
Abstract
While the pathogenesis of Parkinson’s disease is not fully understood, there is increasing evidence that inflammatory responses in the brain are implicated in both disease initiation and progression. The inflammatory process in Parkinson’s disease is, however, not limited to the brain but also involves the gastrointestinal tract. High amounts of cytokines and inflammatory markers are found in the colon of Parkinson’s disease patients and there is now strong epidemiological and genetical evidence linking Parkinson’s disease to inflammatory bowel diseases. Recent findings obtained in both experimental inflammatory bowel diseases and Parkinson’s disease further support a bidirectional link between gastrointestinal inflammation and brain neurodegeneration. Altogether, these observations suggest a role for gastrointestinal inflammation in the initiation and progression of Parkinson’s disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
An accumulating body of literature has emerged over recent years to show that Parkinson’s disease (PD) is not only a disorder of the brain, but also of the gut–brain axis (reviewed in [1]). Gastrointestinal (GI) symptoms occur in almost every PD patient at some point [2] and post-mortem studies have consistently shown that alpha-synuclein aggregates are found in the enteric nervous system (ENS) in almost all cases [3,4,5]. Recent reports have shown that, aside from enteric neuropathology and GI dysfunction, PD patients also exhibit some degree of GI inflammation. The possible link between GI inflammation and PD is further reinforced by observations showing that PD and inflammatory bowel disease (IBD) are genetically and epidemiologically linked. In the first part of this review, we summarize the current knowledge on GI inflammation in PD, while the second part is dedicated to the possible role of GI inflammation in the development of the disease.
PD and IBD are genetically linked
In 2007, Bialecka et al. published the first paper on a possible genetic link between IBD and PD [6]. They focused on the CARD15/NOD2 (caspase recruitment domain-containing protein 15/nucleotide-binding oligomerization domain-containing protein 2), which is associated with Crohn’s disease (CD) and encodes for intracellular signalling molecules that recognize bacterial components and mediate the activation of nuclear factor-kappa B (NF-κB) [7]. They found a higher frequency of 3 CARD15/NOD2 gene variants previously associated with CD in a group of 308 sporadic PD patients when compared to 220 controls [6]. The Leucine-rich repeat kinase 2 (LRRK2) gene, which has emerged as the gene most commonly associated with both familial and sporadic PD, has been subsequently identified by genome-wide association studies as a major susceptibility gene for CD [8]. A recent exome-sequencing study, performed in Ashkenazi Jews by identifying functional gene variants in LRRK2 that conferred either increased risk or protection from both CD and PD, ties even closer these two disorders [9].
PD and IBD are epidemiologically linked
In 1995, based on a short case series, Bihari and Lees suggested that there might be an unrecognized association between PD and ulcerative colitis (UC) [10]. Nothing has been published on this subject for the last 20 years, but the genetic overlap between PD and CD relaunched the debate. While a small monocentric retrospective study did not observe any increased occurrence of CD in PD patients [11], four large nationwide surveys showed that IBD patients had an increased risk of subsequent PD compared to controls [12,13,14,15] (Table 1). After combining the data of these four studies, a recent meta-analysis showed that the overall risk of PD in IBD was significantly higher than in controls (relative risk 1.41, 95% confidence interval 1.19–1.66), with a 28% and 30% increased risk of PD for CD and UC, respectively [16]. Two studies (American and Swedish) reported an increased hazard ratio in both CD and UC groups [13, 15] (Table 1). The Danish study demonstrated that the risk of PD was significantly higher among patients with UC but not with CD, while the Taiwanese one showed the opposite (Table 1). Interestingly, in the American cohort, an almost 80% reduction in the incidence rate of PD was observed among IBD patients who were exposed to anti-TNF therapy compared with those who were not exposed, suggesting that systemic inflammation is involved in the pathogenesis of both diseases [13].
