stroke 11(1):NP16–NP17
Lan B et al (2020) Extract of Naotaifang, a compound Chinese herbal medicine, protects neuron ferroptosis induced by acute cerebral ischemia in rats. J Integr Med 18(4):344–350
Zhao D et al (2023) Mechanisms of ferroptosis in Alzheimer’s Disease and therapeutic effects of natural plant products: a review. Biomed Pharmacother 164:114312
Liu J, Kang R, Tang D (2022) Signaling pathways and defense mechanisms of ferroptosis. Febs j 289(22):7038–7050
Liu H et al (2019) Brain ceruloplasmin expression after experimental Intracerebral Hemorrhage and Protection Against Iron-Induced Brain Injury. Transl Stroke Res 10(1):112–119
Vogt A-CS et al (2021) On Iron Metabolism and its regulation. Int J Mol Sci, 22(9)
Rodríguez-Graciani KM et al (2022) Effects of Ferroptosis on the Metabolome in Cardiac cells: the role of Glutaminolysis. Antioxidants, 11(2)
She X et al (2020) Cross Talk between ferroptosis and cerebral ischemia. Front NeuroSci, 14
Yigitkanli K et al (2017) Increased 12/15-Lipoxygenase leads to widespread brain Injury following global cerebral ischemia. Transl Stroke Res 8(2):194–202
Vatte S, Ugale R (2023) HIF-1, an important regulator in potential new therapeutic approaches to ischemic Stroke. Neurochem Int 170:105605
Luo Q et al (2023) Enriched environment attenuates ferroptosis after cerebral ischemia/reperfusion injury by regulating iron metabolism. Brain Res Bull 203:110778
Chen Y et al (2022) BRD4770 functions as a novel ferroptosis inhibitor to protect against Aortic Dissection. Pharmacol Res, 177
Joshi S et al (2022) Inhibiting Erastin-Induced Ferroptotic Cell death by Purine-Based Chelators. ChemBioChem 23(9):e202100654
Hu X et al (2023) The role of ferroptosis and its mechanism in ischemic Stroke. Exp Neurol 372:114630
Guo H et al (2021) Carthamin yellow improves cerebral ischemia–reperfusion injury by attenuating inflammation and ferroptosis in rats. Int J Mol Med, 47(4)
Millán M et al (2021) Targeting Pro-oxidant Iron with Deferoxamine as a treatment for ischemic Stroke: safety and optimal dose selection in a Randomized Clinical Trial. Antioxid (Basel), 10(8)
Guo X et al (2022) Iron promotes neurological function recovery in mice with ischemic Stroke through endogenous repair mechanisms. Free Radic Biol Med 182:59–72
Jayakumar T et al (2013) Hinokitiol, a natural tropolone derivative, offers neuroprotection from thromboembolic stroke in vivo Evid Based Complement Alternat Med, 2013: p. 840487
Xi J et al (2022) Hinokitiol functions as a ferroptosis inhibitor to confer neuroprotection. Free Radic Biol Med 190:202–215
Lachowicz JI et al (2022) Thymosin β4 is an endogenous Iron Chelator and Molecular Switcher of Ferroptosis. Int J Mol Sci, 23(1)
Huang KJ et al (2019) Assessment of zero-valent iron-based nanotherapeutics for ferroptosis induction and resensitization strategy in cancer cells. Biomater Sci 7(4):1311–1322
Chen X et al (2020) Iron Metabolism in Ferroptosis. Front Cell Dev Biol 8:590226
Lin Z et al (2021) Lipid metabolism in Ferroptosis. Adv Biol (Weinh) 5(8):e2100396
Kagan VE et al (2017) Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13(1):81–90
Ursini F, Maiorino M (2020) Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med 152:175–185
Jia B et al (2023) ACSL4-Mediated ferroptosis and its potential role in Central Nervous System Diseases and injuries. Int J Mol Sci, 24(12)
Guo J, Tuo QZ, Lei P (2023) Iron, ferroptosis, and ischemic Stroke. J Neurochem 165(4):487–520
