
Abstract
The late onset neuropathologies, including Alzheimer’s disease and Parkinson’s disease, have become increasingly prevalent. Their causation has been linked to genetics, gut microbiota dysbiosis (gut dysbiosis), autoimmune diseases, pathogens and exposures to neurotoxins. An alternative explanatory hypothesis is provided for their pathogenesis. Virtually everyone has pervasive daily exposures to neurotoxins, through inhalation, skin contact, direct blood transmission and through the gastrointestinal tract by ingestion. As a result, every individual has substantial and fluctuating neurotoxin blood levels. Two major barriers to neurotoxin entry into the central nervous system are the blood–brain barrier and the intestinal wall, in the absence of gut dysbiosis. Inflammation from gut dysbiosis, induced by antibiotic usage, can increase the intestinal wall permeability for neurotoxins to reach the bloodstream, and also increase the blood–brain barrier permeability to neurotoxins. Gut dysbiosis, including gut dysbiosis caused by antibiotic treatments, is an especially high risk for neurotoxin entry into the brain to cause late onset neuropathologies. Gut dysbiosis has far-reaching immune system and central nervous system effects, and even a transient gut dysbiosis can act in combination with neurotoxins, such as aluminum, mercury, lead, arsenic, cadmium, selenium, manganese, organophosphate pesticides and organochlorines, to reach neurotoxin blood levels that can initiate a late onset neuropathology, depending on an individual’s age and genetic vulnerability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There are several late onset neuropathologies with suspected links to certain biological triggers, such as pathogen infections, immune disorders, autoimmune diseases, abnormalities in gut microbiota (gut dysbiosis), and exposures to neurotoxins, including mercury, aluminum, lead, arsenic, cadmium, selenium, manganese, organophosphate pesticides, and organochlorines. However, while several strong correlations to metal toxins and mechanisms for neuropathologies have been analyzed by separate studies and far larger scale meta-analysis studies, these neuropathologies still need an explanation for their pathogenesis [1, 2]. A pathogenesis hypothesis is proposed, that with some variations, could also apply to several late onset neuropathologies. Alzheimer’s disease and Parkinson's disease will be the focus of discussion as the two most important neurodegenerative disease examples of late onset neuropathologies [1, 2].
Antibiotics and Gut Dysbiosis Initiation
Furthermore, gut microbiota dysbiosis (gut dysbiosis) has been linked to several neuropathologies, including Parkinson's disease and Alzheimer’s disease [3]. It has long been suspected that gut dysbiosis is involved in the pathogenesis of several late onset neuropathlogies, because the gut microbiota have major influences on a host’s immune system and central nervous system (CNS) [3, 4].
For example, one associated symptom of gut dysbiosis is constipation, and it has been long observed that constipation is one of the most common non-motor symptoms of Parkinson’s disease, which may predict future Parkinson’s disease by more than a decade [5, 6]. This gut dysbiosis symptom was incorporated into one widely known hypothesis that Parkinson’s disease starts in the enteric nervous system and spreads to the CNS through the vagus nerve by an unknown prion-like pathogen [5, 6]. Supporting evidence includes the detection of pathological aggregates of the presynaptic neuronal protein α-synuclein in gastrointestinal tissues removed from patients several years before their diagnosis of Parkinson’s disease [5].
Epidemiological studies have provided additional evidence, including the observation that truncal vagotomy (i.e., removing part of the vagus nerve at the gastroesophageal junction to denervate multiple organs, including the stomach, liver, gall bladder, pancreas, small intestine, and proximal colon) decreases the hazard ratio of Parkinson’s disease to 0.59, at least for the first 5–20 years post-surgery [5, 6]. In addition, experimental seeding of α-synuclein preformed fibrils into human α-synuclein SNCA gene model transgenic murine duodenum intestinal walls induced α-synuclein aggregates and gut-to-brain trans-synaptic transportation of preformed fibrils [7]. This was interpreted to be the result of recruitment of endogenous α-synuclein into the sympathetic and parasympathetic propagation of pathological α-synuclein aggregates in a manner similar to human patients [7]. Furthermore, it has also been noted that murine Parkinson’s disease progression is triggered from enteric neuron release of more α-synuclein after murine stomachs were exposed to the neutrotoxic pesticide rotenone that induces parkinsonism [7].
However, another associated symptom of gut dysbiosis is gut inflammation, and it has been hypothesized that, depending on an individual’s age and genetic vulnerability, chronic low-level inflammatory bowel disease is a fundamental cause of Parkinson's disease [8]. Extensive observational evidence indicates that brain inflammation is implicated in disease initiation and disease progression, and the inflammation associated with Parkinson’s disease affects both the brain and the gastrointestinal tract [8]. Increased levels of inflammatory cytokines and other inflammatory markers have been observed in the gastrointestinal tracts of Parkinson’s disease patients and there is extensive epidemiological evidence and genetic evidence that Parkinson’s disease is associated with inflammatory bowel diseases, depending on an individual’s age and genetic vulnerability [8]. Recent observations concerning inflammatory bowel diseases and Parkinson’s disease provide evidence indicating a bidirectional link exists between degenerative brain neuropathologies and gastrointestinal inflammation, and also imply gastrointestinal inflammation involvement as a facilitator in the initiation and progression of late onset neuropathologies, including Parkinson’s disease [8].
There is also considerable evidence that antibiotic exposures can initiate major gut microbiota disruptions, such as inflammatory bowel diseases, including Crohn’s disease, which can affect either the small or large intestines [9]. For example, in the extensively documented case of pediatric patients, a link has been seen between antibiotic treatment exposures (documented through recorded purchases) to childhood gastrointestinal disorders [9]. The odds of Crohn’s disease increased as the number of antibiotic treatment exposures (usages inferred from recorded purchases) increased, especially for seven or more courses of antibiotics, especially for boys [9]. The cephalosporin antibiotics were the most strongly linked antibiotics to the initiation of Crohn’s disease [9].
