
Abstract
Autophagy plays a critical role in epileptic neuronal injury, and recent studies have demonstrated that FAM134B plays an important role in regulating autophagy. However, the effect of FAM134B on epileptic neuronal injury remains unclear. In this study, we investigated the role of FAM134B in neuronal apoptosis and endoplasmic reticulum (ER) stress using the hippocampal neuronal culture model of acquired epilepsy (AE) in vitro. We found that in this model, the level of autophagy significantly increased, indicated by an elevated LC3-II/LC3-I ratio. FAM134B overexpression using lentiviral vectors enhanced autophagy, whereas FAM134B downregulation using lentiviral vectors impaired this process. In addition, the ER Ca2+ concentration was decreased and the intracellular level of reactive oxygen species was increased in this model. FAM134B overexpression was sufficient to reverse these changes. Moreover, FAM134B overexpression attenuated ER stress as shown by a decrease in the expression of C/-EBP homologous protein and glucose-regulated protein 78, and neuronal apoptosis induced by seizure, while FAM134B downregulation caused the opposite effects. Further, pre-treatment with the selective autophagy inhibitor 3-methyladenine abolished the effects of FAM134B on ER stress and neuronal apoptosis. Altogether, we demonstrate that FAM134B is an important regulator of AE-induced ER stress and neuronal apoptosis by controlling autophagy function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autophagy alterations exist in a variety of neurological diseases, including epilepsy (Giorgi et al. 2015). The Family with sequence similarity 134, member B (FAM134B), which contains a reticulon homology domain (RHD) and LC3-interacting region (LIR), is the firstly identified ER autophagy receptor and exerts an important effect on normal cell homeostasis by regulating autophagy (Islam et al. 2018). FAM134B controls ER turnover by specific binding to microtubule-associated protein 1 light chain 3B (MAP1LC3B), which in turn binds to the phagophore membrane via its C-terminal LC3-interacting region (LIR) (Bhaskara et al. 2019). Recent studies have demonstrated that FAM134B-regulated autophagy is crucial for cell survival, especially under conditions of stress (Khaminets et al. 2015). However, the role of FAM134B-regulated autophagy in epileptic neuronal injury is unknown.
The endoplasmic reticulum (ER) is a vital organelle for neuronal survive, responsible for multiple functions including Ca2+ storage, signaling and protein folding, and maturation (Song et al. 2016; Yin et al. 2017). Various cellular stresses such as disturbance of ER Ca2+ homeostasis and oxidative stress can contribute to ER stress (Duan et al. 2017; Song et al. 2016). Moderate ER stress promotes the dissociation of glucose-regulated protein 78 (GRP78) to reduce the number of misfolded proteins and restore cell function (Liu et al. 2018). However, exacerbation of ER stress leads to downstream apoptotic signaling molecules activation such as cysteinyl aspartate-specific proteinase 12 (caspase 12) and C/-EBP homologous protein (CHOP) (Liu et al. 2018; Shimodaira et al. 2014). Growing data suggest an interaction between ER stress and autophagy. It has been recognized that ER stress can activate autophagy to reduce cell stress, while excessive autophagy triggers neuronal death (Niu et al. 2018; Ko et al. 2015). However, the effect of the FAM134B on seizure-induced ER stress is still elusive.
In this study, we evaluated the effects of FAM134B on neuronal apoptosis and ER stress using lentiviral vector-mediated manipulation of its expression in the hippocampal neuronal culture (HNC) model of acquired epilepsy (AE). In addition, we used the selective autophagy inhibitor 3-methyladenine (3-MA) to gain insights into the mechanisms of FAM134B in this context.
Materials and Methods
Primary Hippocampal Neuronal Cultures
All animal protocols were approved by the animal care and use committee of Zhengzhou University, China, confirming that all animal research and experimental operations are strictly in accordance with international guidelines for animal studies.
Primary hippocampal neurons were generated from healthy newborn Sprague–Dawley rats which were purchased from the Animal Center of Zhengzhou University. Dissociated neurons were plated on poly-l-lysine-coated (0.1 mg/ml) culture plates in 6-well plates at 4 × 105 cells per well and grown in planting medium containing neurobasal-A medium, 2% B27, 2 mM glutamine and 10% fetal bovine serum (Biological Industries, Israel) (Hinze et al. 2017). After 4 h of culture, the planting medium was removed and replaced with maintenance medium containing 2% B27, 2 mM glutamine, and neurobasal-A medium. Half of the maintenance medium was regularly refreshed every 2 days. Purity of hippocampal neurons was measured by immunofluorescence and confirmed to be > 95% by quantifying the proportion of positive cells.