Zhao J et al (2022) Activation of SSAT1/ALOX15 Axis aggravates cerebral Ischemia/Reperfusion Injury via triggering neuronal ferroptosis. Neuroscience 485:78–90
Sun S et al (2023) Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct Target Ther 8(1):372
Proneth B, Conrad M (2019) Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ 26(1):14–24
Ren JX et al (2021) Crosstalk between Oxidative Stress and Ferroptosis/Oxytosis in Ischemic Stroke: Possible Targets and Molecular Mechanisms Oxid Med Cell Longev, 2021: p. 6643382
Yang K et al (2022) The mechanism of ferroptosis regulating oxidative stress in ischemic Stroke and the regulation mechanism of natural pharmacological active components. Biomed Pharmacother 154:113611
Cui Y et al (2021) ACSL4 exacerbates ischemic Stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav Immun 93:312–321
Jin Z et al (2023) Astragaloside IV alleviates neuronal ferroptosis in ischemic Stroke by regulating fat mass and obesity-associated-N6-methyladenosine-acyl-CoA synthetase long-chain family member 4 axis. J Neurochem 166(2):328–345
Tuo QZ et al (2022) Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal Transduct Target Ther 7(1):59
Zhan S et al (2023) SATB1/SLC7A11/HO-1 Axis ameliorates ferroptosis in Neuron cells after ischemic Stroke by Danhong Injection. Mol Neurobiol 60(1):413–427
Feng Y et al (2019) Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem Biophys Res Commun 520(3):606–611
Maiorino M, Conrad M, Ursini F (2018) GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and Open issues. Antioxid Redox Signal 29(1):61–74
Shah R, Margison K, Pratt DA (2017) The potency of Diarylamine Radical-Trapping antioxidants as inhibitors of ferroptosis underscores the role of Autoxidation in the mechanism of cell death. ACS Chem Biol 12(10):2538–2545
Jiang X et al (2023) Discovery and optimization of olanzapine derivatives as new ferroptosis inhibitors. Bioorg Chem 133:106393
Ma Y et al (2020) Energy metabolism as a regulator of ferroptosis. Cell Cycle 19(22):2960–2962
Chen Y et al (2022) Ferroptosis: a New Strategy for Cancer Therapy. Front Oncol 12:830561
Poltorack CD, Dixon SJ (2022) Understanding the role of cysteine in ferroptosis: progress & paradoxes. Febs j 289(2):374–385
Hu W et al (2022) Ferroptosis and its role in Chronic Diseases. Cells, 11(13)
Rodríguez-Graciani KM et al (2022) Effects of Ferroptosis on the Metabolome in Cardiac cells: the role of Glutaminolysis. Antioxid (Basel), 11(2)
Sun J et al (2023) Midbrain dopamine oxidation links ubiquitination of glutathione peroxidase 4 to ferroptosis of dopaminergic neurons. J Clin Invest, 133(10)
Shen Z et al (2022) Glutamate excitotoxicity: potential therapeutic target for ischemic Stroke. Biomed Pharmacother 151:113125
Fan G et al (2023) The initiator of neuroexcitotoxicity and ferroptosis in ischemic Stroke: glutamate accumulation. Front Mol Neurosci 16:1113081
Wang H et al (2022) Exogenous glutathione exerts a therapeutic effect in ischemic Stroke rats by interacting with intrastriatal dopamine. Acta Pharmacol Sin 43(3):541–551
Alim I et al (2019) Selenium drives a Transcriptional Adaptive Program to Block Ferroptosis and treat Stroke. Cell 177(5):1262–1279e25
Mirończuk A et al (2021) Selenium, copper, zinc concentrations and Cu/Zn, Cu/Se Molar Ratios in the serum of patients with Acute ischemic Stroke in northeastern Poland-A New Insight into Stroke Pathophysiology. Nutrients, 13(7)
Wang SN et al (2023) Humanized cerebral organoids-based ischemic Stroke model for discovering of potential anti-stroke agents. Acta Pharmacol Sin 44(3):513–523
