
Abstract
Mitophagy plays a key role in epileptic neuronal injury, and recent studies have shown that FUNDC1 plays an important role in regulating mitophagy. However, the specific effect of FUNDC1 on neuronal damage in epilepsy is unknown. In this study, we investigated the role of FUNDC1 in mitophagy and neuronal apoptosis using a hippocampal neuronal culture model of acquired epilepsy (AE) in vitro. We found that mitophagy levels were significantly increased in this model, as indicated by elevated LC3A/B ratios. FUNDC1 overexpression using lentiviral vectors enhanced mitophagy, whereas FUNDC1 down-regulation using lentiviral vectors impaired this process. Overexpression of FUNDC1 significantly decreased AE-induced superoxide anion, enhanced cell viability, reduced oxidative stress, and reduced neuronal apoptosis in epileptic hippocampus, while FUNDC1 down-regulation caused the opposite effect. In conclusion, we demonstrated that FUNDC1 is an important modulator of AE-induced neuronal apoptosis by controlling mitophagy function.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Epilepsy is a serious chronic neurological disease, which leads to transient brain dysfunction due to sudden abnormal discharges of neurons in the brain, which is characterized by occasional, repetitive and spontaneous, and can be accompanied by cognitive dysfunction, anxiety and depression and other well disorders, seriously affecting the lives of loyalists, causing a huge economic burden to families and society [1, 2]. Epidemiological studies have shown that there are currently approximately 65 million patients with epilepsy worldwide [3]. Although there have been a large number of studies on the pathogenesis of epilepsy in recent years, the mechanisms involved in the development of epilepsy have not been clarified. Seizures can damage mitochondria, mainly manifested as mitochondrial cristae swelling and disruption of membrane integrity, while mitochondrial dysfunction can also lead to seizures [4, 5]. Autophagy is also closely related to epilepsy. Studies have confirmed the presence of abnormalities in autophagy in the brain tissue of experimental epileptic animals and epileptic patients [6]. McMahon et al. found a significant decrease in the level of cellular autophagy after knockout of the At97 gene in mice, resulting in recurrent spontaneous seizures in mice [7].
Clinical studies have found that there are a large number of neurons with abnormal firing in the brain of epilepsy patients, as well as neuronal damage and loss caused by epilepsy [8]. As an essential element in life, Mg2+ can block the influx of calcium ions and inhibit depolarization, maintaining the normal function of the central nervous system and neural electrophysiological activity [9]. Studies have shown that more than 90% of hippocampal neurons will show epileptiform discharge after 11 days of culture in normal medium and 3 h of culture after removing Mg2+ [10]. The action potentials of spontaneous and recurrent firing of neurons induced by magnesium-free external fluid are similar to the electrophysiological activity of epileptic patients during seizures. At the same time, the model of neuronal epilepsy induced by magnesia-free external fluid has the advantages of no blood–brain barrier, stable model and long recording time [11, 12]. Therefore, magnesium-free effluent-induced hippocampal neuronal epilepsy model is an ideal in vitro epilepsy model.
Mitophagy, as a selective autophagy, has been considered to be a potential mechanism responsible for regulating mitochondrial quality, mainly through autophagosome formation and autophagosome and lysosome fusion to selectively remove mitochondria with intracellular structural damage and dysfunction or degrade unnecessary normal mitochondria, prevent the accumulation of harmful products such as superoxide anion, while ensuring the normal function of mitochondria, and play a key role in meeting cellular energy needs and maintaining the balance of the intracellular environment. Notably, dysfunctional, mainly defective mitophagy is involved in the pathophysiology of various human diseases, including cardiovascular diseases, neurodegenerative diseases, cancer, autoimmune diseases and aging [13].
FUNDC1, as a mitochondrial outer membrane protein in mammalian cells under hypoxia, is accumulated by interacting with ER-mitochondria contact sites, acting through LIR motifs binding to the autophagy marker LC3. Under hypoxia, FUNDC1 is phosphorylated at tyrosine 18 and serine 13 by SRC kinase and CK2, which reduces its affinity for LC3; however, dephosphorylation by PGAM5 or other yet to be identified phosphatases largely increases its interaction with LC3 or other autophagy genes, thereby initiating mitophagy [14], but the mechanism by which FUNDC1-mediated mitophagy plays a role in the transfer of epileptogenic hippocampal neurons in magnesium-free exterior fluid is not clear.
In this study, we used an in vitro hippocampal neuron epilepsy model to further clarify the role of FUNDC1-mediated mitophagy in the development of epilepsy by intervening FUNDC1 expression by lentiviral re-extraction of Lenti-FUNDC1 and Lenti-FUNDC1-shRNA.