Appendectomy and PD
So far, six studies have investigated the possible association between appendectomy and PD [17,18,19,20,21,22], with the hypothesis that appendectomy would be associated with a reduction in the risk of subsequent PD. This assumption primarily relied on the observation that the vermiform appendix, which is innervated by the vagus nerve [23], contains high amounts of alpha-synuclein [24]. Another plausible hypothesis is that appendectomy might indirectly reduce the incidence of PD by decreasing the risk of IBD [25]. Among these six studies, two observational [18, 20] and three cohorts’ studies found no association between appendectomy and PD risk [17, 19, 21] (Table 2). Much has been written about a recent article, which, by contrast to all other existing studies found that the risk to develop PD was significantly lower (almost 20%) in subjects with appendectomy compared to those without appendectomy: PD incidence was 1.60 per 100,000 person-years (95% confidence interval 1.46–1.75) versus 1.98 (95% confidence interval 1.87–2.10), respectively. They also found that the healthy human appendix contains pathological post-translationally modified forms of alpha-synuclein, suggesting that the appendix may be a preferential site of origin for PD [22]. These are novel and intriguing findings, but further studies are critically needed to determine if the observed post-translational modifications of alpha-synuclein are specific to the appendix or also observed in adjacent GI tract segments such as ileum and colon.
PD patients exhibit GI tract inflammation
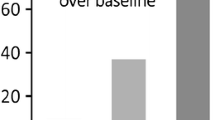
The genetic link between PD and IBD, along with the role of CNS inflammation in the development of PD, prompted us to search for the presence of GI tract inflammation in PD patients. In a seminal study, we analyzed the expression levels of the main pro-inflammatory cytokines (tumor necrosis factor-α, interferon-γ, interleukin-6 and interleukin-1β) in colonic biopsies from 19 PD patients and 14 age-matched healthy controls. We found that the mRNA expression levels of all pro-inflammatory cytokines were significantly elevated in the ascending and descending colon of PD patients when compared to controls [26] (Fig. 1). By contrast, no changes were observed when colonic biopsies from patients with multiple system atrophy and progressive supranuclear palsy were analyzed, suggesting that the observed changes in cytokines expression are specific to PD (Fig. 1). Using a similar approach, we also showed that the expression of the pro-inflammatory enzyme cyclooxygenase-2 (COX-2) was also increased in PD [27]. We were nevertheless struck by the heterogeneity of the amounts of cytokines among PD patients, as some showed levels similar to control subjects while others had up to a sixfold up-regulation. Correlation analyses showed that all pro-inflammatory cytokines were up-regulated within the same patients, suggesting that not all but only a subset of PD patients had an “enteric pro-inflammatory profile” [26]. In addition, the levels of all pro-inflammatory cytokines were negatively correlated with disease duration, but no correlation was observed with GI symptoms, disease severity or cumulative lifetime dose of l-DOPA [26]. Our results were further confirmed by another research group that used a microarray approach to analyze a panel of genes involved in inflammatory pathways in sigmoid biopsies from six PD patients and four controls [28].

Colonic expression of pro-inflammatory cytokines in PD and atypical parkisonism. The mRNA expression levels of TNF-α (a), IFNγ (b), IL-6 (c) and IL-1β (d) are significantly increased in colonic biopsies (descending colon) from PD patients (n = 29) as compared to control subjects (n = 29) (p = 0.0008, p = 0.004, p = 0.003 and p = 0.03, respectively). The mRNA expression levels of TNF-α (a), IFNγ (b), IL-6 (c) and IL-1β (d) are not significantly different between progressive supranuclear palsy (PSP, n = 8) and control subjects (n = 29) (p = 0.74, p = 0.35, p = 0.23, p = 0.28, respectively) and between multiple system atrophy (MSA, n = 6) and controls (n = 29) (p = 0.30, p = 0.12, p = 0.08 and p = 0.14, respectively). Horizontal bars represent the mean. Differences between mRNA expression levels of patients and controls were analyzed by unpaired two-tailed Mann–Whitney test. For all statistical tests, p < 0.05 was deemed significant. *p < 0.05, **p < 0.01 and ***p < 0.001 as compared to controls
The analysis of GI inflammation based on stool specimens has already been successfully used in large cohorts of IBD patients [29]. This logically led Houser et al. to conduct an extensive analysis of immune- and inflammatory-related proteins in the stool of 156 individuals with PD and 110 controls using multiplex immunoassay [30]. They found elevated levels of interleukin-1α, 1β and C-reactive protein in the stool of PD patients. Although not significant, the levels of some up-regulated factors tended to be inversely correlated with age and disease duration [30]. Using a more focused approach, Schwiertz et al. found that calprotectin, a fecal marker of intestinal inflammation, was also significantly increased in the feces of PD patients when compared to controls [31].