Li Z et al (2016) Carvacrol exerts neuroprotective effects Via suppression of the inflammatory response in Middle cerebral artery occlusion rats. Inflammation 39(4):1566–1572
Ileriturk M, Kandemir FM (2023) Carvacrol protects against λ-Cyhalothrin-induced hepatotoxicity and nephrotoxicity by modulating oxidative stress, inflammation, apoptosis, endoplasmic reticulum stress, and autophagy. Environ Toxicol 38(7):1535–1547
Guan X et al (2019) The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation. Life Sci 235:116795
Zhu K et al (2022) Glycyrrhizin Attenuates Hypoxic-Ischemic Brain Damage by Inhibiting Ferroptosis and Neuroinflammation in Neonatal Rats via the HMGB1/GPX4 Pathway Oxid Med Cell Longev, 2022: p. 8438528
Guan X et al (2021) Galangin attenuated cerebral ischemia-reperfusion injury by inhibition of ferroptosis through activating the SLC7A11/GPX4 axis in gerbils. Life Sci 264:118660
Bai X et al (2023) Angong Niuhuang Wan inhibit ferroptosis on ischemic and hemorrhagic Stroke by activating PPARγ/AKT/GPX4 pathway. J Ethnopharmacol 117438
Yuan Y et al (2021) Kaempferol Ameliorates Oxygen-Glucose Deprivation/Reoxygenation-Induced Neuronal Ferroptosis by Activating Nrf2/SLC7A11/GPX4 Axis Biomolecules, 11(7)
Li Y et al (2024) Ferroptosis and endoplasmic reticulum stress in ischemic Stroke. Neural Regen Res 19(3):611–618
Xu H et al (2022) Molecular Mechanism of Epimedium Extract against Ischemic Stroke Based on Network Pharmacology and Experimental Validation Oxid Med Cell Longev, 2022: p. 3858314
Forcina GC, Dixon SJ (2019) GPX4 at the crossroads of lipid homeostasis and Ferroptosis. Proteomics 19(18):e1800311
Yingze Y et al (2022) NOX2-mediated reactive oxygen species are double-edged swords in focal cerebral ischemia in mice. J Neuroinflammation 19(1):184
Mao L et al (2020) Low expression of miR–532–3p contributes to cerebral ischemia/reperfusion oxidative stress injury by directly targeting NOX2. Mol Med Rep 22(3):2415–2423
Yang X et al (2021) Superoxide Dismutase Gene Polymorphism is Associated with ischemic Stroke risk in the China Dali Region Han Population. Neurologist 26(2):27–31
Chen X et al (2020) Astragalin alleviates cerebral ischemia-reperfusion injury by improving anti-oxidant and anti-inflammatory activities and inhibiting apoptosis pathway in rats. BMC Complement Med Ther 20(1):120
Li YX et al (2022) Advances in the research of nano delivery systems in ischemic Stroke. Front Bioeng Biotechnol 10:984424
Liang R et al (2021) Electroacupuncture Preconditioning Reduces Oxidative Stress in the Acute Phase of Cerebral Ischemia-Reperfusion in Rats by Regulating Iron Metabolism Pathways Evid Based Complement Alternat Med, 2021: p. 3056963
Li G et al (2021) Electroacupuncture ameliorates cerebral ischemic Injury by inhibiting Ferroptosis. Front Neurol 12:619043
Lou Y et al (2022) Ferroptosis: a new strategy for traditional Chinese medicine treatment of Stroke. Biomed Pharmacother 156:113806
Duan C et al (2022) Activation of the PPARγ prevents Ferroptosis-Induced neuronal loss in response to Intracerebral Hemorrhage through synergistic actions with the Nrf2. Front Pharmacol 13:869300
Song X, Long D (2020) Nrf2 and ferroptosis: a New Research Direction for neurodegenerative Diseases. Front Neurosci 14:267
Kerins MJ, Ooi A (2018) The roles of NRF2 in modulating Cellular Iron Homeostasis. Antioxid Redox Signal 29(17):1756–1773
Li S et al (2021) Inhibition of ferroptosis by up-regulating Nrf2 delayed the progression of diabetic Nephropathy. Free Radic Biol Med 162:435–449