Materials and Methods
Animal and Experimental Reagents
Healthy Sprague–Dawley rats within 24 h of birth, SPF grade (Zhengzhou University, China). All experimental procedures were conducted in conformity with insti-tutional guidelines for the care and use of laboratory animals in Zhengzhou University(ZZU-LAC20211210[02]). Reagents: magnesium-free external solution (2.5 mmol/L KCl, 145 mmol/L NaCl, 10 mmol/L glucose, 0.002 mmol/L glycine, 2 mmol/L CaCl2 and 10 mmol/L HEPES, pH 7.4) [15, 16]: normal extracellular solution (magnesium free external solution + 1 mmol/L MgCl2),RNA kit (Invitrogen, America); reverse transcription kit(Yeasen, Shanghai,China); TUNEL kit (Roche, Germany); DHE kit (Beyotime, China), TMRE kit (Beyotime, China) NSE antibody (Abcam, UK); FUNDC1 antibody (Sigma, America), LC3B antibody (Abcam,UK); lentiviral empty vector Lenti-pGV, overexpression of FUNDC1 lentiviral empty vector Lenti-FUNDC1, low-expression FUNDC1 lentiviral vector Lenti-FUNDC1-ShRNA (GeneChem, Shanghai,China).
Culture of Primary Hippocampal Neurons
All animal protocols were approved by the Animal Care and Utilization Committee of Zhengzhou University, China, confirming that all animal studies and experimental manipulations were in strict compliance with international animal research guidelines.
The hippocampal tissue of Sprague–Dawley rats with 24 h after birth was isolated under aseptic conditions, the surface blood vessels were removed and cut into tissue blocks and transferred to a Petri dish, digested with 0.125% trypsin and added with implantation culture medium to terminate digestion, the supernatant was discarded, resuspended and washed and added with implantation culture medium to make cells. Neuronal cells were seeded in 6-well cell culture plates previously coated with L-poly at a density of 3.5 × 105cells/mL and cultured in a 37 °C incubator with 5% CO2 for 4 h before the implantation culture medium was replaced with the maintenance culture medium. Maintenance medium was changed in half every 2 days. The purity of hippocampal neuro was detected by immunohistochemical staining, and the next experiment was performed when the purity was > 95%.
Experimental Design
After discarding the maintenance culture medium, rat hippocampal neurons were rinsed three times with magnesium-free external solution and cultured with magnesium-free external solution for 3 h to induce hippocampal neuron epilepsy model.
Primary hippocampal neurons were randomly divided into: (1) control group (CON group): neurons were treated with normal extracellular solution for 3 h; (2) magnesium-free induction group (AE group): neurons were treated with magnesium-free external solution for 3 h: (3) Lenti-pGV group (negative control group): neurons were successfully transfected by Lenti-pGV and continued to be cultured with maintenance medium and treated with magnesium-free external solution for 3 h: (4) Lenti-FUNDC1 group (overexpression FUNDC1 group): neurons were successfully transfected by Lenti-FUNDC1 and continued to be cultured with maintenance medium, and neurons were treated with magnesium-free external solution for 3 h. (5) Lenti-FUNDC1-shRNA group (low expression FUNDC1 group): neurons were successfully transfected by Lenti-FUNDC1-shRNA and continued to be cultured with maintenance medium and treated with magnesium-free external solution for 3 h. In each group, after treatment with normal cell extract or magnesium-free extract for 3 h, culture with maintenance culture medium was continued for 24 h before the next experiment (Table 1).
NSE staining
Neuronal cells were seeded into 24-well plates with cell coverslips for culture, and neuronal purity identification was performed using NSE staining on day 7. The maintenance medium of each well was aspirated and discarded, rinsed twice with PBS buffer, fixed with 4% paraformaldehyde for 30 min, broken with 0.2% Triton X-100 for 5 min, and incubated with 10% goat serum for 1 h. NSE primary antibody (1:200) was added and incubated overnight at 4 °C. The primary antibody was aspirated and discarded, rinsed with PBS, and the secondary antibody was added and incubated for 1 h in the dark. After rinsing with PBS, 10 μL of anti-fluorescence attenuation mounting medium containing DAPI was added to the slide, and the cells were removed from the slide and covered on the slide. Ten fields were randomly selected and observed under a fluorescence microscope to calculate the positive rate of NSE positive cells and take the mean value, that is, neuronal purity.