Experimental Design
Hippocampal neurons were plated in 6-well plates at 4 × 105 cells per well. After 4 h of culture, the planting medium was removed and replaced with maintenance medium. The AE model was induced using a conventional method: hippocampal neurons were incubated in a Mg2+-free solution containing 145 mM NaCl, 2 mM CaCl2, 2.5 mM KCl, 0.002 mM glycine, 10 mM HEPES, and 10 mM glucose, with a pH of 7.4 (Blair et al. 2006; Kiese et al. 2017). After 10 days of culture, primary hippocampal neurons were randomly assigned to the following groups: (1) control group, neurons incubated in normal extracellular fluid for 3 h; (2) AE group, neurons cultured in Mg2+-free solution for 3 h and then cultured in maintenance medium; (3) negative control group, neurons incubated with lenti-pGV for 12 h and then cultured in maintenance medium for 72 h before incubation in Mg2+-free solution for 3 h; (4) lenti-FAM134B (upregulation FAM134B) group, neurons incubated with lenti-FAM134B for 12 h and then cultured in maintenance medium for 72 h before incubation in Mg2+-free solution for 3 h; (5) lenti-FAM134B-shRNA (downregulation FAM134B) group, neurons incubated with lenti-FAM134B-shRNA for 12 h and then cultured in maintenance medium for 72 h before incubation in Mg2+-free solution for 3 h; and (6) 3-MA group, neurons incubated with lenti-FAM134B-shRNA for 12 h, cultured in maintenance medium for 72 h and then incubated in the presence of the autophagy inhibitor 3-MA for 24 h before incubation in Mg2+-free solution for 3 h.
MTT Assay
The methyl tetrazolium (MTT) assay was used to assess cell viability. Briefly, hippocampal neurons were seeded in 96-well plates at a density of 2 × 105 cells/ml. After 10 days of culture, the maintenance medium was removed and replaced with 20 µl of 5 mg/ml MTT solution and cultured at 37 °C for 4 h. Then, 150 µl of DMSO was added to dissolve MTT-formazan precipitates prior to incubation for 4–18 h at 37 °C. Absorbance density values were spectrophotometrically measured at 570 nm to calculate the cell viability.
TUNEL Assay
Apoptosis was determined by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay (Roche Diagnostics, Indianapolis, IN) according to the manufacturer’s instructions. Briefly, coverslips containing primary neurons were processed for 30 min in 4% paraformaldehyde at 37 °C and then for 5 min in 0.1% Triton X-100 at 37 °C. Afterwards, the coverslips containing primary neurons were incubated for 1 h in TUNEL reaction mixture and then with DAPI in the dark at 37 °C for 5 min. Finally, the coverslips containing primary neurons were mounted with glycerol. In each group, a total of 500 neurons were detected and the positive cell proportion was calculated for statistical analyses.
Determination of Ca2+ Concentration in ER
The concentration of ER Ca2+ was assessed by using specific fluorescent probes (Mag-Fluo-AM) according to the instructions providing by manufacturer (Jiemei, Shanghai, China). Neurons were plated in 12-well plates at a density of 2 × 105 cells/ml and rinsed with 500 µl reagent A and then cultured in 300 µl reaction mixture in the dark for 60 min at 37 °C. Neurons were then rinsed with reagent A. The concentration of ER Ca2+ was quantified using a fluorescence microscope (Olympus, Japan).
Measurement of ROS
The level of intracellular reactive oxygen species (ROS) was determined by using a commercial kit (Nanjing Jiancheng Bioengineering Institute, Jiangsu, China) according to the instructions providing by manufacturer. Neurons were incubated with fluorescent probe 2′, 7′-dichlorofluorescin diacetate (DCFH-DA) in the dark for 30 min at 37 °C. Next, the neurons were resuspended in ice-cold Krebs–Ringer solution. The level of ROS was spectrophotometrically measured at a wavelength of 488 nm.