Abdalkader M et al (2018) Targeting Nrf2 to suppress ferroptosis and mitochondrial dysfunction in Neurodegeneration. Front Neurosci 12:466
Wang L et al (2022) Nrf2 regulates oxidative stress and its role in cerebral ischemic Stroke. Antioxid (Basel), 11(12)
Zhang Z et al (2023) Neuroprotective effects of Chrysanthemum morifolium on cerebral ischemia- reperfusion injury contributes to the oxidative stress suppression and related Keap1/Nrf2 pathway. Brain Inj 37(4):269–281
Mazur A et al (2021) Nrf2 as a therapeutic target in ischemic Stroke. Expert Opin Ther Targets 25(3):163–166
Shin EJ et al (2019) Cytoprotective effects of an aqueous extracts from Atrina Pectinate Meat in H(2)O(2)-Induced oxidative stress in a human hepatocyte. Adv Exp Med Biol 1155:661–674
Yang Y et al (2022) Geraniin Protects against Cerebral Ischemia/Reperfusion Injury by Suppressing Oxidative Stress and Neuronal Apoptosis via Regulation of the Nrf2/HO-1 Pathway Oxid Med Cell Longev, 2022: p. 2152746
Chen L et al (2023) Procyanidins Alleviated Cerebral Ischemia/Reperfusion Injury by inhibiting ferroptosis via the Nrf2/HO-1 signaling pathway. Molecules, 28(8)
Li C et al (2022) Ferroptosis contributes to hypoxic-ischemic brain injury in neonatal rats: role of the SIRT1/Nrf2/GPx4 signaling pathway. CNS Neurosci Ther 28(12):2268–2280
Kuo PC et al (2017) -1,2-Dithiole-3-thione as a novel therapeutic agent for the treatment of ischemic Stroke through Nrf2 defense pathway. Brain Behav Immun 3:180–192
Wu C et al (2023) 15, 16-Dihydrotanshinone I protects against ischemic Stroke by inhibiting ferroptosis via the activation of nuclear factor erythroid 2-related factor 2. Phytomedicine 114:154790
Zhang L et al (2024) Review of the therapeutic potential of Forsythiae Fructus on the central nervous system: active ingredients and mechanisms of action. J Ethnopharmacol 319(Pt 2):117275
Hu Q et al (2022) β-Caryophyllene suppresses ferroptosis induced by cerebral ischemia reperfusion via activation of the NRF2/HO-1 signaling pathway in MCAO/R rats. Phytomedicine 102:154112
Deng X et al (2023) Nrf2 and ferroptosis: a New Research Direction for ischemic Stroke. Cell Mol Neurobiol 43(8):3885–3896
Zhou ZX et al (2023) Withaferin A inhibits ferroptosis and protects against intracerebral Hemorrhage. Neural Regen Res 18(6):1308–1315
Tan Q et al (2021) A new ferroptosis inhibitor, isolated from Ajuga Nipponensis, protects neuronal cells via activating NRF2-antioxidant response elements (AREs) pathway. Bioorg Chem 115:105177
Lei L et al (2022) P53 protein and the Diseases in central nervous system. Front Genet 13:1051395
Xu R, Wang W, Zhang W (2023) Ferroptosis and the bidirectional regulatory factor p53. Cell Death Discov 9(1):197
Zhang L et al (2022) Post-translational modifications of p53 in ferroptosis: Novel pharmacological targets for Cancer Therapy. Front Pharmacol 13:908772
Chu B et al (2019) ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol 21(5):579–591
Yan H et al (2022) MDMX elevation by a novel Mdmx-p53 interaction inhibitor mitigates neuronal damage after ischemic Stroke. Sci Rep 12(1):21110
Wu Z et al (2017) LncRNA-N1LR enhances Neuroprotection Against ischemic Stroke probably by inhibiting p53 phosphorylation. Mol Neurobiol 54(10):7670–7685
Yuan M et al (2017) Effects of puerarin combined with conventional therapy on ischemic Stroke. Exp Ther Med 14(4):2943–2946
