
Abstract
Zinc has reported to play a neuroprotective role in the development of spinal cord injury (SCI). The protective mechanism of zinc remains to be uncovered. The aim of the current study was to investigate the neuroprotective mechanism of zinc in the progression of SCI. The C57BL/6J mouse SCI model was established to confirm the protective role of zinc in vivo, while the cellular model was induced in mouse microglial BV2 cells by using lipopolysaccharide (LPS). The expression levels of XIST, miR-374a-5p and NLRP3 inflammasome as well as the autophagy-related proteins were detected using real-time PCR and immunoblotting. Cell viability was assessed by CCK-8 assay. Apoptosis was evaluated by TUNEL staining, flow cytometry, the determination of apoptosis-related proteins. The target relationship was confirmed by luciferase reporter assays. Zinc improved locomotor function in SCI mice and alleviated LPS-induced BV2 cell injuries by inhibiting apoptosis and initiating autophagy processes. XIST and NLRP3 inflammasome was upregulated while miR-374a-5p was downregulated in spinal cords of SCI mice and LPS-treated BV2 cells. All these effects were inhibited by Zinc treatment. XIST knockdown triggered microglial autophagy-mediated NLRP3 inactivation in LPS-induced BV2 cells by regulating miR-374a-5p. Zinc treatment protected BV2 cells from LPS-induced cell injury by the downregulation of XIST. This process might be through autophagy‑mediated NLRP3 inflammasome inactivation by targeting miR-374a-5p. Zinc downregulates XIST and induces neuroprotective effects against SCI by promoting microglial autophagy-induced NLRP3 inflammasome inactivation through regulating miR-374a-5p. Our finding provides novel opportunities for the understanding of zinc-related therapy of SCI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Spinal cord injury (SCI) is a devastating neurological disorder involving the temporary or permanent loss of the motor, sensory and autonomic function [1]. Due to the lack of therapeutic drugs and limited understanding of its pathogenesis, there is no direct and effective treatment for SCI [2, 3]. Therapeutic activation of autophagy plays a protective role in the management of SCI [4].
As an essential trace element, zinc participates in the regulation of various enzymes and proteins, playing a momentous role in diverse physiological and pathological events, such as bone remodeling [5], immunoregulation [6], and endocrine modulation [7]. Recent studies have shown that zinc supplementation can reduce neuronal impairment and promote functional recovery after SCI [8, 9]. More importantly, we have previously demonstrated that zinc exerts a neuroprotective effect against SCI by augmenting microglial autophagy through inhibiting NLRP3 inflammasome activation [10]. However, the neuroprotective mechanism by which zinc mediated autophagy-induced NLRP3 inflammasome inactivation in the progression of SCI needs to be further explored.
XIST is a widely reported long non-coding RNA (lncRNA) in SCI, which has been shown to promote microglial apoptosis, inflammatory response and neuronal apoptosis [11, 12]. According to the bioinformatics analysis, miR-374a-5p was predicted as a downstream target of XIST. In a previous study, Chen et al. reported that miR-374a-5p provided neuroprotective and anti-inflammatory effects in neonatal hypoxic-ischemic encephalopathy (HIE) via the inhibition of NLRP3 inflammasome [13]. To sum up, it is reasonable to speculate that zinc may lead to the silencing of XIST with subsequent upregulation of miR-374a-5p, which in turn inhibit the activation of NLRP3 inflammatory signals, thereby leading to microglial autophagy and SCI amelioration.
Materials and Methods
Animals and Experimental Design
A total of 18 adult 8-week-old C57BL/6J mice (9 males and 9 females) were randomly divided into Sham group, SCI model group, and Zinc-treated group, with 6 animals in each group. Animal experiments were approved by the Animal Care and Use Committees of Jinzhou Medical College (approval number: SYXK2019-0007). The SCI mouse model was established by extradural compression. Briefly, the mice were anesthetized with 10% chloral hydrate (0.3 mL/100 g) and 0.03 mg/kg fentanyl (Sigma-Aldrich, St. Louis, MO, USA) [10, 14], and a laminectomy was performed at the T9-T10 level, followed by routine suture. Sham-operated mice were subjected to the same surgical procedure except spinal contusion. At postoperative 2 h, Zinc gluconate (Biotopped, Beijing, China) with a dose of 30 mg/kg was injected intraperitoneally [8, 10, 15], once daily for 3 days. Simultaneously, mice in the SCI group received an equal volume of isotonic glucose. The motor function was assessed by the Basso, Beattie and Bresnahan (BBB) scores that were graded on a scale of 0–21 [16]. Spinal cord tissues were collected for the following experiments.
