
Abstract
Information processing is onerous. Curiously, active brain tissue does not fully oxidize glucose and instead generates a local surplus of lactate, a phenomenon termed aerobic glycolysis. Why engage in inefficient ATP production by glycolysis when energy demand is highest and oxygen is plentiful? Aerobic glycolysis is associated to classic biochemical effects known by the names of Pasteur, Warburg and Crabtree. Here we discuss these three interdependent phenomena in brain cells, in light of high-resolution data of neuronal and astrocytic metabolism in culture, tissue slices and in vivo, acquired with genetically-encoded fluorescent sensors. These sensors are synthetic proteins that can be targeted to specific cell types and subcellular compartments, which change their fluorescence in response to variations in metabolite concentration. A major site of acute aerobic glycolysis is the astrocyte. In this cell, a Crabtree effect triggered by K+ coincides with a Warburg effect mediated by NO, superimposed on a slower longer-lasting Warburg effect caused by glutamate and possibly by NH4+. The compounded outcome is that more fuel (lactate) and more oxygen are made available to neurons, on demand. Meanwhile neurons consume both glucose and lactate, maintaining a strict balance between glycolysis and respiration, commanded by the Na+ pump. We conclude that activity-dependent Warburg and Crabtree effects in brain tissue, and the resulting aerobic glycolysis, do not reflect inefficient energy generation but the marshalling of astrocytes for the purpose of neuronal ATP generation. It remains to be seen whether neurons contribute to aerobic glycolysis under physiological conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The full oxidation of glucose to CO2 and H2O renders 30–32 ATPs [1]. The alternative to oxidation is the production of lactate, which consumes no oxygen and produces only 2 ATPs. It was therefore surprising to see reports by Marcus Raichle and colleagues showing that evoked neural activity in human subjects is accompanied by glucose consumption in excess of oxygen consumption [2]. This finding was later confirmed by lactate measurements in humans and rodents [3, 4]. As the excess glycolysis occurred in the presence of oxygen, the phenomenon was a puzzling physiological counterpart to the aerobic glycolysis originally described in tumors [5]. Why would the brain shun oxygen and engage in inefficient energy production at the time of its greatest need? We address this question in this article. Sustained aerobic glycolysis, as reported in the developing brain [6] and in proliferating cells [7], will not be discussed here.
Whilst experimenting with yeast in the 1860s, Louis Pasteur observed that glycolysis, then known as fermentation, is acutely suppressed by oxygen [8]. In the 1920s, Otto Warburg christened the phenomenon Pasteursche Reaktion [9], while reporting that it vanishes in tumor slices [5, 10]. The Warburg effect, a term introduced by Efraim Racker in the 1970s [11], has become a prospective therapeutic target in cancer and inflammation [7, 12]. Warburg knew that glucose (but not amino acids and fatty acids) inhibits the respiration of tumors [5]. However, the inhibitory effect of glucose on respiration came to be named after Herbert Crabtree, who published his results (obtained with a Warburg manometer) several years later [13]. We could not find out when was the term Crabtree effect first introduced, but it appears in the literature as early as 1940 [14]. In contrast with the current hype about the Warburg effect, the Crabtree effect remains in relative darkness, except for brewers [15], who take advantage of the ancient evolutionary invention of ethanol as a tactical weapon [16, 17].
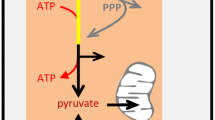
The Pasteur effect is the suppression of glycolysis by mitochondrial oxidative metabolism. The Warburg effect is the inhibition/failure of the Pasteur effect. The Crabtree effect is the suppression of mitochondrial oxidative metabolism by glycolysis, i.e. the mirror image of the Pasteur effect (Table 1). There are other oxygen sinks in mammalian cells but for the sake of brevity, in this review we will refer to mitochondrial oxidative metabolism as respiration. The relationship between these three interdependent effects is depicted in Fig. 1. In a typical mammalian cell, blockage of respiration by anoxia or mitochondrial poisons stimulates glycolysis by a factor of 3 to 10, meaning that the Pasteur effect inhibits glycolysis by 60–90%. The Pasteur effect is thus a major contributor to the balance between glycolysis and respiration (Fig. 1a). The Warburg effect may be understood as a deficit in the capacity of mitochondria to keep glycolysis at bay (Fig. 1b). The Crabtree effect involves a primary increase in glycolytic flux that leads to inhibition of respiration (Fig. 1c). In its original descriptions the increase in glycolysis was secondary to glucose addition [5, 13]. Our group recently reported a variant of the Crabtree effect in astrocytes, in which the inhibition of respiration results not from increased glucose availability but from a primary stimulation of the glycolytic machinery mediated by an extracellular signal [18]. While conceptually different, the Warburg and Crabtree effects look similar as they both involve enhanced relative glucose to oxygen consumption and augmented lactate production. So which of them is responsible for activity-dependent aerobic glycolysis in brain tissue? The answer to that question lies in the metabolic behavior of neurons and astrocytes.