Altogether, these studies provide converging evidence that classic inflammatory processes are overly active in the GI tract in PD patients and also suggest and that GI inflammation is more likely to occur in patients with a short disease duration. These observations are in keeping with the findings obtained in the CNS, which showed that microglia activation occurs early in the disease process.
Gut inflammation in rodents is associated with central dopaminergic neurodegeneration and vice versa
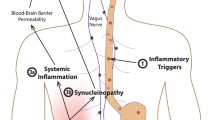
The most widely used mouse model of colitis in rodents employs dextran sodium sulfate (DSS), a chemical colitogen with anticoagulant properties, to induce disease. DSS is a water-soluble, negatively charged sulfated polysaccharide with a highly variable molecular weight ranging from 5 to 1400 kDa. Acute, chronic and relapsing models of intestinal inflammation can be achieved by modifying the concentration of DSS, as well as the frequency of administration and clinical signs of disease appear as soon as one day post-treatment, with an increased expression of the main pro-inflammatory cytokines [32]. Using an acute regimen, Villaran and colleagues were the first to show that rats with DSS-induced colitis were more sensitive to a model of dopaminergic neurodegeneration based on the injection of lipopolysaccharide in the substantia nigra [33]. More recently, another Spanish group observed that chronic treatment with DSS in mice was associated with dopaminergic neuronal death, along with an increased expression of interleukin-1β in the susbtantia nigra [34]. An Italian group from Pisa, by hypothesizing that central dopaminergic degeneration might induce gut inflammation, turned the problem on its head. To this end, they analyzed the modifications of the GI tract in the 6-hydroxydopamine (6-OHDA) model of experimental parkinsonism. This neurotoxin, which does not cross the blood–brain barrier, is classically injected into the substantia nigra and/or in the medial forebrain bundle and produces a massive lesion of nigral dopaminergic cell bodies [35]. The induction of central nigrostriatal dopaminergic degeneration by 6-OHDA was followed after 4 or 8 weeks by bowel inflammation associated with increase in pro-inflammatory cytokine levels (tumor necrosis factor-α and interleukin-1β) and activation of enteric glia [36]. Because a previous study showed that 6-OHDA rats had a decrease in cholinergic neurons of the dorsal motor nucleus of the vagus [37], it is reasonable to hypothesize that the central dopaminergic neurodegeneration triggered by 6-OHDA induces GI tract inflammation through an impairment of the dorsal motor nucleus of the vagus/vagus nerve anti-inflammatory pathway (Fig. 2).

Proposed hypotheses linking GI tract inflammation and PD. a GI inflammation might set off alpha-synuclein expression and aggregation in submucosal neurons and alpha-synuclein pathology would then subsequently spread to the brain via the vagus nerve. b The effects of GI inflammation on the brain could be mediated through systemic inflammation. c Finally, it might be also hypothesized that the dopaminergic denervation in the CNS induces GI tract inflammation through an impairment of the dorsal motor nucleus of the vagus/vagus nerve anti-inflammatory pathway. This figure was created using Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License
How does GI inflammation influence the development of PD?