Hematoxylin and Eosin (HE) Staining
Spinal cord tissues were fixed in the same perfusate at 4 °C for 72 h, and cut into 4 µm thickness per section. PBS was washed and fixed in 4% paraformaldehyde, then dehydrated, embedded and sliced. The sections were HE-stained using standard methods (0.5% hematoxylin for 5 min at 25 °C; 0.5% eosin for 1 min at 25 °C), imaged with a microscope (Olympus, Tokyo; Japan) at ×40 magnification.
TUNEL Staining
The technique of TUNEL staining is useful to evaluate the cell apoptosis in the spinal cords of mice and BV2 cells. Formalin-fixed, paraffin-embedded spinal cords were cut into 5-µm thick sections, de-paraffinized with xylene and dehydrated with gradient ethanol. After washed with PBS, sections were digested by DNase-free Proteinase K for antigen retrieval, incubated with 3% H2O2 in PBS for 20 min at room temperature, and prepared for TUNEL reaction using a commercial kit (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s guidelines. Samples were then counterstained with 4′-6-diamidino-2-phenylindole (DAPI), and the TUNEL-positive cells were counted under a microscope (Olympus, Tokyo; Japan) at ×400 magnification. For cell immunofluorescence staining, BV2 microglial cells were collected and washed with cold PBS. After fixation with 4% paraformaldehyde for 30 min and two washes with PBS for 2 min, the cells were incubated for 1 h with a TUNEL mixture. The TUNEL-stained cells were visualized with a fluorescence microscope.
Cell Culture, Transfection and Treatment
DMEM (Life Technologies, Carlsbad, CA, USA) containing 10% fetal bovine serum was used to incubate mouse microglial BV2 cells purchased from American Type Culture Collection (Manassas, VA, USA). Prior to transfection, BV2 cells were cultured in 6-well plates until they were 90% confluent. The pcDNA3.1-XIST, si-XIST, miR-374a-5p mimic, inhibitor, and corresponding negative controls, synthesized by GenePharma Co., Ltd. (Shanghai, China) were transfected into BV2 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). At 48 h after transfection, transfection efficiency was assessed by fluorescent microscopy and flow cytometry as the frequency of cells positive for green fluorescent. At 48 h posttransfection, BV2 cells were stimulated with LPS (1 µg/mL) combined with ATP (5 mmol/L) for 6 h or treated with zinc gluconate (100 µmol/L) for 6 h.
Luciferase Reporter Assay
The method of luciferase reporter assay is useful to determine the interaction between XIST and miR-374a-5p. Using Lipofectamine 2000, BV2 cells were co-transfected with the wild-type/mutated XIST reporter plasmids and miR-374a-5p mimic or miR-NC. Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA) was used for the luciferase assays.
Quantitative RT-PCR Analysis
Total RNA was extracted from spinal cord tissues and cultured cells using Trizol reagent (Invitrogen) and reversely transcribed to cDNA using Primer-Script TM one step RT-PCR kit (Takara, Shiga, Japan). To quantify the expression of XIST, miR-374a-5p and NLRP3, real time PCR was performed using SYBR Green I real time PCR kit (CoWin Bioscience Co., Beijing, China).
Western Blot
Total proteins were extracted from tissues and cultured cells using Radio-Immunoprecipitation Assay (RIPA) buffer (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Protein concentration was measured by a BCA Protein Assay Kit (Beyotime Biotechnology). Proteins (30 μg) were separated by electrophoresis, and electroblotted onto polyvinylidenedifluoride (PVDF) membranes that were incubated with primary antibodies (1:1000 dilution; all from Cell Signaling Technology, Shanghai, China) targeted to Bax (#14796), Bcl-2 (#4223), Beclin-1 (#3495), P62 (#8025), LC3-I (#4599), LC3-II (#3868), NLRP3 (#15101), IL-1β (#12703), ASC (#13833), caspase-1 (#24232), and loading control GAPDH (#5174) overnight at 4 °C, after blocking the nonspecific binding sites with 5% skim milk. Blots were then incubated with HRP-conjugated secondary antibodies (1:2000 dilution; Cell Signaling Technology).