Zhang X et al (2022) Integrative Analyses of Biomarkers Associated with endoplasmic reticulum stress in ischemic Stroke. Comput Math Methods Med 2022:4212180
Li M et al (2022) Baicalein ameliorates cerebral ischemia-reperfusion injury by inhibiting ferroptosis via regulating GPX4/ACSL4/ACSL3 axis. Chem Biol Interact 366:110137
Ye Z et al (2023) BAICALEIN RELIEVES BRAIN INJURY VIA INHIBITING FERROPTOSIS AND ENDOPLASMIC RETICULUM STRESS IN A RAT MODEL OF CARDIAC ARREST. Shock 59(3):434–441
Xu X, Yan J (2021) β-Caryophyllene may attenuate hyperoxaluria-induced kidney dysfunction in rats by regulating stress marker KIM-1/MCP-1 and NF-κB signaling pathway. J Biochem Mol Toxicol 35(11):e22891
Lv Y et al (2023) Structural insights into FSP1 catalysis and ferroptosis inhibition. Nat Commun 14(1):5933
Nakamura T et al (2023) Phase separation of FSP1 promotes ferroptosis. Nature 619(7969):371–377
Mishima E et al (2022) A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 608(7924):778–783
Zhang S et al (2023) FSP1 oxidizes NADPH to suppress ferroptosis. Cell Res,
Nakamura T et al (2023) Integrated chemical and genetic screens unveil FSP1 mechanisms of ferroptosis regulation. Nat Struct Mol Biol 30(11):1806–1815
Fang Y et al (2022) Discovery of novel diphenylbutene derivative ferroptosis inhibitors as neuroprotective agents. Eur J Med Chem 231:114151
Li W et al (2023) FSP1: a key regulator of ferroptosis. Trends Mol Med 29(9):753–764
Chen X et al (2021) Ferroptosis: machinery and regulation. Autophagy 17(9):2054–2081
Mao C et al (2021) DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593(7860):586–590
Yi YY et al (2020) Preventive effects of Neuroprotective agents in a neonatal rat of Photothrombotic Stroke Model. Int J Mol Sci, 21(10)
Akiyama H et al (2023) Molecular mechanisms of ferroptosis and updates of Ferroptosis Studies in cancers and Leukemia. Cells, 12(8)
Xu L et al (2023) Ferroptosis in life: to be or not to be. Biomed Pharmacother 159:114241
Liu Y et al (2023) The diversified role of mitochondria in ferroptosis in cancer. Cell Death Dis 14(8):519
Chen Y et al (2022) BRD4770 functions as a novel ferroptosis inhibitor to protect against Aortic Dissection. Pharmacol Res 177:106122
Hu Q et al (2022) Blockade of GCH1/BH4 Axis activates Ferritinophagy to mitigate the resistance of Colorectal Cancer to Erastin-Induced ferroptosis. Front Cell Dev Biol 10:810327
Sun X et al (2015) HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 34(45):5617–5625
Liang Y et al (2023) HSPB1 facilitates chemoresistance through inhibiting ferroptotic cancer cell death and regulating NF-κB signaling pathway in Breast cancer. Cell Death Dis 14(7):434
Dai Y, Hu L (2022) HSPB1 overexpression improves hypoxic-ischemic brain damage by attenuating ferroptosis in rats through promoting G6PD expression. J Neurophysiol 128(6):1507–1517
Aolymat I, Hatmal MM, Olaimat AN (2023) The emerging role of heat shock factor 1 (HSF1) and heat shock proteins (HSPs) in Ferroptosis. Pathophysiology 30(1):63–82
Li Q et al (2008) Hsf1 is required for the nuclear translocation of p53 Tumor suppressor. Neoplasia 10(10):1138–1145
Shi X et al (2021) Increased HSF1 promotes infiltration and Metastasis in Cervical Cancer via enhancing MTDH-VEGF-C expression. Onco Targets Ther 14:1305–1315
Jia G et al (2023) HSF1 is a novel prognostic biomarker in high-risk Prostate cancer that correlates with ferroptosis. Discov Oncol 14(1):107
Hayano M et al (2016) Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ 23(2):270–278