In light of the increasing evidence showing that PD patients exhibit gut inflammation, a relatively straightforward scenario for PD pathogenesis has been proposed [38, 39]. In such a model, an unknown pathogen triggers intestinal inflammation, which in turn induces the expression and aggregation of alpha-synuclein in submucosal neurons whose terminal axons are only micrometers away from the gut lumen. The pathological process would then further spread to the brain via the vagal preganglionic innervation of the gut, as this has been already demonstrated for neural tracers [40] (Fig. 2). What are the arguments for and against this scenario? Regarding the role of the vagus nerve, two Scandinavian studies showed that full truncal vagotomy was associated with a decreased risk for subsequent PD, suggesting that vagal innervation may be involved in the development of PD [41, 42]. These cohorts’ findings are supported by the observation that several changes observed in the brain of DSS-treated mice, such as decrease in TH expression and nigral neuronal loss, were partly prevented by vagotomy [34]. When it comes to inflammation-induced spreading of alpha-synuclein from the gut to the brain, things are a bit more complicated. In an attempt to reinforce the possible association between PD and IBD, we analyzed the expression levels and the post-translational modifications of alpha-synuclein in IBD. Although we identified an elevated amount of alpha-synuclein in colonic samples from patients with CD, we were unable to find any pathological changes such as phosphorylation and aggregation known to be associated with PD [43]. In addition, two independent studies performed in both acute and chronic DSS-treated mice found that intestinal inflammation was not associated with any increase in alpha-synuclein expression and phosphorylation [34, 44] and we recently showed that alpha-synuclein in enteric neurons was transcriptionally downregulated by acute inflammation via a p38 signaling pathway [44]. Although far from being definitive, these findings cast a doubt on the direct implication of enteric alpha-synuclein in the effects of GI inflammation in PD. An alternative scenario, which does not involve alpha-synuclein, suggests that the increased intestinal permeability and systemic inflammation are sufficient to lead to blood–brain barrier disruption, brain inflammation and ultimately alteration in brain dopaminergic function [45] (Fig. 2b). Apart from these two “bottom-up” hypotheses, a “top-down” hypothesis in which GI inflammation results from a compromised vagal pathway is also plausible [46] (Fig. 2c).
Why do not all IBD patients develop PD?
All the above arguments support a role for GI inflammation in the pathogenesis of PD. However, the associations between IBD and PD are small in magnitude and one may wonder why only a small subset and not the vast majority of IBD patients will subsequently develop PD and vice versa (for example, the difference observed in the Swedish cohort [15] between IBD and controls corresponds to 1 extra case of PD for every 10,000 IBD patients followed for a year). These findings remind us that the pathogenesis of PD is far from being understood and the movement disorders research field is now questioning the accuracy of considering PD as a single entity [47]. In a recent opinion paper, Brundin’s group proposes that factors contributing to neurodegeneration in PD could be classified into three categories: triggers, facilitators, and aggravators [48]. They first acknowledge that with the exception of relatively rare genetic forms of PD and MPTP intoxication, triggers of PD remain largely unknown. Regarding facilitators, which are defined as factors that potentiate the effects of triggers on the nervous system or spread the pathology to more central parts of the nervous system, the list of potential candidates is a bit longer, including among others systemic inflammation, mitochondrial dysfunction and clearance of neurotoxic proteins [49]. One might therefore posit that GI inflammation in PD is one more facilitator that would explain a small part of the etiologic puzzle of PD.
Conclusions and perspectives: what are the next steps?
It is only in the past few years that neurologists and neuroscientists started to get interested in GI inflammation in PD and, although interesting, the existing research on this topic is still at a rather preliminary stage. The first results obtained in PD patients suggest that GI inflammation is inversely correlated to disease duration [26, 30], but these findings need to be confirmed in larger sample size studies that will enroll drug-naïve patients as well as subjects with isolated REM sleep behavior disorder with a longitudinal evaluation of their gut inflammatory status. In addition to the utilization of cutting-edge technology for a large detection of inflammatory markers, it will be also critical to comprehensively assess the motor and nonmotor features in each patient to determine if PD patients with high levels of enteric inflammation have a more severe disease course and progression or not. The role of LRRK2 in linking GI inflammation, immunity and PD is another avenue to be explored. A recent study showed that transgenic mice overexpressing LRRK2 are more sensitive to DSS-induced colitis and that normalization of LRRK2 activation blocks the release of TNF-α by cultured dendritic cells from CD patients [50]. To explore the role of LRRK2 and gut inflammation, it might be relevant to study neurodegeneration and alpha-synuclein pathology in the ENS and the CNS of these mice and to perform additional experiments, including LRRK2 expression and phosphorylation in GI samples of PD patients. Finally, the recent PET studies by Per Borghammer’s group, showing that it is possible to evaluate the parasympathetic denervation of the GI tract in living PD patients [46], also open new opportunities for a better understanding of the role of GI inflammation in the development of the disease. Combining this approach to a comprehensive analysis of inflammation marker in GI samples or feces would allow us to correlate the degree of vagal denervation to the inflammatory status in the GI tract and thus to confirm or not that GI inflammation might be a key starting event in PD.