QPCR
Total RNA was extracted from hippocampal neurons by columnar extraction, followed by reverse transcription of cDNA using a reverse transcription kit and cDNA to RNA conversion using a QPCR kit with the following primer sequences (Table 2).
CCK8 Assay
CCK8 is a widely used method for cell proliferation and cytotoxicity determination based on water-soluble tetrazolium salts. The maintenance culture medium was removed and CCK8 solution was added in the dark and incubated at 37 °C for 90 min in the dark. The absorbance was measured at 450 nm to calculate cell viability. Please check and confirm the inserted citation of Table 2 is correct. If not, please suggest an alternative citation. Please note that figures and tables should be cited in sequential order in the text.We confirm that the inserted citation of Table 2 is correct.
TMRE Assay
The changes of mitochondrial membrane potential in early stage were determined by TMRE to determine the status of early apoptosis. The culture medium was sucked and discarded, rinsed with PBS, added with TMRE staining working solution in the dark, incubated at 37 °C for 30 min, and rapidly imaged under a fluorescence microscope to maintain the same exposure.
TUNEL Assay
Apoptosis was detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay. The reptile containing primary neurons was fixed with 4% paraformaldehyde for 1 h, and the membrane was broken with 0.2% Triton X-100 for 10 min. Neurons were mixed with TUNEL reaction solution for 1 h at 37 °C and reacted with POD for 1 h at 37 °C, DAB substrate was added, and the reaction was performed for 1 min at room temperature, followed by counterstaining with hematoxylin for several seconds, gradient dehydration for 2 min, xylene transparency for 10 min, and mounting with neutral resin. Apoptotic cells were observed under a fluorescence microscope, and the number of positive cells was calculated.
Determination of Superoxide Anion
Intracellular superoxide anion levels were determined by DHE to determine cellular oxidative stress levels. The culture medium was sucked and discarded, rinsed with PBS, added with DHE staining working solution in the dark, incubated at 37 °C for 30 min, and rapidly imaged under a fluorescence microscope to maintain the same exposure.
Cell Immunofluorescence
Immunofluorescence was used to examine whether the magnesium-free induced in vitro epilepsy model induced Fundc1 recruitment to mitophagy and expression of Fundc1 and LC3B. After fixation with 4% paraformaldehyde for 30 min, the membrane was broken with 0.2% Triton X-100 for 5 min and incubated with 10% goat serum for 1 h, followed by incubation with goat anti-FUNDC1 (1:200; Sigma) and goat anti-LC3B (1:200; Abcam) overnight at 4 °C. The primary antibody was aspirated and discarded, rinsed with PBS, and the secondary antibody was added and incubated for 1 h in the dark. After rinsing with PBS, 10 μL of anti-fluorescence attenuation mounting medium containing DAPI was added to the slide, and the cells were removed from the slide and covered on the slide. Observe under a fluorescence microscope and maintain the same exposure setting.
Infection with Lentiviral Vectors
Lentivirus-transfected neuronal cells Hippocampal neurons were placed on 6-well plates at a density of 3 × 105 cells/mL, and after 5 days of culture, neuronal cells were transfected using Lenti-pGV, Lenti-FUNDC1, and Lenti-FUNDC1-shRNA, respectively, at a multiplicity of infection of 10. After 12 h of culture, the culture medium containing lentiviral vectors was discarded and normal culture medium was added; after 72 h of culture, the infection efficiency was observed using a fluorescence microscope, and the next intervention and detection were performed.
Statistical Processing
All results were expressed as mean ± standard deviation (SD) and analyzed using GraphPad Prism 8 software version 3.0. Diferences between groups were determined by using one-way ANOVA. P values < 0.05 were considered statistically significant.
Results
FUNDC1 Overexpression Attenuated AE-Induced Neuronal Apoptosis
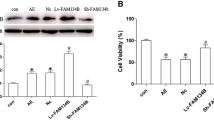
QPCR was used to detect the expression of FUNDC1 to confirm the infection efficiency of the lentiviral vector. Pretreatment with Lenti-FUNDC1 increased FUNDC1 expression and pretreatment with Lenti-FUNDC1-shRNA decreased FUNDC1 expression compared with control (Fig. 1A). Neuronal survival was significantly reduced in the AE group compared with the control group as determined by the CCK8 assay (Fig. 1B). FUNDC1 overexpression using Lenti-FUNDC1 could attenuate AE-induced neuronal death, whereas downregulation of this protein using Lenti-FUNDC1-shRNA exacerbated AE-induced neuronal death (Fig. 1B). Statistically, there was no significant difference between the AE group and negative control groups (Fig. 1B).