Cell Viability Assay
CCK-8 assay was used to detect cell viability. After transfection and treatment, BV2 cells (100 µL) were seeded on 96-well plates at a density of 2000 cells per well, and then 10 µL CCK-8 reagent (Beyotime Biotechnology) was added to each well. After incubation for 1 h, the absorbance which revealed cell viability was detected at the wavelength of 450 nm.
Flow Cytometry Analysis
BV2 cell apoptosis was detected by Annexin V-FITC/PI double staining with flow cytometry. After transfection and treatment, BV2 cells were washed with PBS and fixed with ethyl alcohol at 4 °C for 2 h and stained with FITC Apoptosis Detection kit (BD Biosciences, San Diego, CA, USA) at 4 °C in the dark for 30 min. The apoptotic rate was analyzed by flow cytometer (Beckman Coulter, Fullerton, CA, USA).
Immunofluorescence Staining
BV2 cells were fixed with 4% paraformaldehyde for 30 min, followed by permeabilization with 0.4% Triton X-100 for 5 min at room temperature. Cells were blocked with 5% bovine serum albumin at 37 °C for 25 min and incubated with 50 μg/mL LC3 primary antibody at 4 °C for 24 h. After washed with PBS for three times, the nuclei were stained with DAPI for 3 min at room temperature. Negative control was in the absence of primary antibody. Immunofluorescence was observed and evaluated using a confocal microscope (Olympus).
Statistical Analysis
All data were presented as mean ± standard deviation (SD). Statistical comparison was performed by two-sided Student’s t test or ANOVA using SPSS 19.0 software (IBM, Chicago, IL, USA). p < 0.05 was considered significant.
Results
Zinc Induces Autophagy-Mediated NLRP3 Inactivation Following SCI by Decreasing XIST Expression
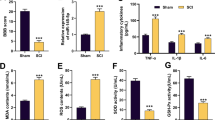
The neurological function was assessed by the BBB scoring system to determine whether zinc could improve locomotor recovery following SCI. As shown in Fig. 1A, the BBB scores were significantly reduced in the SCI group compared with the sham group, which were markedly increased in zinc-treated mice. Additionally, histopathological changes in spinal cord tissues were examined by H&E staining. The congestion, edema and structural damages in the spinal cord of SCI rats were showed to be notably relieved after Zinc treatment (Fig. 1B). To evaluate the effects of zinc on cellular apoptosis caused by SCI, TUNEL staining of the spinal cords and western blot analysis of apoptosis-related proteins were performed. The number of TUNEL-positive cells was observably higher in the SCI group than that in the sham group, while the amount of apoptotic cells was significantly reduced after zinc administration (Fig. 1C). As illustrated in Fig. 1D, zinc reversed Bax upregulation and Bcl-2 downregulation induced by SCI. The expression levels of autophagy-related proteins were analyzed by immunoblotting. The expression of Beclin-1 and the ratio of LC3-II to LC3-I were markedly inhibited, while P62 was significantly increased in SCI mice, compared with those in sham-operated animals. In contrast, zinc administration exhibited an opposite trend (Fig. 1E). Then, the molecular mechanism of zinc during SCI was further identified. As shown in Fig. 1F, zinc treatment mitigated the upregulation of XIST and NLRP3 and the downregulation of miR-374a-5p after SCI. Similarly, the protein expression of NLRP3, IL-1β, ASC and caspase-1 were markedly increased in the spinal cords of SCI mice. Notably, zinc treatment inhibited NLRP3 inflammasome activation as shown by the decreases in the levels of NLRP3, IL-1β, ASC and caspase-1 in SCI mice (Fig. 1G).