Gut dysbiosis has been reported to degrade the intestinal epithelial cells and mucus layers and allow substances to ultimately enter the brain and CNS [10]. Gut dysbiosis allows solutes, from ions to macromolecule proteins and bacterial toxins, to pass through the tight junctions of the intestinal epithelial barrier and the gut vascular barrier by paracellular and transcellular pathways [10]. Thus, specific antibiotics can induce gut dysbiosis that assists the passage of neurotoxins through the walls of the gastrointestinal tract. In contrast, in the absence of gut dybiosis, the ingested neurotoxins in food and water, such as aluminum, would normally enter and exit the gastrointestinal tract without complete absorption through the intestinal wall, which will be discussed in more detail later. Thus, during gut inflammation, gut dysbiosis increases toxin transmission through the intestinal wall into the circulatory system, and such toxin penetration to the circulatory system has been reported [11, 12]. Eventually, the increased neurotoxin level(s) can reach threshold levels that surmount a vulnerable individual’s defenses against the neurotoxin(s) at the blood–brain barrier (BBB) and allow the neurotoxin(s) to initiate Alzheimer’s disease or Parkinson's disease or another late onset neuropathology.
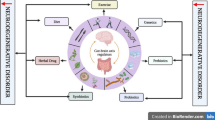
For clarity, Fig. 1 summarizes the main steps of the hypothesized pathogenesis of a late onset neuropathology resulting from a confluence of antibiotics, gut microbiota dysbiosis and neurotoxin exposure.

Provides one proposed path of a neuropathology induced by a gut microbiota dysbiosis that increases levels of circulating neurotoxins and facilitates their passage through the blood brain barrier
The Blood–Brain Barrier
Human BBB formation includes the full integration of astrocytes and the communication between the pericyte cells in the basement membrane of the brain capillaries, the endothelial cells of the brain capillaries and the glycocalyx luminal coating secreted by the endothelial cells to increase the BBB integrity [13,14,15,16]. BBB formation minimizes transcellular transport and maximizes expression of endothelial tight junction proteins, but this is not completed until a considerable time after birth [13,14,15,16]. Pericytes are essential in BBB development and function, and in inhibiting nonspecific transcytosis and leukocyte adhesion molecule expression [15]. Astrocytes do not appear necessary for BBB formation, but they provide dynamic BBB regulation and repair in neurological disease [15]. An age-related decline in the BBB, including age-related pericyte dysfunction, has been extensively reported [15]. If there is any gut dysbiosis, any neurotoxins in the gastrointestinal tract can more easily pass through the intestinal wall into the circulatory system. And if the BBB is incomplete or dysfunctional at the same time, this will facilitate neurotoxin entry into the brain and CNS. However, at any age, with or without BBB completion, a gut dysbiosis for any reason (e.g., initiated by antibiotic usage) is a significant factor that facilitates neurotoxins in reaching the brain and CNS to cause late onset neuropathologies [17].
Gut Dysbiosis and Neuropathologies
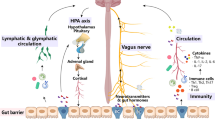
One question is how far-reaching are the consequences for the CNS from a gut dysbiosis? An individual’s neurological function and behavior, including depression, anxiety, and social behavior, is affected by certain microbes in the individual’s gut microbiota, due to signals to the CNS by means of several neural, endocrine, and immune pathways [3, 4]. The communication between the gut microbiota and the CNS is known as the “microbiota-gut-brain axis” or the “microbiome-gut-brain axis” [3, 4]. For example, most inflammatory cytokines produced by immune cells in the gut can penetrate the BBB [4]. Upon reaching the CNS and brain, these cytokines can activate neuron and glial cell receptors and activate microglia, the macrophages in the brain [4].
Gut microbiota also assist the regulation the “hypothalamic–pituitary–adrenal axis” (HPA axis) [3]. In addition, gut microbiota also assist the synthesis of neuromodulatory metabolites for normal brain functionality, such as dopamine, serotonin, kynurenine, glutamate, N-acetyl aspartate, epinephrine (adrenaline), and norepinephine, histamine, branched chain amino acids, tryptophan precursors and metabolites, short-chain fatty acids (acetate, propionate, butyrate), and γ-aminobutyric acid [3]. In addition, there is neural communication with the CNS through the autonomic nervous system, including the sympathetic and parasympathetic nervous system, such as communications between the gut microbiota and the vast number of enteric neurons embedded in the lining of the gastrointestinal tract [3, 18]. The bidirectional communications between the brain, the gut microbiota, the enteric nervous system, the spinal and vagus nerve pathways, the sympathetic and parasympathetic nervous system has been called a microbiota-gut-brain “connectome” [18].
Furthermore, there are additional consequences from gut dysbiosis population reductions in helpful bacteria, and population increases in the pathogenic fungal pathogens, including Candida albicans and pathogenic bacteria of the bacterial families such as Enterobacteriaceae, that can also induce intestinal inflammation by activation of CD4 TH1 cells and TH17 cells [19]. And there is considerable neuron and T cell crosstalk, even in the gastrointestinal tract [19]. For example, an anti-inflammatory reflex through vagus efferent nerves can ultimately reach certain T cells, choline acetyltransferase T cells, that can produce acetylcholine, recognized by the acetylcholine receptor on splenic macrophages, and can thereby reduce gastrointestinal inflammation by reducing splenic macrophage production of tumor necrosis factor-α (TNF-α) [19]. Intestinal inflammation can affect peripheral immune cell reactions by elevating pro-inflammatory cytokines levels in the blood, such as TNF-α, interleukin-1 (IL-1), interleukin-6 (IL-6), and interferon-γ, which are suspected of causing neuroinflammatory, neurodegenerative diseases, including Parkinson’s disease [20].
Gut microbiota can influence CNS neuroinflammation by three pathways: (1) by moderating CNS resident immune cells, such as microglia and astrocytes, by production of short-chain fatty acids (SCFA), including SCFA butyrate, and indole derivatives; (2) by increasing peripheral immune responses through production of several inflammatory cytokines, including IL-17 released by gastrointestinal tract TH17 cells; and (3) by increasing immune cell entry into the CNS by TH17 cell production of endothelial nitric oxide synthase (eNOS), which induces CNS endothelial cells to increase BBB permeability [20, 21].
Table 1 summarizes some major pathways for gut dysbiosis to impact the brain and CNS to induce late onset neuropathologies.