Pasteur, Warburg and Crabtree. a The Pasteur effect is the tonic inhibition of glycolysis by mitochondrial respiration that is abrogated by anoxia. b The Warburg effect is the weakening of the Pasteur effect, leading to lactate production despite the presence of oxygen. c The Crabtree effect is the inhibition of mitochondrial respiration by augmented glycolysis, also leading to lactate production. Note that the Warburg and Crabtree effects may not be distinguished without detailed knowledge of biochemical events involved
Neurons
Neurons are the main energy consumers of brain tissue, accounting for > 90% of the ATP turnover triggered by activity [19]. Substantial evidence in vitro and in vivo indicates that neurons consume both glucose and lactate, the latter produced by neighboring astrocytes upon neuronal prompting [20,21,22,23,24]. The transfer of lactate from astrocytes to neurons is termed Astrocyte-to-Neuron Lactate Shuttle (ANLS), a phenomenon first proposed by Luc Pellerin and Pierre Magistretti in the 1990s [25] and since characterized by multiple experimental approaches [20,21,22,23,24]. ANLS is an evolutionary conserved phenomenon, an extreme version of which occurs in Drosophila melanogaster [26, 27]. However, its existence is not universally accepted [28,29,30].
Attempts to investigate the impact of synaptic activity on the metabolism of cultured neurons using prolonged bath application of glutamate or glutamate receptor agonists have produced contrasting results. While glutamate was found to inhibit glucose transport [31] and glycolysis [32], engaging NMDA receptors led to glycolysis stimulation [33]. Thanks to the availability of genetically-encoded fluorescent sensors for metabolites, it has recently been possible to look into these phenomena with improved temporal resolution. Even short exposures of neurons to glutamate and NMDA provoke metabolic stress, with glutamate having the most dramatic effect [34,35,36]. As a more physiological activation by electrical stimulation, which did not perturb ATP or ATP/ADP, resulted in robust glycolytic stimulation [36], it seems likely to us that inhibition of neuronal glucose transport and glycolysis by glutamate represent pathological events, akin to the generalized shutdown of metabolism observed in multiple systems under metabolic stress [37]. Two other studies based on genetically-encoded sensors gave more direct information on the balance between glycolysis and respiration. In the first study, hippocampal granule cells in acute tissue slices responded to afferent stimulation (60 pulses distributed over 3 s) with transient increases in cytosolic NADH/NAD+ ratio and lactate [38], pointing to stronger activation of glycolysis relative to respiration. However, the neuronal lactate surge was insensitive to blockage of the lactate transporters [38], suggesting that there was no influx or efflux of lactate and that therefore the activity-dependent extracellular lactate surge observed in vivo [4, 39] originates in another cell type, i.e. astrocytes. In the second study, fluxes were measured with transport-stop protocols. Exposure of cultured hippocampal neurons to a short theta burst (40 pulses distributed over 11 s) elicited a strong stimulation of both glucose consumption (200%) and mitochondrial pyruvate consumption (300%), but did not change cytosolic pyruvate or lactate [36]. This shows that the balance between glycolysis and respiration withstood the change in flux regime. Glycolysis and mitochondria were proposed to be synchronized by a mechanism involving the Na+/K+ ATPase pump independently of adenine nucleotides and Ca2+ [36]. In this same study, tetanic stimulation (600 pulses over 30 s) caused ATP depletion and inhibition of mitochondrial pyruvate consumption, indicative of mitochondrial collapse. This failure coincided with a large increase in intramitochondrial Ca2+, which is also observed in neurons exposed to toxic glutamate levels. Measurements in different types of neurons at varying levels of activation will be needed to ascertain the conditions under which the balance between glycolysis and respiration breaks down. The nature of the stimulation protocol is relevant, as hinted by the sensitivity of long term potentiation (LTP) and long term depression (LTD) to the specific arrangement of pulse stimulation [40]. Our working model at this stage is that at rest and at moderate levels of activation, neurons consume glucose and also lactate from astrocytes (more below), whereas at supraphysiological stimulation (e.g. excitotoxicity), mitochondria fail and neurons start to produce lactate. A fine balance between glycolysis and respiration in these cells is ensured by shared control of both pathways by the Na+/K+ pump [36]. It is not known how could the Na+ pump, which is a surface protein, exert control over the metabolism of mitochondria, most of which lie hundreds of nanometers away. On top of this, there is a robust Pasteur effect evidenced by the strong response of neuronal glycolysis to metabolic and oxidative stress [41]. It remains to be seen whether neurons contribute to aerobic glycolysis in brain tissue under physiological conditions and how much of the incremental glucose consumption of active neurons is diverted to the pentose-phosphate pathway, which does not generate ATP but antioxidant power [42].