Zinc induces autophagy-mediated NLRP3 inactivation following SCI by decreasing XIST expression. C57BL/6J mice were divided into sham group, SCI group and SCI + Zinc group (n = 6 per group). A BBB scores were evaluated. B Representative images of H&E staining at ×40 magnification in spinal cord tissues. C TUNEL staining was performed in spinal cords. D, E The protein levels of Bax, Bcl-2, Beclin-1, P62 and LC3-II/I ratio in spinal cords were determined by western blotting. F XIST, miR-374a-5p and NLRP3 levels in spinal cords were detected by qRT-PCR. G The protein levels of NLRP3, IL-1β, ASC and caspase-1 in spinal cords were determined by western blotting. Data were expressed as mean ± SD and each experiment was performed at least 3 independent times. **p < 0.01
Zinc Downregulates XIST to Alleviate LPS-Induced Autophagy Inhibition in Microglia
Similar effects of zinc were observed in mouse microglial BV2 cells treated with LPS as in SCI mice. Cell viability was restricted, while zinc resulted in increased cell viability (Fig. 2A). The results of TUNEL assay (Fig. 2B) showed that the proportion of TUNEL-positive cells significantly increased after LPS stimulation, whereas the percentage of cell death was reduced upon treatment with zinc. The typical graphs and statistical data of flow cytometry (Fig. 2C) were consistent with the results of TUNEL assay. Immunofluorescence analysis revealed that during treatment with LPS, the proportion of LC3-positive BV2 cells decreased significantly, while zinc treatment induced the increase of LC3-positive BV2 cells (Fig. 2D). Similar to the changes of apoptosis- and autophagy-related proteins observed in vivo, the expression of Bax and P62 were elevated, while the protein levels of Bcl-2, Beclin-1 and LC3-II/I ratio were downregulated in LPS-treated BV2 cells, and zinc inhibited these detrimental alterations (Fig. 2E, F). As indicated in Fig. 2G, H, LPS induced the upregulation of XIST and NLRP3 at mRNA and protein levels, as well as the protein levels of IL-1β, ASC and caspase-1 and suppressed the mRNA levels of miR-374a-5p, which were all reversed by zinc treatment.

Zinc downregulates XIST to alleviate LPS-induced autophagy inhibition in microglia. Mouse microglial BV2 cells were exposed to LPS (1 µg/mL) for 6 h or treated with zinc gluconate (100 µmol/L) for 6 h. After LPS stimulation or zinc treatment, cell viability (A), quantitative analysis of the proportion of TUNEL positive cells (B), the proportion of total apoptotic cells by flow cytometry assay (C), immunofluorescence analysis of LC3 expression (D), the protein expression of apoptosis- and autophagy-related markers (E–F), the expression of XIST, miR-374a-5p and NLRP3 (G) and the protein expression of NLRP3, IL-1β, ASC and caspase-1 (H) were examined using CCK-8 assay, flow cytometry, western blotting, qRT-PCR and western blotting, respectively. Data were expressed as mean ± SD and each experiment was performed at least 3 independent times. **p < 0.01
XIST Knockdown Attenuates LPS-Induced Microglial Activation by Inducing Autophagy
In order to clarify the functional role of XIST in LPS-induced BV2 cell injury, XIST knockdown was confirmed in BV2 cells by transfection with si-XIST. Fluorescent microscopy and flow cytometry were used to evaluate the efficiency of transfection. The results demonstrated that approximately 70.1% BV2 cells could express GFP after transfection with GFP vector (Fig. S1). The relative expression level of XIST was assessed by qRT-PCR, and a significant downregulation of XIST was seen in si-XIST group (Fig. 3A). And the downregulation of XIST overturned LPS-induced XIST and NLRP3 upregulation and miR-374a-5p downregulation in BV2 cells (Fig. 3B). Meanwhile, XIST knockdown led to a decrease in the protein levels of NLRP3, IL-1β, ASC and caspase-1 in LPS-treated BV2 cells (Fig. 3C). The inhibition of cell viability (Fig. 3D) and the enhancement of apoptotic cells (Fig. 3E, F) in LPS-treated BV2 cells were both reversed by XIST silencing. XIST knockdown exhibited enhanced LC3 puncta accumulation compared to cells treated with LPS (Fig. 3G). As expected, LPS-induced Bax upregulation and Bcl-2 downregulation was diminished by XIST inhibition (Fig. 3H). Beyond that, XIST depletion promoted Beclin-1 expression and the conversion of LC3-I into LC3-II, but declined the protein levels of P62 in LPS-treated BV2 cells (Fig. 3I).