Immunofluorescence
Immunofluorescence was used to examine whether seizures induce the recruitment of FAM134B into autophagosomes. Coverslips containing primary neurons were fixed for 30 min in 4% paraformaldehyde at 37 °C and then permeabilized for 5 min in 0.1% Triton X-100 at 37 °C. Next, the neurons were blocked for 1 h in 10% goat serum at 37 °C and then incubated with rabbit anti-FAM134B (1:100; Abcam, USA) and mouse anti-LC3B (1:200; CST, USA) at 4 °C overnight, followed by incubation with dye-labeled secondary antibodies at 37 °C for 1 h. The expression of FAM134B and LC3B was observed under a confocal microscope (Zeiss, Germany).
Western Blotting
Hippocampal neurons were scraped on ice in 80 μl of RIPA buffer-containing protease inhibitor. Each sample was added a quarter volume of 5 × loading buffer and boiled at 100 °C for 5 min. These proteins were separated using 12% SDS-PAGE and stained with rabbit anti-β-actin (1:1000; CST, USA), mouse anti-CHOP (1:500; CST, USA), rabbit anti-GRP78 (1:500; Abcam, USA), rabbit anti-caspase12 (1:1000; CST, USA), and mouse anti-LC3B (1:1000; CST, USA) at 4 °C overnight, followed by incubation with secondary antibodies at 37 °C for 1 h. Analysis was performed using Image J software.
Infection with Lentiviral Vectors
Hippocampal neurons were seeded in 6-well plates at a density of 2 × 105 cells/ml. After 5 days of culture, cells were infected with lenti-FAM134B, lenti-FAM134B-shRNA, or lenti-pGV (Jikai, Shanghai, China) at a multiplicity of infection (MOI) of 15. The medium containing lentiviral vectors was removed and replaced with maintenance medium after 12 h. Infection efficiency was observed using a fluorescence microscope (Olympus, Japan) and was verified by Western blot analysis of the expression of FAM134B after 72 h of culture.
Statistical Analysis
All results are expressed as the mean ± standard deviation (SD) and were analyzed using SPSS software version 17.0. Differences between groups were determined by using one-way ANOVA and Newman–Keuls test. A P-value of < 0.05 was considered statistically significant.
Results
FAM134B Overexpression Attenuates AE-Induced Neuronal Apoptosis
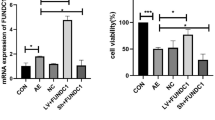
The infection efficiency of lentiviral vectors was verified by Western blot analysis of the expression of FAM134B. Relative to the control group, pre-treatment with lenti-FAM134B increased the FAM134B expression and pre-treatment with lenti-FAM134B-shRNA elevated the expression of FAM134B (Fig. 1a). Neuronal viability, as measured by MTT assay, was significantly decreased in the AE group as compared with the control group (Fig. 1b). Overexpression of FAM134B using lenti-FAM134B attenuated AE-induced neuronal death, while downregulation of this protein using lenti-FAM134B-shRNA exacerbated AE-induced neuronal death (Fig. 1b). Statistically, there were no significant differences between the AE group and the negative control group (Fig. 1b).

Effect of FAM134B on hippocampal neuronal damage induced by acquired epilepsy (AE). a The infection efficiency of lentiviral vectors was analyzed by Western blot analysis of the expression of FAM134B. Hippocampal neurons were only incubated with lenti-FAM134B and lenti-FAM134B-shRNA for 12 h and then cultured in maintenance medium for 72 h. b Cell viability, assessed by MTT assay. Hippocampal neurons were incubated with lenti-FAM134B, lenti-FAM134B-shRNA or lenti-pGV for 12 h and then cultured in maintenance medium for 72 h before exposure to Mg2+-free solution. Data are expressed as mean ± SD of six independent experiments. *P < 0.05 compared to the control group, #P < 0.05 compared to the AE group
In addition, the apoptosis rate of cells was assessed by TUNEL assay. Neuronal apoptosis was significantly increased in the AE group (Fig. 2). However, FAM134B overexpression attenuated AE-induced neuronal apoptosis and FAM134B downregulation exacerbated this process (Fig. 2). The difference between the AE group and the negative control group was not statistically significant (Fig. 2).