References
Chapelet G, Leclair-Visonneau L, Clairembault T et al (2018) Can the gut be the missing piece in uncovering PD pathogenesis? Parkinsonism Relat Disord. https://doi.org/10.1016/j.parkreldis.2018.11.014
Edwards LL, Quigley EM, Pfeiffer RF (1992) Gastrointestinal dysfunction in Parkinson’s disease: frequency and pathophysiology. Neurology 42:726–732
Beach TG, Adler CH, Sue LI et al (2010) Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol 119:689–702. https://doi.org/10.1007/s00401-010-0664-3
Gelpi E, Navarro-Otano J, Tolosa E et al (2014) Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov Disord 29:1010–1018. https://doi.org/10.1002/mds.25776
Wakabayashi K, Takahashi H, Takeda S et al (1988) Parkinson’s disease: the presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol 76:217–221
Bialecka M, Kurzawski M, Klodowska-Duda G et al (2007) CARD15 variants in patients with sporadic Parkinson’s disease. Neurosci Res 57:473–476. https://doi.org/10.1016/j.neures.2006.11.012
Maeda S, Hsu L-C, Liu H et al (2005) Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science 307:734–738. https://doi.org/10.1126/science.1103685
Umeno J, Asano K, Matsushita T et al (2011) Meta-analysis of published studies identified eight additional common susceptibility loci for Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis 17:2407–2415. https://doi.org/10.1002/ibd.21651
Hui KY, Fernandez-Hernandez H, Hu J et al (2018) Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aai7795
Bihari K, Lees AJ (1987) Cigarette smoking, Parkinson’s disease and ulcerative colitis. J Neurol Neurosurg Psychiatry 50:635
Fujioka S, Curry SE, Kennelly KD et al (2017) Occurrence of Crohn’s disease with Parkinson’s disease. Parkinsonism Relat Disord 37:116–117. https://doi.org/10.1016/j.parkreldis.2017.01.013
Lin J-C, Lin C-S, Hsu C-W et al (2016) Association between parkinson’s disease and inflammatory bowel disease: a Nationwide Taiwanese Retrospective Cohort Study. Inflamm Bowel Dis 22:1049–1055. https://doi.org/10.1097/MIB.0000000000000735
Peter I, Dubinsky M, Bressman S et al (2018) Anti-tumor necrosis factor therapy and incidence of parkinson disease among patients with inflammatory bowel disease. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2018.0605
Villumsen M, Aznar S, Pakkenberg B et al (2018) Inflammatory bowel disease increases the risk of Parkinson’s disease: a Danish nationwide cohort study 1977–2014. Gut. https://doi.org/10.1136/gutjnl-2017-315666
Weimers P, Halfvarson J, Sachs MC et al (2019) Inflammatory bowel disease and Parkinson’s disease: a Nationwide Swedish Cohort Study. Inflamm Bowel Dis 25:111–123. https://doi.org/10.1093/ibd/izy190
Zhu F, Li C, Gong J et al (2019) The risk of Parkinson’s disease in inflammatory bowel disease: a systematic review and meta-analysis. Dig Liver Dis 51:38–42. https://doi.org/10.1016/j.dld.2018.09.017
Marras C, Lang AE, Austin PC et al (2016) Appendectomy in mid and later life and risk of Parkinson’s disease: a population-based study. Mov Disord 31:1243–1247. https://doi.org/10.1002/mds.26670
Mendes A, Gonçalves A, Vila-Chã N et al (2015) Appendectomy may delay Parkinson’s disease onset. Mov Disord 30:1404–1407. https://doi.org/10.1002/mds.26311
Palacios N, Hughes KC, Cereda E et al (2018) Appendectomy and risk of Parkinson’s disease in two large prospective cohorts of men and women. Mov Disord 33:1492–1496. https://doi.org/10.1002/mds.109