Effect of FUNDC1 on hippocampal neuronal injury induced by acquired epilepsy (AE). A The infection efficiency of lentiviral vectors was analyzed by QPCR analysis of FUNDC1 expression. B Hippocampal neurons were incubated with Lenti-FUNDC1 and Lenti-FUNDC1-shRNA only for 12 h, and then cultured in maintenance medium for 72 h. Cell survival was measured by CCK8 assay. Hippocampal neurons were incubated with Lenti-FUNDC1, Lenti-FUNDC1-shRNA, or Lenti-pGV for 12 h and cultured in magnesium-free solution for 72 h. Data are presented as mean ± standard deviation of three independent experiments. *P < 0.05 compared with AE group. ***P < 0.001 compared with AE group. ****P < 0.0001 compared with AE group
Early mitochondrial membrane potential was determined by TMRE to determine early apoptosis, and normal cells showed bright red fluorescence after TMRE staining. Neuronal apoptosis was significantly increased in the AE group (Fig. 2A). However, overexpression of FUNDC1 attenuated AE-induced neuronal apoptosis, whereas downregulation of FUNDC1 exacerbated this process (Fig. 2A). There was no significant difference between the AE group and negative control group (Fig. 2A).

Effect of FUNDC1 on early apoptosis of hippocampal neurons induced by acquired epilepsy (AE). A Apoptosis of hippocampal neurons was detected by TMRE. Decreased red fluorescence was observed in all AE-negative controls, while FUNDC1 overexpression reduced AE-induced apoptosis in the Lenti-FUNDC1 group. In Lenti-FUNDC1-shRNA, down-regulation of FUNDC1 exacerbated AE-induced neuronal apoptosis. Data are presented as mean ± standard deviation of three independent experiments. *P < 0.05 compared with AE group. B Apoptosis of hippocampal neurons was detected by TUNEL assay. Apoptotic neurons (positive cells) were observed in the AE negative control group, while FUNDC1 overexpression in the Lenti-FUNDC1 group protected against AE-induced neuronal apoptosis. In Lenti-FUNDC1-shRNA, down-regulation of FUNDC1 exacerbated AE-induced neuronal apoptosis. Data are presented as mean ± standard deviation of three independent experiments. *P < 0.05 compared with AE group. **P < 0.01 compared with AE group. ****P < 0.0001 compared with AE group
The apoptosis cell was measured by TUNEL assay. Neuronal apoptosis was significantly increased in the AE group (Fig. 2B). However, overexpression of FUNDC1 attenuated AE-induced neuronal apoptosis, whereas downregulation of FUNDC1 exacerbated this process (Fig. 2B). There was no significant difference between the AE group and negative control group (Fig. 2B).
In this study, overexpression of FUNDC1 significantly reduced the apoptosis of hippocampal neurons in epileptic models, and had a protective effect on hippocampal neurons in vitro, while knockdown FUNDC1 did the opposite, suggesting that FUNDC1 may protect hippocampal neurons by inhibiting apoptosis pathways.
FUNDC1 Overexpression Enhanced AE-Induced Mitophagy
LC3A/B ratio was significantly higher in the AE group than in the control group (Fig. 3). Relative to the AE group, overexpression of FUNDC1 increased the LC3A/B ratio, whereas its downregulation decreased this ratio (Fig. 3). No difference was found between the AE group and negative control group (Fig. 3). To further evaluate the effect of FUNDC1 on mitophagy, neurons were immunohistochemically stained for FUNDC1 and LC3B. We found that the expression of these proteins was increased in AE compared with controls (Fig. 3).

Effect of FUNDC1 on LC3A/B ratio in acquired epilepsy (AE). Quantitative analysis of LC3A/B ratio in hippocampal neurons. by QPCR Data are presented as mean ± standard deviation of three independent experiments. *P < 0.05 compared with AE group. ****P < 0.0001 compared with AE group
Overexpression of FUNDC1 further increased LC3B expression (Fig. 4). In contrast, down-regulation of FUNDC1 reduced LC3B expression (Fig. 4). There was no significant difference between the AE group and negative control group (Fig. 4). Together, these data suggest that mitophagy levels are regulated by FUNDC1.

Effect of FUNDC1 on acquired epilepsy (AE)-induced immunostaining for FUNDC1 and LC3B, blue is the nucleus, red is the marker protein (A is FUNDC1, B is LC3B). Representative images of FUNDC1 and LC3B immunofluorescence in hippocampal neurons. Data are presented as mean ± standard deviation of three independent experiments. *P < 0.05 compared with AE group. **P < 0.01 compared with AE group ***P < 0.001 compared with AE group. ****P < 0.0001 compared with AE group
Our study found that compared with the CON group, the levels of autophagy markers were increased in the AE group, suggesting enhanced autophagy levels in magnesium-free induced hippocampal neurons.