Yu H et al (2017) Ferroptosis, a new form of cell death, and its relationships with tumourous Diseases. J Cell Mol Med 21(4):648–657
Zheng J, Conrad M (2020) The metabolic underpinnings of Ferroptosis. Cell Metab 32(6):920–937
Zhu Z et al (2021) Protective Properties of the Extract of Chrysanthemum on Patients with Ischemic Stroke J Healthc Eng, 2021: p. 3637456
Zimmermann C et al (2004) Antioxidant status in acute Stroke patients and patients at Stroke risk. Eur Neurol 51(3):157–161
Xu L et al (2022) Edaravone dexborneol protects cerebral ischemia reperfusion injury through activating Nrf2/HO-1 signaling pathway in mice. Fundam Clin Pharmacol 36(5):790–800
Xu J et al (2021) Edaravone Dexborneol Versus Edaravone alone for the treatment of Acute ischemic Stroke: a phase III, Randomized, Double-Blind, comparative trial. Stroke 52(3):772–780
Kobayashi S et al (2019) Effect of Edaravone on neurological symptoms in real-world patients with Acute ischemic Stroke. Stroke 50(7):1805–1811
Liu W et al (2022) Edaravone ameliorates cerebral ischemia-reperfusion Injury by Downregulating Ferroptosis via the Nrf2/FPN pathway in rats. Biol Pharm Bull 45(9):1269–1275
Rothstein JD (2017) Edaravone: a new drug approved for ALS. Cell 171(4):725
Peng Z (2023) Efficacy and safety of Agatroban in improving the prognosis of ischemic Stroke patients. Am J Transl Res 15(9):5699–5706
Tian S et al (2023) The neuroprotective effect of Dl-3-n-butylphthalide in epileptic rats via inhibiting endoplasmic reticulum stress. Folia Neuropathol 61(2):185–195
Ma Y et al (2023) Effects of Dl-3-n-butylphthalide on cognitive functions and blood-brain barrier in chronic cerebral hypoperfusion rats. Naunyn Schmiedebergs Arch Pharmacol 396(11):3207–3220
Lu JD et al (2023) Butylphthalide protects against ischemia-reperfusion injury in rats via reducing neuron ferroptosis and oxidative stress. J Investig Med 71(6):623–633
Zhang H et al (2022) DL-3-n-butylphthalide (NBP) alleviates poststroke cognitive impairment (PSCI) by suppressing neuroinflammation and oxidative stress. Front Pharmacol 13:987293
Zhang X et al (2022) Keap1-Nrf2/ARE signal pathway activated by butylphthalide in the treatment of ischemic Stroke. Am J Transl Res 14(4):2637–2646
He J et al (2023) Effects of NBP on postoperative cognitive dysfunction in rats via Nrf 2/ARE pathway Aging (Albany NY), 15(1): p. 276–286
Ye Z et al (2023) Dl-3-n-butylphthalide activates Nrf2, inhibits ferritinophagy, and protects MES23.5 dopaminergic neurons from ferroptosis. Chem Biol Interact 382:110604
Zhang H et al (2023) A comparative study of the neuroprotective effects of dl-3-n-butylphthalide and edaravone dexborneol on cerebral ischemic Stroke rats. Eur J Pharmacol 951:175801
Crapper McLachlan DR et al (1991) Intramuscular desferrioxamine in patients with Alzheimer’s Disease. Lancet 337(8753):1304–1308
Moujalled D, Strasser A, Liddell JR (2021) Molecular mechanisms of cell death in neurological Diseases. Cell Death Differ 28(7):2029–2044
Zhang Y, He ML (2017) Deferoxamine enhances alternative activation of microglia and inhibits amyloid beta deposits in APP/PS1 mice. Brain Res 1677:86–92
Guo C et al (2013) Intranasal deferoxamine reverses iron-induced memory deficits and inhibits amyloidogenic APP processing in a transgenic mouse model of Alzheimer’s Disease. Neurobiol Aging 34(2):562–575
Devos D et al (2014) Targeting chelatable iron as a therapeutic modality in Parkinson’s Disease. Antioxid Redox Signal 21(2):195–210