Yilmaz R, Bayram E, Ulukan Ç et al (2017) Appendectomy history is not related to Parkinson’s disease. J Parkinsons Dis 7:347–352. https://doi.org/10.3233/JPD-171071
Svensson E, Horváth-Puhó E, Stokholm MG et al (2016) Appendectomy and risk of Parkinson’s disease: a nationwide cohort study with more than 10 years of follow-up. Mov Disord 31:1918–1922. https://doi.org/10.1002/mds.26761
Killinger BA, Madaj Z, Sikora JW et al (2018) The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aar5280
Altschuler SM, Escardo J, Lynn RB, Miselis RR (1993) The central organization of the vagus nerve innervating the colon of the rat. Gastroenterology 104:502–509
Gray MT, Munoz DG, Gray DA et al (2014) Alpha-synuclein in the appendiceal mucosa of neurologically intact subjects. Mov Disord 29:991–998. https://doi.org/10.1002/mds.25779
Russel MG, Dorant E, Brummer RJ et al (1997) Appendectomy and the risk of developing ulcerative colitis or Crohn’s disease: results of a large case-control study. South Limburg Inflammatory Bowel Disease Study Group. Gastroenterology 113:377–382
Devos D, Lebouvier T, Lardeux B et al (2013) Colonic inflammation in Parkinson’s disease. Neurobiol Dis 50:42–48. https://doi.org/10.1016/j.nbd.2012.09.007
Pochard C, Leclair-Visonneau L, Coron E et al (2018) Cyclooxygenase 2 is upregulated in the gastrointestinal tract in Parkinson’s disease. Mov Disord 33:493–494. https://doi.org/10.1002/mds.27237
Perez-Pardo P, Dodiya HB, Engen PA et al (2018) Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut. https://doi.org/10.1136/gutjnl-2018-316844
Eeckhaut V, Machiels K, Perrier C et al (2013) Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut 62:1745–1752. https://doi.org/10.1136/gutjnl-2012-303611
Houser MC, Chang J, Factor SA et al (2018) Stool immune profiles evince gastrointestinal inflammation in Parkinson’s disease. Mov Disord 33:793–804. https://doi.org/10.1002/mds.27326
Schwiertz A, Spiegel J, Dillmann U et al (2018) Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson’s disease. Parkinsonism Relat Disord. https://doi.org/10.1016/j.parkreldis.2018.02.022
Eichele DD, Kharbanda KK (2017) Dextran sodium sulfate colitis murine model: an indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J Gastroenterol 23:6016–6029. https://doi.org/10.3748/wjg.v23.i33.6016
Villarán RF, Espinosa-Oliva AM, Sarmiento M et al (2010) Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: potential risk factor in Parkinson’s disease. J Neurochem 114:1687–1700. https://doi.org/10.1111/j.1471-4159.2010.06879.x
Garrido-Gil P, Rodriguez-Perez AI, Dominguez-Meijide A et al (2018) Bidirectional neural interaction between central dopaminergic and gut lesions in Parkinson’s disease models. Mol Neurobiol. https://doi.org/10.1007/s12035-018-0937-8
Blandini F, Armentero M-T, Martignoni E (2008) The 6-hydroxydopamine model: news from the past. Parkinsonism Relat Disord 14(Suppl 2):S124–S129. https://doi.org/10.1016/j.parkreldis.2008.04.015
Pellegrini C, Fornai M, Colucci R et al (2016) Alteration of colonic excitatory tachykininergic motility and enteric inflammation following dopaminergic nigrostriatal neurodegeneration. J Neuroinflamm 13:146. https://doi.org/10.1186/s12974-016-0608-5
Zheng L-F, Wang Z-Y, Li X et al (2011) Reduced expression of choline acetyltransferase in vagal motoneurons and gastric motor dysfunction in a 6-OHDA rat model of Parkinson’s disease. Brain Res 1420:59–67. https://doi.org/10.1016/j.brainres.2011.09.006