An increased BBB permeability will decrease the ability of the BBB to block blood-borne neurotoxins, such as aluminum, mercury, organophosphates, organochlorines, etc., from entering the brain. Depending on the neurotoxin blood levels, an increased BBB permeability, from even a transient gut dysbiosis, can permit a destructive dose of neurotoxins to enter a brain and accumulate to cause brain cell damage and initiate a neurodegenerative disease [21]. In addition, there is increasing evidence that a gut dysbiosis increases the intestinal permeability to allow more toxins to penetrate the intestinal wall and enter the circulatory system to elevate systemic inflammation [22].
Major Neurotoxins and Their Consequences
There are several metals and chemicals that are neurotoxic, including aluminum, mercury, the organophosphate pesticides, the organochlorines, etc. [1, 17, 23,24,25]. This section will first focus on aluminum, because of aluminum's extensive toxicity, the pervasive human exposures to aluminum and its presence in blood, and because aluminum ions have been detected by certain highly sensitive analytical techniques in the brains of individuals that suffered from various neuropathologies, including Alzheimer's disease, as well as other neurodegenerative diseases [1, 2, 17, 23].
Aluminum Exposures
Aluminum exposures for humans are amazingly pervasive. Aluminum sources include several types of air pollution from coal burning, motor vehicles and smoking, water pollution, and foods that contain considerable aluminum [2, 23]. Aluminum exposures for individuals also include cosmetics, antiperspirants, sunscreens and sunblocks, pharmaceuticals (e.g., antacids, buffered analgesics, anti-diarrheal drugs, anti-ulcer drugs, etc.), pesticides, various types of foods, including food prepared in aluminum pans, aluminum foil, parchment paper, baked goods made with baking powder containing aluminum and food additives including aluminum-based food colorings, and pottery [2, 23]. Aluminum blood levels are also highly elevated by hemodialysis, and aluminum is ingested daily from drinking water that is treated for deflocculation using aluminum sulfates and aluminum chlorides [23].
Gastrointestinal absorption after ingestion is a major contributor to aluminum accumulation in individuals, and aluminum absorption from water is usually greater than aluminum absorption from food [23]. Food absorption of aluminum is affected by an individual’s age, stomach contents and acidity, and the type of aluminum compound, as well as the presence of aluminum absorption inhibitors, including polyphenols, phytates, iron, phosphates and silicon; and the presence of aluminum absorption enhancers, including citrate, maltol, lactate and fluorides [23]. Aluminum in ingested food and water passing through the gastrointestinal tract is normally little absorbed; some estimates of non-absorption rates range from 90 to 99% [23, 24]. However, aluminum is completely absorbed from hemodialysis and hypodermic injections, such as from injections of aluminum salt adjuvants in vaccines designed to stimulate the immune system’s T cells and B cells [2, 23].
It has been reported that 90% of the aluminum circulating in the blood is transported by transferrin (an iron-transporter protein) and the remaining aluminum circulating in the blood is transported by albumin and citrate, with healthy blood levels in blood serum ranging between one to three micrograms per liter, and with ten times higher levels in hemodialysis patients [23]. Aluminum is known to reach the brain and cerebrospinal fluid, and it can penetrate the placenta and reach a fetus, and reach children through milk from lactating mothers [23]. Elevated intestinal absorption of aluminum contributes to aluminum accumulation in various organs, whereas the kidneys excrete about 95% of the blood aluminum by elimination in the urine as aluminum citrate, and the remainder is eliminated in feces, sweat, hair, nails, etc. [23].
The effects of aluminum’s toxicity are extensive, such as oxidative stress; inflammation in the lungs, intestines, heart and testis; immunosuppression by inducing lymphocyte dysfunction; denaturation and alterations of proteins; enzyme stimulation or inhibition; metabolic impairments; and genotoxicity with reduced cell proliferation and differentiation [1, 2, 23, 24]. Furthermore, aluminum causes amyloid formation; causes estrogenic effects on breast cancer cells; causes fetal defects by teratogenesis; inhibits mineral intake by altering intestinal and cellular mineral uptake; induces apoptosis and tissue necrosis; and damages cell membrane permeability and receptor functions [1, 2, 23, 24]. Aluminum also induces endocrine disruptions; interferes with cartilage and bone formation and mineralization; causes hypertension, ischemic strokes, and thrombosis; causes contact allergies; and interferes with vitamin D biological functions in the intestines [1, 2, 23, 24].
Aluminum salts can activate the NLRP3 inflammasome by several pathways, such as phagosome destabilization, lysosome acidification and by elevated reactive oxygen species (ROS) levels [1, 23]. Damage by oxidative stress through increased lipid peroxidation and depleted anti-oxidant defenses has been reported in the prefrontal cortex, cerebellum, hippocampus and brainstem of brains [1]. Chronic aluminum exposure impairs cellular anti-oxidant defenses by reducing cellular levels of glutathione transferase, peroxidase, catalase, superoxide dismutase and glutathione (GSH) [1]. Aluminum oral ingestion damages intestinal epithelial cells and elevates intestinal inflammation and thereby elevates intestinal barrier permeability [1]. Aluminum’s neurotoxicity is mainly due to its ability to induce oxidative stress and mitochondrial dysfunction in brain and CNS cells, both directly and indirectly, by interference with calcium homeostasis in mitochondrial functions in brain and CNS cells [1].
Aluminum exposure can cause iron homeostasis disruption leading to elevated iron levels [23]. Oxidative stress and injury, mediated by iron, is therefore enhanced by aluminum [23]. Elevated concentrations of cellular iron can increase cellular oxidative damage and are linked to the pathogenesis of neurodegenerative disorders including Alzheimer’s disease, discussed below [23].
Aluminium can induce microglia to release neuroinflammatory, pro-inflammatory cytokines TNF-α and IL-6, and release cytokine-inducible nitric oxide synthase (iNOS or NOS-2), nitric oxide (NO) and reactive oxygen species (ROS) [25, 26]. This is interesting, since there is widely reported that microglia, using cytokines like TNF-α, play an important role in brain development by controlling processes, including synaptic pruning, synaptic plasticity, synaptogenesis, neuronal development and other neurogenesis processes [27]. Microglial dysfunction and/or priming initiated by aging, immune challenges, inflammatory events or other brain changes, which interfere with processes including synaptic pruning and neuronal proliferation, have been linked to the initiation of neuropathologies [27,28,29,30].