Effect of FAM134B on hippocampal neuron apoptosis induced by acquired epilepsy (AE). TUNEL assay was used to assess hippocampal neuron apoptosis. Both the AE group and the negative control group exhibit apoptotic neurons (positive cells), whereas in the lenti-FAM134B group, FAM134B overexpression protects neurons against apoptosis induced by AE. In the lenti-FAM134B-shRNA group, FAM134B downregulation exacerbates AE-induced neuronal apoptosis. Data are expressed as mean ± SD of six independent experiments. *P < 0.05 compared to the AE group
FAM134B Overexpression Augments Autophagy Induced by AE
The ratio of LC3-II/LC3-I was significantly increased in the AE group as compared to the control group (Fig. 3). Relative to the AE group, FAM134B overexpression enhanced the ratio of LC3-II/LC3-I, whereas its downregulation decreased this ratio (Fig. 3). No differences were found between the AE and negative control groups (Fig. 3).

Effect of FAM134B on LC3-II/LC3-I ratio induced by acquired epilepsy (AE). Western blot and quantitative analysis of LC3-II/LC3-I ratio in hippocampal neurons. Data are expressed as mean ± SD of six independent experiments. *P < 0.05 compared to the control group, #P < 0.05 compared to the AE group
In order to further assess the effects of FAM134B on autophagy, neurons were co-immunostained for FAM134B and LC3B. We found more extensive colocalization of these proteins in the AE group as compared with controls, and FAM134B overexpression further increased this elevated colocalization (Fig. 4). On the contrary, downregulation of FAM134B decreased the colocalized areas of FAM134B and LC3B (Fig. 4). No significant differences were found between the AE group and negative control group (Fig. 4). Altogether, these data indicate that the level of autophagy is regulated by FAM134B.

Effect of FAM134B on co-immunostained for FAM134B and LC3B induced by acquired epilepsy (AE). Representative images of FAM134B and LC3B immunofluorescence in hippocampal neurons. Colocalization of FAM134B with LC3B was determined using the Pearson's coefficient. Data are expressed as mean ± SD of six independent experiments. *P < 0.05 compared to the control group, #P < 0.05 compared to the AE group
FAM134B Overexpression Attenuates ER Ca2+ Release and Intracellular ROS Production Induced by AE
As shown in Fig. 5, compared with the control group, the ER Ca2+ concentration was reduced and the intracellular level of ROS was increased in the AE group. This was rescued by FAM134B overexpression, which significantly increased the ER Ca2+ concentration and diminished the intracellular ROS level as compared with the AE group (Fig. 5). In contrast, FAM134B downregulation had the opposite effect, exacerbating the defects observed in response to AE. The AE group showed no differences compared with the negative control group (Fig. 5).

Effect of FAM134B on changes of endoplasmic reticulum (ER) Ca2+ concentration and intracellular reactive oxygen species (ROS) production induced by acquired epilepsy (AE). a ER Ca2+ concentration in hippocampal neurons. b Levels of ROS in hippocampal neurons. Data are expressed as mean ± SD of six independent experiments. *P < 0.05 compared to the control group, #P < 0.05 compared to the AE group
FAM134B Overexpression Attenuates AE-Induced ER Stress
We further found that relative to controls, AE caused a significant increase in the GRP78 and CHOP expression (Fig. 6). While FAM134B knockdown further raised these expression levels, its overexpression decreased the GRP78 and CHOP levels compared to that in the AE group (Fig. 6). No significant differences were found between the AE group and negative control group (Fig. 6). These results suggest that ER stress induced by AE is regulated by FAM134B expression.

Effect of FAM134B on glucose-regulated protein 78 (GRP78) and C/-EBP homologous (CHOP) expression induced by acquired epilepsy (AE). Western blot and quantitative analysis of GRP78 and CHOP expression in hippocampal neurons. Data are expressed as mean ± SD of six independent experiments. *P < 0.05 compared to the control group, #P < 0.05 compared to the AE group
Inhibition of Autophagy by 3-MA Abolishes the Protective Effects of FAM134B
To further confirm whether FAM134B exerts effects on apoptosis and ER stress through induced autophagy, we took advantage of the selective autophagy inhibitor 3-MA. As shown in Fig. 7, in the AE group, the GRP78 and CHOP levels were increased compared to the control group, and FAM134B overexpression reversed this effect. Interestingly, pre-treatment with 3-MA significantly abolished the effects of FAM134B on neuronal apoptosis and ER stress, indicating that the observed effects were specific to FAM134B (Fig. 7). The AE group and the negative control group showed no statistically significant differences (Fig. 7).