Lema Tomé CM, Tyson T, Rey NL et al (2013) Inflammation and α-synuclein’s prion-like behavior in Parkinson’s disease—is there a link? Mol Neurobiol 47:561–574. https://doi.org/10.1007/s12035-012-8267-8
Houser MC, Tansey MG (2017) The gut-brain axis: is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? NPJ Parkinsons Dis 3:3. https://doi.org/10.1038/s41531-016-0002-0
Walter GC, Phillips RJ, Baronowsky EA, Powley TL (2009) Versatile, high-resolution anterograde labeling of vagal efferent projections with dextran amines. J Neurosci Methods 178:1–9. https://doi.org/10.1016/j.jneumeth.2008.11.003
Liu B, Fang F, Pedersen NL et al (2017) Vagotomy and Parkinson disease: a Swedish register-based matched-cohort study. Neurology 88:1996–2002. https://doi.org/10.1212/WNL.0000000000003961
Svensson E, Horváth-Puhó E, Thomsen RW et al (2015) Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol 78:522–529. https://doi.org/10.1002/ana.24448
Prigent A, Lionnet A, Durieu E et al (2019) Enteric alpha-synuclein expression is increased in Crohn’s disease. Acta Neuropathol 137:359–361. https://doi.org/10.1007/s00401-018-1943-7
Prigent A, Gonzales J, Durand T et al (2018) Acute inflammation down-regulates alpha-synuclein expression in enteric neurons. J Neurochem. https://doi.org/10.1111/jnc.14656
Guan Q, Zhang J (2017) Recent advances: the imbalance of cytokines in the pathogenesis of inflammatory bowel disease. Mediat Inflamm 2017:4810258. https://doi.org/10.1155/2017/4810258
Fedorova TD, Seidelin LB, Knudsen K et al (2017) Decreased intestinal acetylcholinesterase in early Parkinson disease: an 11C-donepezil PET study. Neurology 88:775–781. https://doi.org/10.1212/WNL.0000000000003633
Greenland JC, Williams-Gray CH, Barker RA (2019) The clinical heterogeneity of Parkinson’s disease and its therapeutic implications. Eur J Neurosci 49:328–338. https://doi.org/10.1111/ejn.14094
Johnson ME, Stecher B, Labrie V et al (2019) Triggers, facilitators, and aggravators: redefining Parkinson’s disease pathogenesis. Trends Neurosci 42:4–13. https://doi.org/10.1016/j.tins.2018.09.007
Zou W, Pu T, Feng W et al (2019) Blocking meningeal lymphatic drainage aggravates Parkinson’s disease-like pathology in mice overexpressing mutated α-synuclein. Transl Neurodegener 8:7. https://doi.org/10.1186/s40035-019-0147-y
Takagawa T, Kitani A, Fuss I et al (2018) An increase in LRRK2 suppresses autophagy and enhances Dectin-1-induced immunity in a mouse model of colitis. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aan8162
Acknowledgements
Work in our laboratory was supported by Institut de France (Fondation NRJ), France Parkinson, CECAP (Comité d’Entente et de Coordination des Associations de Parkinsoniens), ADPLA (Association des Parkinsoniens de Loire Atlantique), FFPG (Fédération française des groupements parkinsoniens) and Parkinsoniens de Vendée.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors report no disclosure relevant to the research covered in this article.
Rights and permissions
About this article

Cite this article
Rolli-Derkinderen, M., Leclair-Visonneau, L., Bourreille, A. et al. Is Parkinson’s disease a chronic low-grade inflammatory bowel disease?. J Neurol 267, 2207–2213 (2020). https://doi.org/10.1007/s00415-019-09321-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-019-09321-0