Aluminum’s neurotoxic effects also affect astrocytes, since astrocytes (protoplasmic astrocytes, fibrous astrocytes, and two types of radial astroglia) have major roles in brain development as crucial components of the glia limitans, together with pericytes and endothelial cells, for regulating the BBB between the bloodstream and brain parenchyma [31]. In addition, astrocytes regulate processes in synaptic transmission, neuronal migration, synaptogenesis, and very likely assist oligodendrocytes in neuronal myelination [31, 32]. Glial cell numbers equal or surpass the number of neurons, and glial cells are critical to brain neuronal circuit development and maintenance [33]. The glutamate removal function of astrocytes that is critical for neurons is impaired by neurotoxins, because the metal transporter functions of astrocytes, designed for the transport of zinc and iron, also result in astrocytes becoming major targets for several neurotoxic metals, including manganese, lead, aluminum and mercury [32]. In addition, there are reports that inhibited astrocyte functions have an essential role in the pathogenesis of neuropathologies, such as Alzheimer’s disease [31,32,33].
Table 2 summarizes some significant toxic effects from aluminum exposures.
The Late Onset Neuropathologies
Significant gut dysbiosis has been linked to several late onset neuropathologies, including Parkinson’s disease and Alzheimer’s disease [3]. Gut microbiota have profound effects on the metabolism and the maintenance of a host's immune system and central nervous system (CNS), and it has been proven that gut microbiota can induce several effects on murine behavior [3]. In fact, the administration of antibiotic cocktails to laboratory animals has been extensively utilized to disturb the gut microbiota of laboratory animals, with demonstrated effects on the anxiety and sociability of the laboratory animals [3].
Alzheimer’s Disease from Aluminum Exposures
Alzheimer's disease has symptoms of a marked and progressive deterioration of several regions of the brain involved with cognitive function and memory [29]. Excessive microglial activation and microglial induced neuroinflammation have a major role in neurodegeneration and Alzheimer’s disease [29]. In Alzheimer’s disease, aluminum can play a neurotoxic role by activating microglia to release neuroinflammatory cytokines TNF-α and IL-6, iNOS, NOS-2, NO and ROS [24,25,26]. Atrophied astrocytes and their reduction of synaptic transmission, connectivity and neuronal survival; and reactive astrocytes and their release of proinflammatory cytokines and iNOS; also have a major role in Alzheimer's disease [32].
Several papers have established a strong correlation between the levels of aluminum in drinking water and the incidence of Alzheimer’s disease throughout the world, including the United Kingdom, Canada, Norway and France [1]. Aluminum has been detected by some sensitive techniques in brain plaques and neurofibrillary tangles [1]. Aluminum has also been detected in its binding to critical parts of Alzheimer's disease affected brains, such as the hippocampus [1].
Age Increases the Risk of Alzheimer’s Disease from Aluminum Exposures
Age matters, and with age neurotoxins can build up in the brain and CNS faster than they can be removed. As discussed earlier, the kidneys excrete about 95% of the blood aluminum by elimination in the urine as aluminum citrate, and the remainder is eliminated in feces, sweat, hair, nails, etc. [23]. Unfortunately, aluminum levels in the brain generally increase with age [25], so this more plausibly indicates that aluminum removal mechanisms from the body degrade with age, or less plausibly indicates that aluminum absorption by the body increases with age. In either case, as the aluminum levels in the brain increase, the likelihood of aluminum damage to neurons and astrocytes will increase as previously discussed [23,24,25,26]. An age-related increase in brain damage from increasing levels of other neurotoxins (e.g., mercury, etc.) should also be possible, if the rate of elimination from the body decreases with age and the rate of elimination cannot keep up with the rate of absorption into the body. The extensive and persistently severe neurotoxic effects of mercury are the next topic of discussion.
Alzheimer’s Disease from Mercury Exposures
Mercury exposures have also been linked to Alzheimer’s disease, and to plaques, beta amyloid protein, neurofibrillary tangles, and phosphorylated tau proteins observed in the brains of Alzheimer’s disease victims [34]. Mercury is reportedly ten times more toxic to neurons than lead, and more neurotoxic than cadmium, manganese, aluminum and iron [34]. Mercury exposures can include elemental mercury that is easily absorbed by inhalation, inorganic mercury ions that have low gastrointestinal absorption, and organic mercury, such as methyl mercury or ethyl mercury, that have quick absorption by skin, lungs, kidney, heart, and gastrointestinal absorption [35]. Oral bacteria have been reported to convert inorganic mercury from dental amalgams into methyl mercury [35]. Mercury has a significant binding affinity to sulfhydryl (thiol) groups in amino acids, proteins, erythrocytes, crucial enzymes and antioxidants (e.g., N-acetylcysteine, α-lipoic acid, glutathione, metallothioneine, etc.), and these effects add to the toxic and neurotoxic consequences from mercury [34, 35]. Glutathione is a critical intracellular and mitochondrial antioxidant in reducing oxidative stress, inflammation and cardiovascular diseases [35].
Table 3 summarizes significant toxic effects from mercury exposures.
Mercury bio-accumulates in the brain, liver, kidneys and muscles, resulting in damage to the brain, lungs, kidneys, nervous system, immune system, heart and cardiovascular system [35]. Dietary exposures of mercury are literally increasing every year, since mercury levels in fish muscle tissues have been increasing every year, such as an observed annual mercury increase averaging 3.8% each year since 1998 in North Pacific Ocean yellowfin tuna near Hawaii [36]. This is primarily the result of the burning of mercury-bearing fossil fuels that have increased mercury levels in ocean water shallower than 1000 meters by an average of ~ 3% per year since at least 1995 [36].
Dental amalgams are also a major mercury exposure risk for most individuals, especially since the relatively low mercury vapor emitting low copper content dental amalgams were superseded in the 1970s by much higher mercury vapor emitting high copper dental amalgams [37]. Mercury vapor emissions from the high copper dental amalgams has been greatly increased, from 3 to 62 times higher (at least 10 times higher) than the mercury vapor emissions from the highest emitters of the low copper dental amalgams [37]. Dental amalgam mercury vapor emissions are enhanced by even minor abrasions from chewing, polishing, or from a minor increase in dental amalgam temperature after consuming hot beverages or hot food [37]. Dental amalgams constantly emit un-ionized mercury vapor that can be inhaled, enter the bloodstream through the lung alveoli and easily pass through the BBB to the brain [34]. Because of this, since 1991 dental amalgams have been considered the largest source of mercury for most individuals [34].