Selective autophagy inhibitor 3-methyladenine (3-MA) abolishes the protective effects of FAM134B. a Western blot and quantitative analysis of GRP78 and CHOP in hippocampal neurons. b Cell viability, analyzed using MTT assay. Data are expressed as mean ± SD of six independent experiments. *P < 0.05 compared to the control group, #P < 0.05 compared to the AE group, **P < 0.05 compared to the lenti-FAM134B group
Discussion
Recent studies have implicated that autophagy plays a crucial role in various neurodegenerative diseases and FAM134B plays a functional role in autophagy. (Cai et al. 2019; Niu et al. 2018). However, the effects of FAM134B-regulated autophagy on ER stress and neuronal apoptosis induced by AE have not been investigated. In this study, we found that AE can induce ER dysfunction, which is characterized by the depletion of its Ca2+ storage and increased intracellular ROS levels. ER dysfunction was also evident through a significant elevation in ER stress and autophagy levels. Importantly, FAM134B overexpression attenuated ER stress and neuronal apoptosis, while FAM134B knockdown led to opposite effects. Moreover, selective inhibition of autophagy abolished the protective effects of FAM134B on ER stress and apoptosis. Altogether, our results indicate that FAM134B exerts neuroprotective effects on seizure-induced ER stress and apoptosis, and that this is likely based on its regulation of autophagy.
Autophagy is an important process for maintaining intracellular homeostasis. Many studies have demonstrated that autophagy modification can alleviate the results of epileptic seizures (Attia et al. 2019). Here, we found that in the HNC model of AE, autophagy increased. In addition, FAM134B overexpression enhanced seizure-induced autophagic activity, as evidenced by increases in the ratio of LC3-II/LC3-I and in the area of colocalization of FAM134B and LCB, whereas FAM134B downregulation resulted in the opposite. These observations indicate that FAM134B plays a crucial role in regulating AE-induced autophagy.
The ER is the most important Ca2+ storage organelle. Various cellular stresses, including oxidative stress and the depletion of Ca2+ from ER stores, can trigger ER stress responses (Dejeans et al. 2010; You et al. 2016). An increase in the amount of Ca2+ released from the ER can boost the generation of ROS, which causes a positive feedback loop leading to further calcium release and stress (Bhandary et al. 2012). Our results demonstrate that the ER Ca2+ concentration significantly decreases and the intracellular ROS level increases in the HNC model of AE. These data suggest that seizures can contribute to the disruption of ER homeostasis by causing an increase in Ca2+ release from the ER and an elevated oxidative stress level. We observed that FAM134B overexpression significantly attenuated these two processes, while its downregulation further exacerbated them. These results suggest that FAM134B contributes to the recovery of ER function through regulating Ca2+ concentration within this organelle and by limiting oxidative stress.
Disruption of ER homeostasis, such as the depletion of Ca2+ from the ER, can interfere with protein folding and transport, thus contributing to ER stress (Concannon et al. 2008). Moderate ER stress can improve cell survival through the unfolded protein response (UPR) (Shi et al. 2016), which is mediated by ER transmembrane receptors, including IRE1, PERK, and ATF6. Activation of these receptors upregulates the expression of ER chaperones, such as GRP78 (You et al. 2016). On the other hand, severe and/or prolonged ER stress promotes the activation of pro-apoptotic factors such as CHOP (Lei et al. 2017). In our study, we found that FAM134B overexpression inhibited GRP78 and CHOP expression as well as neuronal apoptosis, while FAM134B knockdown evoked the opposite. These results indicate that FAM134B alleviates neuronal ER stress and apoptosis.
Several studies have implicated that autophagy is closely related to ER stress and that autophagy plays an important role in cell survival and cell death (Ding et al. 2007; Giorgi et al. 2015; Song et al. 2017). Autophagy can inhibit apoptosis by eliminating damaged organelles and misfolded/unfolded proteins, inhibiting caspase activation, and clearing sequestosome 1 (SQSTM1)/p62, thereby protecting cells that are under ER stress. In contrast, dysregulation of autophagy may contribute to several pathological conditions by causing cell death (Ogata et al. 2006; Xu et al. 2017). In this study, we demonstrated that selective inhibition of autophagy using 3-MA significantly abolished the effects of FAM134B on ER stress and neuronal apoptosis. This is consistent with previous studies indicating that autophagy induced by ER stress can reduce cell damage, and that FAM134B inhibition contributes to the misfolding or aggregation of proteins leading to neuronal injury (Ciechomska et al. 2013; Khaminets et al. 2015; Song et al. 2017). Taken together, our results suggest that FAM134B is involved in promoting autophagy function to relieve ER stress and prevent neuronal apoptosis.