FUNDC1 Overexpression Attenuated AE-Induced Intracellular Superoxide Anion Production
Compared with the control group, the level of intracellular superoxide anion was increased in the AE group. FUNDC1 overexpression significantly reduced intracellular superoxide anion levels compared with the AE group (Fig. 5). In contrast, downregulation of FUNDC1 had the opposite effect, exacerbating the defects observed in the AE response. There was no significant difference the AE group and negative control group (Fig. 5).

Effect of FUNDC1 on intracellular superoxide anion production in acquired epilepsy (AE) cells. Levels of superoxide anion in hippocampal neurons. Data are presented as mean ± standard deviation of three independent experiments. *P < 0.05 compared with AE group
In this study, overexpression of FUNDC1 significantly reduced AE-induced superoxide anion and the level of intracellular oxidative stress, while knockdown of FUNDC1 significantly reduced the level of intracellular oxidative stress. These results suggest that FUNDC1 can protect hippocampal neurons by reducing mitochondrial oxidative stress.
Discussion
Over the years, many scholars have conducted a large number of clinical studies and animal experiments on the pathogenesis of epilepsy, but the etiology of epilepsy is very complex, and its pathogenesis still needs to be further clarified. Increased extracellular glutamate, a neurotransmitter and energy metabolite, is a biochemical hallmark of epileptic tissues in animals and humans and is thought to cause neurotoxicity and seizures [17, 18]. Excitatory neurotransmitters such as glutamate act on the postsynaptic membrane, increasing its depolarization, leading to mitochondrial damage in neurons, and inhibiting mitophagy response [19,20,21]. Thus, mitophagy and oxidative stress play an important role in the pathological process of epilepsy. Lemasters formally proposed the term "mitophagy" in 2005 to describe the selective degradation process of mitochondria by autophagy. Since then, the molecular mechanism of selective mitophagy has attracted extensive attention. Mitophagy is an important pathway for mitochondria to maintain quality. As a mitochondrial outer membrane protein, FUNDC1 is a novel mitophagy receptor, which can mediate mitophagy under hypoxia by binding to LC3. In this study, QPCR and cellular immunofluorescence were used to detect the increase of FUNDC1 and autophagy markers in epileptogenic hippocampal neurons in vitro, indicating that the increased expression of FUNDC1 may promote mitophagy in the process of epileptogenesis. In order to investigate the exact role of FUNDC1 in epilepsy, we used the lentiviral vectors overexpressing FUNDC1 and knocking down FUNDC1 respectively to establish an in vitro magnesium-free epileptic model of hippocampal neurons by interfering with the expression of FUNDC1. The mitophagy indexes, oxidative stress and apoptosis-related indexes of the cells were measured in groups.
In recent years, more and more studies have found that mitophagy plays an important regulatory role in the seizure process of epilepsy. Autophagy marker protein LC3B and mitochondrial marker TOMM20 were used to detect the autophagy level in the hippocampus of patients with refractory temporal lobe epilepsy [22], and it was found that mutations in the gene encoding mTORC1 autophagy inhibitor were associated with increased susceptibility to epilepsy [23]. In addition, neurotoxicity is also involved in epileptic seizures. For example, excitatory neurotransmitters such as glutamate act on the postsynaptic membrane, increasing its depolarization, leading to continuous influx of synaptic cleft, mitochondrial damage, and blocking mitophagy response [19, 20, 24]. Focal Cortex Dysplasia (FCD) is a common cause of severe epilepsy in children, and the over-activation of mTOR and accumulation of P62 play an important role in the pathogenesis of FCD [25]. Rapamycin is an inducer of mitophagy and an inhibitor of mTOR, which can prevent the further development of epilepsy in the early stage, and late treatment can reduce the seizure frequency of epileptic mice [26, 27].
Mitophagy is a process of autophagy degradation mediated by mitochondria and lysosomes. This process is used to remove damaged and dysfunctional mitochondria from cells [28, 29]. It allows the removal of dysfunctional mitochondria in cells and is important for the maintenance of normal mitochondrial function [30]. Studies have shown that mitophagy is closely related to the pathogenesis of epilepsy. Early studies have shown that the loss of mitochondrial membrane potential is a signal of mitophagy [31, 32]. We found that the levels of autophagy markers increased in the AE group compared with the CON group, suggesting that the level of autophagy was enhanced in magnesium-free hippocampal neurons.