XIST knockdown attenuates LPS-induced microglial activation by inducing autophagy. BV2 cells were transfected with si-XIST or si-NC, which were then exposed to 1 µg/mL of LPS for 6 h. A QRT-PCR analysis was performed to confirm the transfection efficiency. After LPS stimulation or XIST knockdown, the expression of XIST, miR-374a-5p and NLRP3 (B) and the protein expression of NLRP3, IL-1β, ASC and caspase-1 (C), cell viability (D), quantitative analysis of the proportion of apoptotic cells by TUNEL assay (E), and flow cytometry assay (F), immunofluorescence analysis of LC3 expression (G), the protein expression of apoptosis-associated proteins (H), and autophagy-related markers (I) were analyzed by using the corresponding method. Data were expressed as mean ± SD and each experiment was performed at least 3 independent times. **p < 0.01
XIST Sponges miR-374a-5p to Affect Microglial Autophagy via NLRP3 Inflammasome Activation
Our study has found that XIST harbored putative binding sites for miR-374a-5p predicted by the online DIANA tools (Fig. 4A). As depicted in Fig. 4B, the results of luciferase assay revealed that miR-374a-5p mimic decreased the luciferase activity in XIST-WT co-transfected system, conversely, this XIST-MUT scarcely responded to miR-374a-5p overexpression. Next, we explored whether XIST was involved in LPS-induced BV2 cell injury through regulating miR-374a-5p. Confirmed by qRT-PCR, transfection with miR-374a-5p inhibitor resulted in miR-374a-5p knockdown in BV2 cells (Fig. 4C). Likewise, miR-374a-5p silencing antagonized XIST knockdown-induced miR-374a-5p upregulation, and the alleviation of NLRP3, IL-1β, ASC and caspase-1 expression levels in LPS-treated BV2 cells, as shown in Fig. 4D, E. Moreover, miR-374a-5p inhibition prevented XIST knockdown-induced promotion of cell viability (Fig. 4F) and the decrease of apoptotic cells (Fig. 4G, H) in LPS-treated BV2 cells. MiR-374a-5p inhibitor also decreased LC3 puncta accumulation (Fig. 4I). In addition, miR-374a-5p inhibitor impaired XIST knockdown-induced Bax downregulation and Bcl-2 upregulation induced by in LPS-treated BV2 cells (Fig. 4J). Besides, miR-374a-5p inhibitor abrogated the suppression of P62 expression, and the increased protein levels of Beclin1 and LC3-II/I ratio caused by XIST knockdown in LPS-treated BV2 cells (Fig. 4K).

XIST sponges miR-374a-5p to affect microglial autophagy via NLRP3 inflammasome activation. A The putative binding sites between XIST and miR-374a-5p were shown according to online DIANA tools. B Dual-luciferase reporter assay examined the relative luciferase activity of reporter vectors containing WT/MUT-XIST in BV2 cells when co-transfection with miR-374a-5p mimics or miR-NC. C Expression level of miR-374a-5p was detected by RT-qPCR in BV2 cells when transfection with miR-374a-5p inhibitor or inhibitor NC. After LPS stimulation or the knockdown of XIST or miR-374a-5p, the expression of miR-374a-5p and NLRP3 (D) and the protein expression of NLRP3, IL-1β, ASC and caspase-1 (E), cell viability (F), quantitative analysis of the proportion of apoptotic cells by TUNEL assay (G), and flow cytometry assay (H), immunofluorescence analysis of LC3 expression (I), the protein expression of apoptosis-associated proteins (J) and autophagy-related markers (K) were analyzed by using the corresponding method. Data were expressed as mean ± SD and each experiment was performed at least 3 independent times. **p < 0.01
Zinc Enhances Microglial Autophagy in LPS-Induced BV2 Cells by Regulating XIST-Mediated miR-374a-5p/NLRP3 Axis
XIST was overexpressed in BV2 cells by transfecting XIST vector. The results of fluorescent microscopy and flow cytometry demonstrated that more than 70% BV2 cells could express GFP (Fig. S2). The transfection efficiency was further confirmed by qRT-PCR (Fig. 5A). In LPS-irritated BV2 cells, the ectopic expression of XIST changed a series of expression patterns induced by zinc treatment by upregulating XIST and NLRP3 inflammasome activation and downregulating miR-374a-5p expression (Fig. 5B, C). Prominently, restoration of XIST expression impaired the protective effects of zinc in LPS-damaged BV2 cells by decreasing cell viability (Fig. 5D), facilitating apoptosis as evidenced by the increased apoptotic cells (Fig. 5E, F) and increased Bax expression and Bcl-2 inhibition (Fig. 5G), and inhibiting autophagy via decreasing the number of LC3 puncta (Fig. 5H), and the expression of the ratio of LC3II/I and Beclin-1 and decreasing the expression of P62 (Fig. 5I).