Autopsies have determined that there is a definite correlation between the levels of inorganic mercury in the brain and blood and the number of dental surfaces filled with dental amalgams [38]. Furthermore, the half-life of inorganic mercury in the brain has also been estimated to be ~ 20 years [38]. Direct links between dental amalgams and neuropathologies have also been reported [38]. A recent study compared elderly individuals with and without dental amalgams and determined a higher odds ratio for Alzheimer's disease (1.105, more specifically 1.07 for men and 1.132 for women, involving over 200,000 individuals over 65 years old in Taiwan) [38]. Another recent study also compared elderly individuals with and without dental amalgams and determined a higher hazard ratio of 1.583 for Parkinson’s disease (involving over 20,000 individuals in Taiwan) [38].
Figure 2 provides one proposed chain reaction path of causation for Alzheimer’s disease, from the beginning of gut microbiota dysbiosis to the final stage of Alzheimer’s disease.

Provides one proposed chain reaction path of causation for Alzheimer’s disease
In summary, since the 1970s there has been a considerable elevation in oral mercury vapor exposure from the utilization of high copper dental amalgams in individuals who have received dental fillings. If combined with the yearly increase in mercury exposure from fish consumption observed since at least the 1990s, these two sources by themselves alone could explain much of the age adjusted increase documented by Medicare data in U.S. adults over 65 for Alzheimer’s disease (5.79% in 1998 to 9.03% in 2013) and kidney disease (5.9% in 1998 to 18.3% in 2013) [39]. But it should be noted that there can be major contributions to late onset neuropathologies from other neurotoxic metals besides mercury; including lead, arsenic, cadmium, selenium and manganese [15, 40,41,42].
Lead
Lead poisoning has been widespread and long reported, but it has become somewhat less common since inorganic lead exposure from the environment has decreased in countries that have banned lead additives to gasoline and paints and lead emissions [40]. Lead exposure is primarily from inhalation of lead particles, ingestion of lead compounds, or from water carried by lead pipes [40]. Divalent lead ions can cross the BBB and cell membranes by mimicking divalent calcium ions, divalent iron ions or divalent zinc ions [40]. Lead causes oxidative stress, mitochondrial dysfunctions, disrupts beneficial selenium dependent processes, and reduces nitric oxide synthase activity in brain cells [40]. Lead bio-accumulates in the brain hippocampus, amygdala, and choroid plexus [40]. Postnatal lead exposure has been linked to neurological disorders, mental retardation, nerve damage, Alzheimer’s disease and Parkinson’s disease [40].
Arsenic
Arsenic exposure is most commonly caused by drinking groundwater containing inorganic arsenic [40]. High level arsenic exposure can cause encephalopathy, and long-term low level arsenic exposure can cause peripheral neuropathologies outside the CNS [40]. Arsenic concentrations from 5 to 50 parts per billion in water have caused children to have degraded cognitive function, verbal abilities, long-term memory and motor skills [40]. Inorganic arsenic creates oxidative stress, especially in mitochondria, from the generation of ROS and lipid peroxidation; and arsenic can also replace phosphate in several metabolic pathways [40].
Cadmium
Cadmium is a neurotoxin with exposure pathways similar to lead, because it can be inhaled from air or tobacco smoke, ingested from foods that have accumulated high levels of cadmium, and its ions can mimic divalent calcium, copper and zinc ions to cross cell membranes [40]. Cadmium is also similar in its neurotoxic effects to mercury in that it has a strong binding affinity to sulfhydryl (thiol) groups on crucial enzymes and antioxidants (e.g., glutathione, etc.), and causes oxidative damage (e.g., by disruptions to detoxification of peroxides), especially for neurons and oligodendrocytes [40].
Selenium
Selenium has a narrow beneficial range, but at significantly higher levels it can act as a neurotoxin [41]. Selenium is important to the body as a chelating antioxidant, and in combination with proteins can remove harmful aluminum, arsenic, lead, manganese and mercury from the body and the brain [41]. The ratio of selenium to mercury in various fish species can also determine the overall harm from consumption of specific fish species [41]. Selenium is mainly ingested and selenium compounds in relatively high concentrations can react with glutathione and other thiol proteins or enzymes to form superoxide anions leading to widespread oxidative stress and apoptosis, necrosis, or necroptosis, contributing to selenium’s cellular toxicity [42]. The different selenium chemical species have various neurotoxic effects, especially for motor neurons [43]. Inorganic tetravalent selenium ions in selenite and inorganic hexavalent selenium ions in selenate may have more than 40 times higher neurotoxicity than organic selenium [43]. Chronic low-level exposure to inorganic selenium has been linked to amyotrophic lateral sclerosis (ALS) and Parkinson’s disease [43].
Manganese
Manganese exposure is usually through food ingestion or from inhalation from mining dust, smelting, welding or exposures to pesticides containing manganese [40]. Manganese is a widely reported neurotoxin [40]. Manganese accumulates in cell mitochondria, and increases iron accumulation, which induces ROS formation and oxidative damage to brain cells, and particularly in astrocytes that results in neuron excitotoxicity [40]. Manganese also assists creation of hydrogen peroxide (H2O2), inhibits oxidative phosphorylation and interferes with adenosine triphosphate (ATP) production [36]. Manganese can also initiate manganism, which includes symptoms of dystonia (muscle spasms) and symptoms similar to Parkinson’s disease, which is the next topic [40].
Parkinson’s Disease
Parkinson’s disease is the second-leading late onset neuropathology after Alzheimer’s disease [44]. Parkinson’s disease is characterized by a reduction in brain’s dopaminergic neurons in the substantia nigra, with a dopamine depletion that results in primary motor symptoms of resting tremor, bradykinesia (i.e., slowness of movement), muscle rigidity and postural instability [44]. Parkinson’s disease’s depletion of dopamine in the human brain’s substantia nigra is also associated with increased activity by the monoamine oxidase B (MAO-B) enzyme which degrades dopamine, and because of this MAO-B inhibitors are used in treating Parkinson’s disease [45].