Liddell JR, Hilton JBW, Crouch PJ (2023) Cu(II) (atsm) significantly decreases microglial reactivity in patients with sporadic Amyotrophic Lateral Sclerosis. Neuropathol Appl Neurobiol 49(5):e12938
Southon A et al (2020) Cu(II) (atsm) inhibits ferroptosis: implications for treatment of neurodegenerative Disease. Br J Pharmacol 177(3):656–667
Fang X et al (2019) Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A 116(7):2672–2680
Radadiya PS et al (2021) Ciclopirox olamine induces ferritinophagy and reduces cyst burden in polycystic Kidney Disease. JCI Insight, 6(8)
Imai T et al (2021) Deferasirox, a trivalent iron chelator, ameliorates neuronal damage in hemorrhagic Stroke models. Naunyn Schmiedebergs Arch Pharmacol 394(1):73–84
Hu X et al (2023) Effects of edaravone dexborneol on neurological function and serum inflammatory factor levels in patients with acute anterior circulation large vessel occlusion Stroke. Transl Neurosci 14(1):20220312
Xu J et al (2019) Safety and efficacy of Edaravone Dexborneol versus edaravone for patients with acute ischaemic Stroke: a phase II, multicentre, randomised, double-blind, multiple-dose, active-controlled clinical trial. Stroke Vasc Neurol 4(3):109–114
Falcione S et al (2023) Sex differences in Thrombin Generation in patients with Acute ischemic Stroke. Transl Stroke Res
Zeng L et al (2023) Animal models of ischemic Stroke with different forms of Middle cerebral artery occlusion. Brain Sci, 13(7)
Jin ZL et al (2023) Ring finger protein 146-mediated long-chain fatty-acid-coenzyme A ligase 4 ubiquitination regulates ferroptosis-induced neuronal damage in ischemic Stroke. Neuroscience,
Doll S et al (2017) ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13(1):91–98
Chen Y et al (2023) The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis 14(3):205
Guo M et al (2023) Engineering Ferroptosis inhibitors as inhalable nanomedicines for the highly efficient treatment of Idiopathic Pulmonary Fibrosis. Bioeng (Basel), 10(6)
Zheng S et al (2023) Proton pump inhibitor-enhanced nanocatalytic ferroptosis induction for stimuli-responsive dual-modal molecular imaging guided cancer radiosensitization. Acta Biomater 162:72–84
Fukuta T et al (2017) Combination therapy with liposomal neuroprotectants and tissue plasminogen activator for treatment of ischemic Stroke. Faseb j 31(5):1879–1890
Funding
This review was supported by the Natural Science Foundation of Yan’an University (YDBK2020-28), Shaanxi Provincial Health Research (2022E010), and Natural Science Foundation of Shaanxi Province(2023-JC-QN-0125 to X-LL). And this review was supported by the Shaanxi Provincial Education Department Scientific Research Program Funded (22JK0612) and (23JK0731 to Y.X.).
Author information
Authors and Affiliations
Contributions
Lei Zhang was responsible for writing and revising the text. Xin Yue Bai was responsible for the preparation of figures and revisions of this article. Ke Yao Sun was responsible for the post-writing and revisions of this article. Xuan Li and Zhao Qi Zhang were responsible for data collection. Yi Ding Liu and Yang Xiang were responsible for revising the article. Xiao Long Liu was responsible for conceptualizing, funding, and guiding the article. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Conflict of Interest
There are no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, L., Bai, X.Y., Sun, K.Y. et al. A New Perspective in the Treatment of Ischemic Stroke: Ferroptosis. Neurochem Res 49, 815–833 (2024). https://doi.org/10.1007/s11064-023-04096-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-023-04096-3