FUNDC1 mediates selective mitophagy through the interaction between LIR and LC3 to be coupled to the core autophagy mechanism [33]. LC3B is a well-known marker that is now widely used to monitor autophagy. The number of LC3B is significantly correlated with the number of autophagosomes [34,35,36]. Previous studies have shown that autophagy activity is related to different seizure types [37, 38]. Studies have shown that autophagy is increased in mice induced by kfucine or pilarpine during seizures, and LC3B-positive autophagic vacuoles accumulate in the hippocampus of mice after repeated seizures [39, 40]. These damaged mitochondria can be removed by mitochondrial protective mechanisms (decreased ATP synthase, membrane depolarization) [41,42,43]. FUNDC1 binds to LC3 through its LIR domain to form an autophagic bilayer that wraps around mitochondria and induces mitophagy. The resulting mitophagy inhibits apoptosis and protects neurons [44]. The change of FUNDC1 phosphorylation level promotes mitophagy and thus affects cell apoptosis. These results suggest that mitochondrial damage may activate mitophagy in rTLE, thereby removing damaged mitochondria, and the resulting mitophagy inhibits apoptosis and protects neurons. Our results were consistent with the above studies: compared with the CON group, LC3A/B and LC3B were increased, superoxide anion levels were increased, and cell viability was decreased in the AE group, which suggested that the level of autophagy, damaged mitochondria, cellular oxidative stress and apoptosis were increased in magnesia-free hippocampal neurons. Moreover, overexpression of FUNDC1 increased the level of mitophagy in neurons and inhibited the level of intracellular oxidative stress and apoptosis, which played a protective role in neurons. In general, insufficient mitophagy can lead to accumulation of damaged mitochondria, resulting in increased excitability and neuronal death in rTLE neurons, which may be involved in the mechanism of epileptogenesis [21].
This study showed that overexpression of FUNDC1 significantly increased the total amount of autophagy marker LC3B in magnesia-free epileptic hippocampal neurons. At the same time, we also found that overexpression of FUNDC1 could significantly reduce AE-induced superoxide anion, intracellular oxidative stress, and apoptosis of epileptic hippocampal neurons. Knockdown of FUNDC1 has the opposite effect, inhibiting the expression of FUNDC1, reducing the level of mitophagy, accumulating damaged mitochondria, increasing the level of apoptosis and oxidative stress, and promoting neuronal death. This suggests that FUNDC1 may protect hippocampal neurons by promoting mitophagy, reducing mitochondrial oxidative stress and inhibiting apoptosis pathway.
In addition, increasing evidence has shown a link between autophagy and various physiological and pathological environments, and suggests that autophagy is involved in the treatment of a variety of human diseases [45,46,47]. As a novel mitophagy receptor, FUNDC1 has been reported to be involved in regulating mitophagy and mitochondrial dynamics, and plays an important role in maintaining the structure and function of mitochondria [48]. Targeting FUNDC1 has been proposed as a new avenue to develop therapeutic interventions for a variety of human diseases, including cardiovascular diseases, metabolic syndrome, cancer, and COPD [48]. However, the exact mechanism of FundC1-mediated mitophagy and the specific role of FUNDC1 in epilepsy remain undefined,therefore, more studies are needed to elucidate the role of FUNDC1 in mitochondrial dynamics and regulation of mitophagy, and to elucidate its role in different diseases. Clearly, further studies on FUNDC1 are still needed to reveal the specific mechanism of mitophagy, which could also help to provide new therapeutic strategies for various diseases. A data availability statement is mandatory for publication in this journal. Please confirm that this statement is accurate, or provide an alternative.We confirm that the data availability statement is accurate.
In conclusion, our study found that FUNDC1 can alleviate mitophagy, apoptosis and oxidative stress of hippocampal neurons induced by epileptic seizure process, and the underlying mechanism may be related to the regulation of mitophagy. In future experiments, we also need to explore the role of FUNDC1 in animal epilepsy models, so as to provide new treatment strategies for epilepsy patients.
Data Availability
Enquiries about data availability should be directed to the authors.