The most strongly linked causes of Parkinson’s disease include inhalation or ingestion of pesticides or other neurotoxins, brain trauma and injury, aging, drugs, and genetic influences [44]. Many studies have established that Parkinson’s disease is associated with farming occupations, rural living, and the drinking of well-water [46,47,48]. There is strong evidence linking Parkinson’s disease with exposures to paraquat, rotenone and other organochlorines [47, 48]. Parkinson’s disease has been linked to exposure to some agricultural organohalogen and organochlorine chemicals in the Agricultural Health Study (AHS) of licensed pesticide applicators and their spouses in Iowa and North Carolina [48].
Parkinson's disease results from multiple types of cellular dysfunctions, including mitochondrial dysfunction [49], lysosomal dysfunction contributing to α-synuclein accumulation [50], proteasomal dysfunction [51], calcium homeostasis disorders [52], innate and adaptive immune system induced neuroinflammation [53], α-synuclein transmission and aggregation [54], and oxidative stress [55]. For example, paraquat causes neuronal damage by generating toxic superoxide free radicals, inducing α-synuclein upregulation, α-synuclein aggregate formation and microglial activation [47]. And neuroinflammation, oxidative stress (including stress from reactive oxygen species created by increased brain levels of the monoamine oxidase B enzyme) and mitochrondrial dysfunction can be caused by ingestion of extremely neurotoxic chemicals which are strongly linked to Parkinson's disease [44,45,46].
Even very low level exposures of extremely neurotoxic chemicals can cause damage. For example, chronic low-level exposure levels (1 mg/kg) and medium exposure levels (10 mg/kg) of paraquat in mice will cause age-dependent damage to neurons in the prefrontal cortex, hippocampus and to dopaminergic neurons in the mesencephalon (midbrain), including the substantia nigra pars compacta [56]. The effects of chronic low-level exposures to humans appear to parallel this damage, because cumulative lifetime exposure to paraquat increases the incidence of Parkinson’s disease in farm workers [56, 57].
Figure 3 provides one proposed chain reaction path of causation for Parkinson’s disease, from the beginning of gut microbiota dysbiosis to the final stage of Parkinson’s disease.

Provides one proposed chain reaction path of causation for Parkinson’s disease
Neuropathologies from Organophosphate and Organochlorine Exposures
Many of the organophosphate pesticides (including chlorphyrifos, diazinon, malathion, parathion, etc.) and organochlorines {including the polychlorinated biphenyls, and the pesticides dieldrin, endosulfan, heptachlor, dichlorodiphenyltrichloroethane (DDT), dichlorodiphenyldichloroethylene (DDE), chlordane, etc.} have been banned for several decades [17]. However, they are still being used in several countries and applications, including food exports, and even in developed countries that long banned their use, individuals are still bio-accumulating these neurotoxins each year and increasing their blood serum levels, at least in reports as recent as 2015 [17]. Organophosphate and organochlorine exposures, such as pesticides and herbicides, have also been linked to late onset neuropathologies such as Parkinson's disease [17].
Future Directions
The blood–brain barrier and the intestinal wall, in the absence of gut dysbiosis, are two major barriers to neurotoxin entry into the central nervous system. Extensive antibiotic usage can induce inflammation from gut dysbiosis, and this can increase the intestinal wall permeability for neurotoxins to pass through and enter the bloodstream, and increase the blood–brain barrier permeability to neurotoxins. Even a transient gut dysbiosis can act in combination with neurotoxins, including aluminum, mercury, lead, arsenic, cadmium, selenium, manganese, organophosphates, organochlorines, and other neurotoxins, to initiate a neuropathology. In summary, several neuropathologies can be initiated in individuals as a result of gut dysbiosis, either long-term or frequently transiently induced by certain antibiotic treatments, acting together with pervasive daily exposures to neurotoxins, depending on an individual’s age and genetic vulnerability. The increasing occurrence of the major neuropathologies can be significantly reduced by a minimization of gut dybiosis by elimination of the usage of certain specific antibiotics, a comprehensive minimization of aluminum exposures, the minimization of mercury exposures by a total prohibition on the use of silver-mercury dental amalgams in dental applications, and a reduction of organophosphate and organochlorine exposures by a more widely enforced world-wide restriction on several neurotoxic herbicides and pesticides.
Data Availability
Not applicable.
Code Availability
Not applicable.
Abbreviations
- SNCA:
-
Synuclein, Alpha
- GI:
-
Gastrointestinal
- CNS:
-
Central nervous system
- HPA:
-
Hypothalamic–pituitary–adrenal
- GABA:
-
γ-Aminobutyric acid
- IgG:
-
Immunoglobulin G
- CD4:
-
Cluster of differentiation 4
- TH1:
-
Helper 1 T cells
- TH17:
-
Helper 17 T cells
- IL-1:
-
Interleukin-1
- IL-6:
-
Interleukin-6
- IL-17:
-
Interleukin-17
- TNF-α:
-
Tumor necrosis factor-α
- eNOS:
-
Endothelial nitric oxide synthase
- BBB:
-
Blood–brain barrier
- ROS:
-
Reactive oxygen species
- GSH:
-
Glutathione
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxygen synthase
- iNOS:
-
Inducible nitric oxygen synthase
- DDT:
-
Dichlorodiphenyltrichloroethane
- DDE:
-
Dichlorodiphenyldichloroethylene
References