In conclusion, our study provides evidence that FAM134B attenuates seizure-induced apoptosis and ER stress in hippocampal neurons, and the underlying mechanism is, at least, in part, based on the regulation of autophagy. Although further studies are needed to unravel the specific mechanisms underlying FAM134B-mediated regulation of autophagy in AE, our results pave the way for the development of novel therapeutic strategies for the treatment of epilepsy.
References
Attia GM, Elmansy RA, Elsaed WM (2019) Neuroprotective effect of nilotinib on pentylenetetrazol-induced epilepsy in adult rat hippocampus: involvement of oxidative stress, autophagy, inflammation, and apoptosis. Folia Neuropathol 57(2):146–160
Bhandary B, Marahatta A, Kim HR, Chae HJ (2012) An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int J Mol Sci 14:434–456
Bhaskara RM, Grumati P, Garcia-Pardo J, Kalayil S, Covarrubias-Pinto A, Chen W, Kudryashev M, Dikic I, Hummer G (2019) Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective ER-phagy. Nat Commun 10:2370
Blair RE, Deshpande LS, Sombati S, Falenski KW, Martin BR, DeLorenzo RJ (2006) Activation of the cannabinoid type-1 receptor mediates the anticonvulsant properties of cannabinoids in the hippocampal neuronal culture models of acquired epilepsy and status epilepticus. J Pharmacol Exp Ther 317:1072–1078
Cai M, Zhao J, Liu Q, Wang X, Wang Y (2019) FAM134B improves preadipocytes differentiation by enhancing mitophagy. Biochim Biophys Acta Mol Cell Biol Lipids 1864:158508
Ciechomska IA, Gabrusiewicz K, Szczepankiewicz AA, Kaminska B (2013) Endoplasmic reticulum stress triggers autophagy in malignant glioma cells undergoing cyclosporine a-induced cell death. Oncogene 32:1518–1529
Concannon CG, Ward MW, Bonner HP, Kuroki K, Tuffy LP, Bonner CT, Woods I, Engel T, Henshall DC, Prehn JH (2008) NMDA receptor-mediated excitotoxic neuronal apoptosis in vitro and in vivo occurs in an ER stress and PUMA independent manner. J Neurochem 105:891–903
Dejeans N, Tajeddine N, Beck R, Verrax J, Taper H, Gailly P, Calderon PB (2010) Endoplasmic reticulum calcium release potentiates the ER stress and cell death caused by an oxidative stress in MCF-7 cells. Biochem Pharmacol 79:1221–1230
Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM (2007) Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem 282:4702–4710
Duan XC, Wang W, Feng DX, Yin J, Zuo G, Chen DD, Chen ZQ, Li HY, Wang Z, Chen G (2017) Roles of autophagy and endoplasmic reticulum stress in intracerebral hemorrhage-induced secondary brain injury in rats. CNS Neurosci Ther 23:554–566
Giorgi FS, Biagioni F, Lenzi P, Frati A, Fornai F (2015) The role of autophagy in epileptogenesis and in epilepsy-induced neuronal alterations. J Neural Transm (Vienna) 122:849–862
Hinze SJ, Jackson MR, Lie S, Jolly L, Field M, Barry SC, Harvey RJ, Shoubridge C (2017) Incorrect dosage of IQSEC2, a known intellectual disability and epilepsy gene, disrupts dendritic spine morphogenesis. Transl Psychiatry 7:e1110
Islam F, Chaousis S, Wahab R, Gopalan V, Lam AK (2018) Protein interactions of FAM134B with EB1 and APC/beta-catenin in vitro in colon carcinoma. Mol Carcinog 57:1480–1491
Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N et al (2015) Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522:354–358
Kiese K, Jablonski J, Hackenbracht J, Wrosch JK, Groemer TW, Kornhuber J, Blümcke I, Kobow K (2017) Epigenetic control of epilepsy target genes contributes to a cellular memory of epileptogenesis in cultured rat hippocampal neurons. Acta Neuropathol Commun 5:79