Zinc enhances microglial autophagy in LPS-induced BV2 cells by regulating XIST-mediated miR-374a-5p/NLRP3 axis. A Expression level of XIST was detected by RT-qPCR in BV2 cells when overexpression of XIST. After the overexpression of XIST or the treatment with LPS or zinc, the expression of XIST, miR-374a-5p and NLRP3 (B) and the protein expression of NLRP3, IL-1β, ASC and caspase-1 (C), cell viability (D), quantitative analysis of the proportion of apoptotic cells by TUNEL assay (E), and flow cytometry assay (F), the protein expression of apoptosis-associated proteins (G), fluorescent images of LC3 puncta (H) and autophagy-related markers (I) were analyzed by using the corresponding method. Data were expressed as mean ± SD and each experiment was performed at least 3 independent times. **p < 0.01
Discussion
The present study generated a novel finding that the XIST/miR-374a-5p axis accounts for the neuroprotective action of zinc via inhibiting NLRP3 inflammasome activation and promoting microglial autophagy.
Zinc has been regarded as a potential therapeutic candidate for SCI due to its diverse beneficial actions. For example, zinc treatment alleviates neuronal apoptosis after SCI by increasing the expression of G-CSF secreted by microglia/macrophages [15]. Zinc elicits a neuroprotective action against apoptosis by inducing neuronal autophagy through AMPK/mTOR signaling pathway in mice with SCI [17]. Furthermore, zinc exerts antioxidative effects in SCI by activating Nrf2/HO-1 pathway and inhibiting NLRP3 inflammation [8]. However, the associated mechanisms are still largely unexplored. Indeed, excessive activation of NLRP3 inflammasome was considered as a pivotal contributor to the development of SCI [18, 19]. Herein, in consistent with our preceding study, we found that zinc induced microglial autophagy and attenuated NLRP3 inflammasome activation in mice with SCI and BV2 microglial cells exposed to LPS [10]. In agreement, several reports have demonstrated the inhibitory effects of zinc on NLRP3 inflammasome activation [20, 21].
LncRNA XIST has previously been implicated in controlling the neuronal and microglial apoptosis, inflammatory reaction, and angiogenesis after SCI [11, 12, 22]. Herein, the downregulation of XIST evoked by zinc treatment was observed in spinal cords of mice with SCI and LPS-impaired BV2 cells. Further research divulged the influence of XIST silencing in protecting BV2 cells against LPS-triggered injury by increasing autophagy and inhibiting NLRP3 inflammasome activation. Of note, XIST overexpression distinctly overturned the protective activities of zinc in LPS-damaged BV2 cells. These data indicated that XIST might participate in the process of zinc protecting BV2 cells from LPS-triggered cell damage.
A large quantity of evidence has uncovered the potential roles of several miRNAs in mediating neuroprotective effects in SCI [23,24,25]. A previous publication demonstrated that miR-374a-5p is essential for the inhibition of OGD-induced neuronal apoptosis via PTEN/PI3K pathway in HIE [26]. Additionally, an interesting evidence uncovered that miR-374a-5p negatively regulated NLRP3 inflammasome by targeting Smad6 in HIE [13]. Our results displayed that miR-374a-5p was decreased in SCI mice and LPS-damaged BV2 cells, which was abolished by zinc. Moreover, we found that XIST reversely regulated the expression of miR-374a-5p. Overexpression of XIST further enhanced NLRP3 inflammasome activity in LPS-treated BV2 cells by regulating the expression of miR-374a-5p. Taken together, we deduced that the neuroprotective effect of zinc might be partially ascribed to the downregulation of NLRP3 inflammasome and the promotion of autophagy activation via regulating XIST/miR-374a-5p.