Morris G, Puri BK, Frye RE (2017) The putative role of environmental aluminum in the development of chronic neuropathology in adults and children. How strong is the evidence and what could be the mechanisms involved? Metab Brain Dis 32(5):1335–1335
Alasfar RH, Isaifan RJ (2021) Aluminum environmental pollution: the silent killer. Environ Sci Pollut Res Int 28(33):44587–44597. https://doi.org/10.1007/s11356-021-14700-0
Cryan JF, O’riordan KJ, Cowan CSM, Sandhu KV, Bastiaanssen TFS, Boehme M, Codagnone MG et al (2019) The microbiota-gut-brain axis. Physiol Rev 99:1877–2013
Zhu S, Jiang Y, Xu K, Cui M, Ye W, Zhao G, Jin L, Chen X (2020) The progress of gut microbiome research related to brain disorders. J Neuroinflammation 17(1):25. https://doi.org/10.1186/s12974-020-1705-z
Skjærbæk C, Knudsen K, Horsager J, Borghammer P (2021) Gastrointestinal dysfunction in Parkinson’s disease. J Clin Med 10(3):493. https://doi.org/10.3390/jcm10030493
Liu B, Fang F, Pedersen NL, Tillander A, Ludvigsson JF, Ekbom A, Svenningsson P, Chen H, Wirdefeldt K (2017) Vagotomy and Parkinson disease: a Swedish register-based matched-cohort study. Neurology 88(21):1996–2002. https://doi.org/10.1212/WNL.0000000000003961
Van Den Berge N, Ferreira N, Gram H, Mikkelsen TW, Alstrup AKO, Casadei N, Tsung-Pin P, Riess O, Nyengaard JR, Tamgüney G, Jensen PH, Borghammer P (2019) Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol 138(4):535–550. https://doi.org/10.1007/s00401-019-02040-w
Rolli-Derkinderen M, Leclair-Visonneau L, Bourreille A, Coron E, Neunlist M, Derkinderen P (2020) Is Parkinson’s disease a chronic low-grade inflammatory bowel disease? J Neurol 267:2207–2213. https://doi.org/10.1007/s00415-019-09321-0
Ramirez J, Guarner F, Bustos Fernandez L, Maruy A, Sdepanian VL, Cohen H (2020) Antibiotics as major disruptors of gut microbiota. Front Cell Infect Microbiol 10:572912. https://doi.org/10.3389/fcimb.2020.572912
Antonini M, Conte M, Sorini C, Falcone M (2019) How the interplay between the commensal microbiota, gut barrier integrity, and mucosal immunity regulates brain autoimmunity. Front Immunol 10:1937
Lau WL, Savoj J, Nakata MB, Vaziri ND (2018) Altered microbiome in chronic kidney disease: systemic effects of gut-derived uremic toxins. Clin Sci 132(5):509–522
Obrenovich MEM (2018) Leaky gut, leaky brain? Microorganisms 6(4):107
Coelho-Santos V, Shih AY (2020) Postnatal development of cerbrovascular structure and neurogliovascular unit. Wiley Interdiscip Rev Dev Biol 9(2):e363
Galea I (2021) The blood-brain barrier in systemic infection and inflammation. Cell Mol Immunol. https://doi.org/10.1038/s41423-021-00757-x
Profaci CP, Munji RN, Pulido RS, Daneman R (2020) The blood-brain barrier in health and disease: important unanswered questions. J Exp Med 217(4):e20190062. https://doi.org/10.1084/jem.20190062
Hussain B, Fang C, Chang J (2021) Blood-brain barrier breakdown: an emerging biomarker of cognitive impairment in normal aging and dementia. Front Neurosci 15:688090. https://doi.org/10.3389/fnins.2021.688090
Kern JK, Geier DA, Homme KG, King PG, Bjorklund G, Chirumbolo S, Geier MR (2017) Developmental neurotoxicants and the vulnerable male brain: a systematic review of suspected neurotoxicants that disproportionally affect males. Acta Neurobiol Exp 77(4):269–296
Gershon MD, Margolis KG (2021) The gut, its microbiome, and the brain: connections and communications. J Clin Invest 131(18):e143768. https://doi.org/10.1172/JCI143768
Jacobson A, Yang D, Vella M, Chiu IM (2021) The intestinal neuro-immune axis: crosstalk between neurons, immune cells, and microbes. Mucosal Immunol 14(3):555–565. https://doi.org/10.1038/s41385-020-00368-1
Janakiraman M, Krishnamoorthy G (2018) Emerging role of diet and microbiota interactions in neuroinflammation. Front Immunol 9:2067
Eshraghi RS, Davies C, Iyengar R, Perez L, Mittal R, Eshraghi AA (2021) Gut-Induced inflammation during development may compromise the blood-brain barrier and predispose to autism spectrum disorder. J Clin Med 10(1):27. https://doi.org/10.3390/jcm10010027
Fukui H (2016) Increased intestinal permeability and decrease barrier function: does it really influence the risk of inflammation? Inflamm Intest Dis 1(3):135–145
Igbokwe IO, Igwenagu E, Igbokwe NA (2019) Aluminum toxicosis; a review of toxic actions and effects. Interdiscip Toxicol 12(2):45–70
Tietz T, Lenzner A, Kolbaum AE, Zellmer S, Riebeling C, Gürtler R, Jung C, Kappenstein O, Tentschert J, Giulbudagian M, Merkel S, Pirow R, Lindtner O, Tralau T, Schäfer B, Laux P, Greiner M, Lampen A, Luch A, Wittkowski R, Hensel A (2019) Aggregated aluminium exposure: risk assessment for the general population. Arch Toxicol 93(12):3503–3521. https://doi.org/10.1007/s00204-019-02599-z
Bondy SC (2016) Low levels of aluminum can lead to behavioral and morphological changes associated with Alzheimer’s disease and age-related neurodegeneration. Neurotoxicology 52:222–229. https://doi.org/10.1016/j.neuro.2015.12.002
Skalny AV, Aschner M, Jiang Y, Gluhcheva YG, Tizabi Y, Lobinski R, Tinkov AA (2021) Molecular mechanisms of aluminum neurotoxicity: update on adverse effects and therapeutic strategies. Adv Neurotoxicol 5:1–34. https://doi.org/10.1016/bs.ant.2020.12.001
Tremblay MÈ (2021) Microglial functional alteration and increased diversity in the challenged brain: insights into novel targets for intervention. Brain Behav Immun Health 16:100301. https://doi.org/10.1016/j.bbih.2021.100301
Anderson SR, Vetter ML (2019) Developmental roles of microglia: a window into mechanisms of disease. Dev Dyn 248(1):98–117. https://doi.org/10.1002/dvdy.1
Hoeijmakers L, Heinen Y, van Dam AM, Lucassen PJ, Korosi A (2016) Microglial priming and Alzheimer’s disease: a possible role for (early) immune challenges and epigenetics? Front Hum Neurosci 10:398