Ko AR, Kim JY, Hyun HW, Kim JE (2015) Endoplasmic reticulum (ER) stress protein responses in relation to spatio-temporal dynamics of astroglial responses to status epilepticus in rats. Neuroscience 307:199–214
Lei Y, Wang S, Ren B, Wang J, Chen J, Lu J, Zhan S, Fu Y, Huang L, Tan J (2017) CHOP favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS ONE 12:e0183680
Liu L, Zhang Y, Wang Y, Peng W, Zhang N, Ye Y (2018) Progesterone inhibited endoplasmic reticulum stress associated apoptosis induced by interleukin-1beta via the GRP78/PERK/CHOP pathway in BeWo cells. J Obstet Gynaecol Res 44:463–473
Niu Q, Chen J, Xia T, Li P, Zhou G, Xu C, Zhao Q, Dong L, Zhang S, Wang A (2018) Excessive ER stress and the resulting autophagic flux dysfunction contribute to fluoride-induced neurotoxicity. Environ Pollut 233:889–899
Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K et al (2006) Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 26:9220–9231
Shi S, Tan P, Yan B, Gao R, Zhao J, Wang J, Guo J, Li N, Ma Z (2016) ER stress and autophagy are involved in the apoptosis induced by cisplatin in human lung cancer cells. Oncol Rep 35:2606–2614
Shimodaira Y, Takahashi S, Kinouchi Y, Endo K, Shiga H, Kakuta Y, Kuroha M, Shimosegawa T (2014) Modulation of endoplasmic reticulum (ER) stress-induced autophagy by C/EBP homologous protein (CHOP) and inositol-requiring enzyme 1alpha (IRE1alpha) in human colon cancer cells. Biochem Biophys Res Commun 445:524–533
Song Q, Gou WL, Zhang R (2016) FAM3A attenuates ER stress-induced mitochondrial dysfunction and apoptosis via CHOP-Wnt pathway. Neurochem Int 94:82–89
Song S, Tan J, Miao Y, Li M, Zhang Q (2017) Crosstalk of autophagy and apoptosis: involvement of the dual role of autophagy under ER stress. J Cell Physiol 232:2977–2984
Xu X, Huang E, Tai Y, Zha X, Chen X, Chen C, Chen R, Liu C, Lin Z, Wang H et al (2017) Nupr1 modulates methamphetamine-induced dopaminergic neuronal apoptosis and autophagy through CHOP-Trib3-mediated endoplasmic reticulum stress signaling pathway. Front Mol Neurosci 10:203
Yin Y, Sun G, Li E, Kiselyov K, Sun D (2017) ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res Rev 34:3–14
You WT, Zhou T, Ma ZC, Liang QD, Xiao CR, Tang XL, Tan HL, Zhang BL, Wang YG, Gao Y (2016) Ophiopogonin D maintains Ca2+ homeostasis in rat cardiomyocytes in vitro by upregulating CYP2J3/EETs and suppressing ER stress. Acta Pharmacol Sin 37:368–381
Acknowledgements
We are grateful for the support by the academy of medical sciences of Zhengzhou University translational medicine platform.
Funding
This study was supported by grants from National Natural Science Foundation of China (Grant Nos. 81971214, 81701272) Provincial Ministry Co-construction Project from Medical Scientific and Technological Research Program of Henan Province (Grant No. SB201902011) and Training plan for young backbone teachers of Zhengzhou University (Grant No. 2019ZDGGJS056).
Author information
Authors and Affiliations
Contributions
NX and CW contributed to conception and design of the study; NX, YL (Yajun Lian), and HZ performed the statistical analysis; YL (Yingjiao Li), YL (Yujuan Li), LD, and XM performed the experiments; NX and YL (Yingjiao Li) wrote the manuscript. All authors contributed to manuscript revision and read and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xie, N., Li, Y., Wang, C. et al. FAM134B Attenuates Seizure-Induced Apoptosis and Endoplasmic Reticulum Stress in Hippocampal Neurons by Promoting Autophagy. Cell Mol Neurobiol 40, 1297–1305 (2020). https://doi.org/10.1007/s10571-020-00814-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-020-00814-5