References
Kaneko Y, Pappas C, Malapira T, Vale F, Tajiri N, Borlongan C (2017) Extracellular HMGB1 modulates glutamate metabolism associated with kainic acid-induced epilepsy-like hyperactivity in primary rat neural cells. Cell Physiol Biochem 41:947–959
Lotze MT, Tracey KJ (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5:331–342
Das A, Cash SS, Sejnowski TJ (2020) Heterogeneity of preictal dynamics in human epileptic seizures. IEEE Access 1:1–1
Chuang Y, Chang A, Lin J, Hsu S, Chan S (2004) Mitochondrial dysfunction and ultrastructural damage in the hippocampus during kainic acid–induced status epilepticus in the rat. Epilepsia 45:1–10
Qiu XX, Cao LL, Yang X, Chi ZF (2013) Alteration of mitochondrial fission and fusion in hippocampus of rats with status epilepticus. J Shandong Univ Health Sci
Yasin SA, Ali AM, Tata M, Picker S (2013) mTOR-dependent abnormalities in autophagy characterize human malformations of cortical development: evidence from focal cortical dysplasia and tuberous sclerosis. Acta Neuropathol 126:207–218
Mcmahon J, Huang X, Yang J, Komatsu M, Yue Z, Qian J, Zhu X, Huang Y (2012) Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J Neurosci 32:15704
Wu Y, Liu D, Song Z (2015) Neuronal networks and energy bursts in epilepsy. Neuroscience 287:175–186. https://doi.org/10.1016/j.neuroscience.2014.06.046
Igelstrom KM, Shirley CH, Heyward PM (2011) Low-magnesium medium induces epileptiform activity in mouse olfactory bulb slices. J Neurophysiol 106:2593–2605. https://doi.org/10.1152/jn.00601.2011
Bijak M (1995) Inhibitory effect of neuropeptide y on epileptiform activity in the frontal cortex and hippocampus in vitro. Pol J Pharmacol 47:461–463
Gloveli T, Albrecht D, Heinemann U (1995) Properties of low Mg2+ induced epileptiform activity in rat hippocampal and entorhinal cortex slices during adolescence. Brain Res Dev Brain Res 87:145–152. https://doi.org/10.1016/0165-3806(95)00069-p
Heinemann U, Zhang CL, Eder C (1993) Entorhinal cortex-hippocampal interactions in normal and epileptic temporal lobe. Hippocampus 3:89–97
Wang L, Shen HM (2020) Seeing is believing: a novel tool for quantitating mitophagy. Cell Res 30:715–716
Roperto S, Russo V, Falco FD, Rosati A, Roperto F (2019) FUNDC1-mediated mitophagy in bovine papillomavirus-infected urothelial cells. Vet Microbiol 234:1–10
Blair RE, Deshpande LS, Sombati S, Falenski KW, Martin BR, DeLorenzo RJ (2006) Activation of the cannabinoid type-1 receptor mediates the anticonvulsant properties of cannabinoids in the hippocampal neuronal culture models of acquired epilepsy and status epilepticus. J Pharmacol Exp Ther 317:1072–1078. https://doi.org/10.1124/jpet.105.100354
Kiese K, Jablonski J, Hackenbracht J, Wrosch JK, Groemer TW, Kornhuber J, Blumcke I, Kobow K (2017) Epigenetic control of epilepsy target genes contributes to a cellular memory of epileptogenesis in cultured rat hippocampal neurons. Acta Neuropathol Commun 5:79. https://doi.org/10.1186/s40478-017-0485-x
Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F et al (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189:211–221. https://doi.org/10.1083/jcb.200910140
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298. https://doi.org/10.1371/journal.pbio.1000298
Bockler S, Westermann B (2014) Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev Cell 28:450–458. https://doi.org/10.1016/j.devcel.2014.01.012
Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW (2015) The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60:7–20. https://doi.org/10.1016/j.molcel.2015.08.016
Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA et al (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell 56:360–375. https://doi.org/10.1016/j.molcel.2014.09.007
Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC (2010) Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 12:747–757. https://doi.org/10.1038/ncb2078
Tooze SA, Yoshimori T (2010) The origin of the autophagosomal membrane. Nat Cell Biol 12:831–835. https://doi.org/10.1038/ncb0910-831
Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y et al (2013) Autophagosomes form at ER-mitochondria contact sites. Nature 495:389–393. https://doi.org/10.1038/nature11910
Jeong SY, Seol DW (2008) The role of mitochondria in apoptosis. BMB Rep 41:11–22. https://doi.org/10.5483/bmbrep.2008.41.1.011
Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N (2012) Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci 125:1488–1499. https://doi.org/10.1242/jcs.094110
Xie Z, Klionsky DJ (2007) Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9:1102–1109. https://doi.org/10.1038/ncb1007-1102
Gelmetti V, De Rosa P, Torosantucci L, Marini ES, Romagnoli A, Di Rienzo M, Arena G, Vignone D, Fimia GM, Valente EM (2017) PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 13:654–669. https://doi.org/10.1080/15548627.2016.1277309