Edmonson CA, Ziats MN, Rennert OM (2016) A non-inflammatory role for microglia in autism spectrum disorders. Front Neurol 7:9
Nutma E, van Gent D, Amor S, Peferoen LAN (2020) Astrocyte and oligodendrocyte cross-talk in the central nervous system. Cells 9(3):600. https://doi.org/10.3390/cells9030600
Verkhratsky A, Parpura V (2016) Astrogliopathology in neurological, neurodevelopmental and psychiatric disorders. Neurobiol Dis 85:254–261
Khakh BS, Sofroniew MV (2015) Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci 18(7):942–952
Siblerud R, Mutter J, Moore E, Naumann J, Walach H (2019) A hypothesis and evidence that mercury may be an etiological factor in Alzheimer’s disease. Int J Environ Res Public Health 16(24):5152
Genchi G, Sinicropi MS, Carocci A, Lauria G, Catalono A (2017) Mercury exposure and heart diseases. Int J Environ Res Public Health 14(1):74
Drevnick PE, Lamborg CH, Horgan MJ (2015) Increase in mercury in Pacific yellow fin tuna. Environ Toxicol Chem 34:931–934
Bengtsson UG, Hylander LD (2017) Increased mercury emissions from modern dental amalgams. Biometals 30(2):277–283
Jirau-Colón H, González-Parrilla L, Martinez-Jiménez J, Adam W, Jiménez-Velez B (2019) Rethinking the dental amalgam dilemma: an integrated toxicological approach. Int J Environ Res Public Health 16(6):1036
Akushevich I, Kravchenko J, Yashkin AP, Yashin AI (2018) Time trends in the prevalence of cancer and non-cancer diseases among older U.S. adults: medicare-based analysis. Exp Gerontol 110:267–276
Garza-Lombó C, Posadas Y, Quintanar L, Gonsebatt ME, Franco R (2018) Neurotoxicity linked to dysfunctional metal ion homeostatis and xenobiotic metal exposure: redox signaling and oxidative stress. Antioxid Redox Signal 28(18):1669–1703
Schofield K (2017) The metal neurotoxins: an important role in current human neural epidemics? Int J Environ Res Public Health 14(12):1511
Misra S, Boylan M, Selvam A, Spallholz JE, Björnstedt M (2015) Redox-active selenium compounds—from toxicity and cell death to cancer treatment. Nutrients 7(5):3536–3556
Vinceti M, Filippini T, Wise LA (2018) Environmental selenium and human health: an update. Curr Environ Health Rep 5(4):464–485. https://doi.org/10.1007/s40572-018-0213-0
Prasad EM, Hung SY (2020) Behavioral tests in neurotoxin-induced animal models of Parkinson’s disease. Antioxidants 9(10):1007
Yeung AWK, Georgieva MG, Atanasov AG, Tzvetkov NT (2019) Monamine oxidases (MAOs) as privileged molecular targets in neuroscience: research literature analysis. Front Mol Neurosci 12:143
Powers R, Lei S, Anandhan A, Marshall DD, Worley B, Cerny RL, Dodds ED, Huang Y, Panayiotidis MI, Pappa A, Franco R (2017) Metabolic investigations of the molecular mechanisms associated with Parkinson’s disease. Metabolities 7(2):22
Nandipati S, Litvan I (2016) Environmental exposures and Parkinson’s disease. Int J Environ Res Public Health 13(9):881
Kamel F, Tanner CM, Umbach DM, Hoppin JA, Alavanja MCR, Blair A, Comyns K et al (2007) Pesticide exposure and self-reported Parkinson’s disease in the agricultural health study. Am J Epidemiol 165(4):364–374
Prasuhn J, Davis RL, Kumar KR (2021) Targeting mitochondrial impairment in Parkinson’s disease: challenges and opportunities. Front Cell Dev Biol 5(8):615461. https://doi.org/10.3389/fcell.2020.615461
Minakaki G, Krainc D, Burbulla LF (2020) The convergence of alpha-synuclein, mitochondrial, and lysosomal pathways in vulnerability of midbrain dopaminergic neurons in Parkinson’s disease. Front Cell Dev Biol 8:580634. https://doi.org/10.3389/fcell.2020.580634
Bentea E, Verbruggen L, Massie A (2017) The proteasome inhibition model of Parkinson’s disease. J Parkinsons Dis 7(1):31–63
Ureshino RP, Erustes AG, Bassani TB, Wachilewski P, Guarache GC, Nascimento AC, Costa AJ, Smaili SS, Pereira GJDS (2019) The interplay between Ca2+ signaling pathways and neurodegeneration. Int J Mol Sci 20(23):6004. https://doi.org/10.3390/ijms20236004
Zaman V, Shields DC, Shams R, Drasites KP, Matzelle D, Haque A, Banik NL (2021) Cellular and molecular pathophysiology in the progression of Parkinson’s disease. Metab Brain Dis 36(5):815–827. https://doi.org/10.1007/s11011-021-00689-5
Henderson MX, Trojanowski JQ, Lee VM-Y (2019) α-Synuclein pathology in Parkinson’s disease and related α-synucleinopathies. Neurosci Lett 709:134316
Trist BG, Hare DJ, Double KL (2019) Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 18(6):e13031. https://doi.org/10.1111/acel.13031
Rudyk CA, McNeill J, Prowse N, Dwyer Z, Farmer K, Litteljohn D, Caldwell W, Hayley S (2017) Age and chronicity of administration dramatically influenced the impact of low dose paraquat exposure on behavior and hypothalmic-pituitary-adrenal activity. Front Aging Neurosci 9:222
Rudyk C, Dwyer Z, McNeill J, Salmaso N, Farmer K, Prowse N, Hayley S (2019) Chronic unpredictable stress influenced the behavioral but not the neurodegenerative impact of paraquat. Neurobiol Stress 11:100179
Acknowledgements
There are no acknowledgements.
Funding
No funding was received for this article.
Author information
Authors and Affiliations
Contributions
Kevin Roe is the sole author of all contributions.
Corresponding author
Ethics declarations
Conflict of interest
The author has no potential conflicts of interest.
Ethical Approval
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Kevin Roe—Retired.
Rights and permissions
About this article

Cite this article
Roe, K. An Alternative Explanation for Alzheimer’s Disease and Parkinson’s Disease Initiation from Specific Antibiotics, Gut Microbiota Dysbiosis and Neurotoxins. Neurochem Res 47, 517–530 (2022). https://doi.org/10.1007/s11064-021-03467-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-021-03467-y