In summary, the results showed that zinc produced the neuroprotection following SCI owing to autophagy-inducing properties conferred by its ability to inhibit XIST/miR-374a-5p-mediated NLRP3 inflammasome activation.
Data Availability
All data generated or analyzed during this study are included in this published article.
Abbreviations
- SCI:
-
Spinal cord injury
- LPS:
-
Lipopolysaccharide
- lncRNA:
-
Long non-coding RNA
- HIE:
-
Hypoxic-ischemic encephalopathy
- BBB:
-
Beattie and Bresnahan
- DAB:
-
Diaminobenzidine
- SD:
-
Standard deviation
References
Schwab JM, Maas AIR, Hsieh JTC, Curt A (2018) Raising awareness for spinal cord injury research. Lancet Neurol 17:581–582
Jendelova P (2018) Therapeutic strategies for spinal cord injury. Int J Mol Sci 19:3200
Venkatesh K, Ghosh SK, Mullick M, Manivasagam G, Sen D (2019) Spinal cord injury: pathophysiology, treatment strategies, associated challenges, and future implications. Cell Tissue Res 377:125–151
Zhu N, Ruan J, Yang X, Huang Y, Jiang Y, Wang Y, Cai D, Geng Y, Fang M (2020) Triptolide improves spinal cord injury by promoting autophagy and inhibiting apoptosis. Cell Biol Int 44:785–794
Huang T, Yan G, Guan M (2020) Zinc homeostasis in bone: zinc transporters and bone diseases. Int J Mol Sci 21:1236
Wessels I, Maywald M, Rink L (2017) Zinc as a gatekeeper of immune function. Nutrients 9(12):1286
Cooper-Capetini V, de Vasconcelos DAA, Martins AR, Hirabara SM, Donato J Jr, Carpinelli AR, Abdulkader F (2017) Zinc supplementation improves glucose homeostasis in high fat-fed mice by enhancing pancreatic beta-cell function. Nutrients 9(10):1150
Li D, Tian H, Li X, Mao L, Zhao X, Lin J, Lin S, Xu C, Liu Y, Guo Y, Mei X (2020) Zinc promotes functional recovery after spinal cord injury by activating Nrf2/HO-1 defense pathway and inhibiting inflammation of NLRP3 in nerve cells. Life Sci 245:117351
Wang Y, Su R, Lv G, Cao Y, Fan Z, Wang Y, Zhang L, Yu D, Mei X (2014) Supplement zinc as an effective treatment for spinal cord ischemia/reperfusion injury in rats. Brain Res 1545:45–53
Lin JQ, Tian H, Zhao XG, Lin S, Li DY, Liu YY, Xu C, Mei XF (2020) Zinc provides neuroprotection by regulating NLRP3 inflammasome through autophagy and ubiquitination in a spinal contusion injury model. CNS Neurosci Ther 27(4):413–425
Gu S, Xie R, Liu X, Shou J, Gu W, Che X (2017) Long coding RNA XIST contributes to neuronal apoptosis through the downregulation of AKT phosphorylation and is negatively regulated by miR-494 in rat spinal cord injury. Int J Mol Sci 18(4):732
Zhao Q, Lu F, Su Q, Liu Z, Xia X, Yan Z, Zhou F, Qin R (2020) Knockdown of long noncoding RNA XIST mitigates the apoptosis and inflammatory injury of microglia cells after spinal cord injury through miR-27a/Smurf1 axis. Neurosci Lett 715:134649
Chen Z, Hu Y, Lu R, Ge M, Zhang L (2020) MicroRNA-374a-5p inhibits neuroinflammation in neonatal hypoxic-ischemic encephalopathy via regulating NLRP3 inflammasome targeted Smad6. Life sciences 252:117664
Cui Y, Wang Y, Li G, Ma W, Zhou XS, Wang J, Liu B (2018) The Nox1/Nox4 inhibitor attenuates acute lung injury induced by ischemia-reperfusion in mice. PloS One 13:e0209444
Li X, Chen S, Mao L, Li D, Xu C, Tian H, Mei X (2019) Zinc improves functional recovery by regulating the secretion of granulocyte colony stimulating factor from microglia/macrophages after spinal cord injury. Front Mol Neurosci 12:18