MacVicar TD, Mannack LV, Lees RM, Lane JD (2015) Targeted siRNA screens identify ER-to-mitochondrial calcium exchange in autophagy and mitophagy responses in RPE1 cells. Int J Mol Sci 16:13356–13380. https://doi.org/10.3390/ijms160613356
Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, Giorgi C, Pinton P (2018) Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69:62–72. https://doi.org/10.1016/j.ceca.2017.05.003
Bouman L, Schlierf A, Lutz AK, Shan J, Deinlein A, Kast J, Galehdar Z, Palmisano V, Patenge N, Berg D et al (2011) Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ 18:769–782. https://doi.org/10.1038/cdd.2010.142
Fessler E, Eckl EM, Schmitt S, Mancilla IA, Meyer-Bender MF, Hanf M, Philippou-Massier J, Krebs S, Zischka H, Jae LT (2020) A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 579:433–437. https://doi.org/10.1038/s41586-020-2076-4
Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, Wiita AP, Xu K, Correia MA, Kampmann M (2020) Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 579:427–432. https://doi.org/10.1038/s41586-020-2078-2
Deus CM, Yambire KF, Oliveira PJ, Raimundo N (2020) Mitochondria-lysosome crosstalk: from physiology to neurodegeneration. Trends Mol Med 26:71–88. https://doi.org/10.1016/j.molmed.2019.10.009
Plotegher N, Duchen MR (2017) Crosstalk between lysosomes and mitochondria in parkinson’s disease. Front Cell Dev Biol 5:110. https://doi.org/10.3389/fcell.2017.00110
Wong YC, Kim S, Peng W, Krainc D (2019) Regulation and function of mitochondria-lysosome membrane contact sites in cellular homeostasis. Trends Cell Biol 29:500–513. https://doi.org/10.1016/j.tcb.2019.02.004
Bose A, Beal MF (2019) Mitochondrial dysfunction and oxidative stress in induced pluripotent stem cell models of Parkinson’s disease. Eur J Neurosci 49:525–532. https://doi.org/10.1111/ejn.14264
Wang Y, Liu N, Lu B (2019) Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci Ther 25:859–875. https://doi.org/10.1111/cns.13140
Sekine S (2020) PINK1 import regulation at a crossroad of mitochondrial fate: the molecular mechanisms of PINK1 import. J Biochem 167:217–224. https://doi.org/10.1093/jb/mvz069
Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G (2005) Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet 14:3477–3492. https://doi.org/10.1093/hmg/ddi377
Kravic B, Harbauer AB, Romanello V, Simeone L, Vogtle FN, Kaiser T, Straubinger M, Huraskin D, Bottcher M, Cerqua C et al (2018) In mammalian skeletal muscle, phosphorylation of TOMM22 by protein kinase CSNK2/CK2 controls mitophagy. Autophagy 14:311–335. https://doi.org/10.1080/15548627.2017.1403716
Lazarou M, Jin SM, Kane LA, Youle RJ (2012) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22:320–333. https://doi.org/10.1016/j.devcel.2011.12.014
Yamano K, Youle RJ (2013) PINK1 is degraded through the N-end rule pathway. Autophagy 9:1758–1769. https://doi.org/10.4161/auto.24633
Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M et al (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 3:1016. https://doi.org/10.1038/ncomms2016
Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M et al (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2:120080. https://doi.org/10.1098/rsob.120080
Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, Matsuda N (2015) Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol 209:111–128. https://doi.org/10.1083/jcb.201410050
Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ (2006) Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy 2:39–46. https://doi.org/10.4161/auto.2229
Elmore SP, Qian T, Grissom SF, Lemasters JJ (2001) The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J 15:2286–2287. https://doi.org/10.1096/fj.01-0206fje
Acknowledgements
We thank the Academy of Medical Sciences of Zhengzhou University for supporting the translational medicine platform.
Funding
This study was supported by a grant from the National Natural Science Foundation of China (Grant No: 81771397).
Author information
Authors and Affiliations
Contributions
Authors YZ and YL participated in the conception and design of the study; XL performed the statistical analysis; YZ, HZ, YC, HS, RF performed the experiments; YZ wrote the manuscript. All authors contributed to the revision of the manuscript and read and approved the submitted version.
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were ethically approved.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, Y., Lian, Y., Lian, X. et al. FUNDC1 Mediated Mitophagy in Epileptic Hippocampal Neuronal Injury Induced by Magnesium-Free Fluid. Neurochem Res 48, 284–294 (2023). https://doi.org/10.1007/s11064-022-03749-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-022-03749-z