All AH, Al Nashash H, Mir H, Luo S, Liu X (2020) Characterization of transection spinal cord injuries by monitoring somatosensory evoked potentials and motor behavior. Brain Res Bull 156:150–163
Lin S, Tian H, Lin J, Xu C, Yuan Y, Gao S, Song C, Lv P, Mei X (2020) Zinc promotes autophagy and inhibits apoptosis through AMPK/mTOR signaling pathway after spinal cord injury. Neurosci Lett 736:135263
Jiang W, Li M, He F, Zhou S, Zhu L (2017) Targeting the NLRP3 inflammasome to attenuate spinal cord injury in mice. J Neuroinflamm 14:207
Zhang M, Wang L, Huang S, He X (2020) MicroRNA-223 targets NLRP3 to relieve inflammation and alleviate spinal cord injury. Life Sci 254:117796
Fan Y, Zhang X, Yang L, Wang J, Hu Y, Bian A, Liu J, Ma J (2017) Zinc inhibits high glucose-induced NLRP3 inflammasome activation in human peritoneal mesothelial cells. Mol Med Rep 16:5195–5202
Zhang L, Chu W, Zheng L, Li J, Ren Y, Xue L, Duan W, Wang Q, Li H (2020) Zinc oxide nanoparticles from Cyperus rotundus attenuates diabetic retinopathy by inhibiting NLRP3 inflammasome activation in STZ-induced diabetic rats. J Biochem Mol Toxicol 34:e22583
Cheng X, Xu J, Yu Z, Xu J, Long H (2020) LncRNA Xist contributes to endogenous neurological repair after chronic compressive spinal cord injury by promoting angiogenesis through the miR-32-5p/Notch-1 axis. Front Cell Dev Biol 8:744
Chen S, Wei J, Huang L, Feng B, Guo W (2020) MiRNA-194-5p inhibits inflammatory response after spinal cord injury via regulating TRAF6. Minerva Med 111:603–606
Sun F, Li SG, Zhang HW, Hua FW, Sun GZ, Huang Z (2020) MiRNA-411 attenuates inflammatory damage and apoptosis following spinal cord injury. Eur Rev Med Pharmacol Sci 24:491–498
Zhou CL, Li F, Wu XW, Cong CL, Liu XD, Tian J, Zheng WZ, Yan JL (2020) Overexpression of miRNA-433-5p protects acute spinal cord injury through activating MAPK1. Eur Rev Med Pharmacol Sci 24:2829–2835
Jiang F, Yang M, Wu C, Wang J (2019) Potential roles of miR-374a-5p in mediating neuroprotective effects and related molecular mechanism. J Mol Neurosci 69:123–132
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81671907 and 81871556).
Author information
Authors and Affiliations
Contributions
XGZ and JFS conceived and designed the experiments; YJY and SL analyzed and interpreted the results of the experiments; JQL and XFM performed the experiments; all authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
11064_2021_3441_MOESM1_ESM.tif
Supplementary file1 Knockdown of XIST by transfection with si-XIST vector. (A) Cells showing green fluorescent represented BV2 cells with successfully transfected vector, detected by fluorescent microscopy. (B) After transfection of si-XIST for 48h, more than 70% BV2 cells could express green fluorescent, detected by flow cytometry (TIF 1928 kb)
11064_2021_3441_MOESM2_ESM.tif
Supplementary file2 Overexpression of XIST by transfection with XIST overexpression plasmid. (A) Cells showing green fluorescent represented BV2 cells with successfully transfected plasmid, detected by fluorescent microscopy. (B) After transfection of XIST overexpression plasmid for 48h, more than 70% BV2 cells could express green fluorescent, detected by flow cytometry (TIF 1924 kb)
Rights and permissions
About this article

Cite this article
Zhao, X., Sun, J., Yuan, Y. et al. Zinc Promotes Microglial Autophagy Through NLRP3 Inflammasome Inactivation via XIST/miR-374a-5p Axis in Spinal Cord Injury. Neurochem Res 47, 372–381 (2022). https://doi.org/10.1007/s11064-021-03441-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-021-03441-8