Astrocytes
Astrocytes are net lactate producers as shown by animal experiments in culture, in slices and in vivo [20, 21, 23, 24]. The robust glycolytic phenotype of these cells is partly explained by stabilization of the master regulator of glycolysis, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3); [42, 43]. PFKFB3 is the enzyme that generates fructose-2,6-biphosphate, a potent allosteric activator of the glycolytic enzyme 6-phosphofructokinase-1 (PFK1). Such is the strength of glycolysis in astrocytes that they are still able to export lactate when bathed in 10 mM lactate (Valdebenito R. and Barros L.F., unpublished data). Contributing to this vectoriality is the expression in astrocytes of a lactate channel gated by extracellular lactate that can export even against a concentration gradient, using membrane potential as the driving force [44]. Pannexin hemichannels may also contribute to vectorial lactate export from astrocytes [45].
Astrocytic glycolysis is sensitive to several neuronal signals, acting through different mechanisms over different spatial and temporal domains. Stimulation by glutamate is mediated by the Na+/glutamate cotransporter and the Na+ pump [25], peaks at 10–20 min and leaves the cell in a stimulated state long after removal of glutamate [46]. It is accompanied by stimulation of the glucose transporter GLUT1, also via the Na+/glutamate cotransporter and the Na+ pump [46,47,48]. Glutamate is oxidized by astrocytes [49], but in the short term its effect on respiration [50, 51] is smaller than its effect on glycolysis, as evidenced by a strong lactate production [25, 46]. Throughout the brain, postsynaptic activity is kept low by tonic GABA-mediated inhibition despite ongoing glutamate release. This means that astrocytes are exposed to some glutamate even if neuronal energy demand, chiefly postsynaptic, is low. Considering the sluggish time course of glycolytic modulation by glutamate, its permanency upon glutamate removal and the fact that astrocytic glutamate uptake does not necessarily correlate with neuronal energy demand, it seems plausible that astrocytes integrate phasic glutamate signals into a sustained metabolic signal, which primes them to receive phasic information of postsynaptic energy demand, for example via extracellular K+. This tonic function may also extend to oligodendroglia, where glutamate facilitates glycolysis and lactate release through slow GLUT1 translocation to the cell surface mediated by NMDA receptors [52]. Exposure of astrocytes to glutamate results in rapid ATP depletion [18, 51, 53] but the stimulation of glycolysis develops much later so there does not seem to be a fast mechanistic link between the two phenomena. One point to be considered is the manner of glutamate application. We have discussed how bath application of glutamate or glutamate agonists to neurons results in inordinate Ca2+ increases and metabolic stress. This raises the issue of whether the astrocytic ATP depletion observed upon bath application of glutamate is, or is not, a physiological phenomenon. At any rate, the aerobic glycolysis induced by glutamate in astrocytes may well be regarded as a Warburg effect (Fig. 1b).
A more faithful second-to-second reporter of neuronal energy demand is extracellular K+. Active dendrites release K+ equimolarly with their uptake of Na+, which is in turn directly proportional to the ATP demand of the Na+ pump. Using microelectrodes and microdialysis, average extracellular K+ in the central nervous system was measured at 2.5–3 mM under sleep and anesthesia, rising to 4 mM in the awake state [54] and up to 6 mM under physiological stimulation [55]. The tiny size of the brain interstice (20 nm) implies that the μm electrodes create a third space that dampens fluctuations and that local extracellular K+ variations are even larger than recorded [56]. Early investigation of the metabolic effects of K+ on astrocytes using radioactive 2-deoxyglucose found small or no effects on glycolysis even at 50 mM [57,58,59]. However, with the advent of genetically-encoded sensors and their improved temporal resolution, it was possible to observe a strong, immediate stimulation of glucose consumption, even at 4 mM extracellular K+ [46]. The stimulation of glucose consumption by K+ requires a functional Na+ pump [46] and is driven by the Na+/bicarbonate cotransporter NBCe1, leading to intracellular alkalinization [60,61,62]. In addition, the NBCe1, acting through the bicarbonate-sensitive adenylyl cyclase, mediates the mobilization of glycogen in response to extracellular K+ [63] and, according to cytosolic NADH measurements, is also involved in the metabolic effects of glutamate and ATP [62]. Extracellular K+ contributes further to aerobic glycolysis in its activation of the astrocytic lactate channel [44], leading to cytosolic lactate depletion and release of product inhibition of glycolysis [64]. Exposure of astrocytes to K+ resulted in elevated ATP levels and inhibition of respiration [18, 51], showing that aerobic glycolysis induced by K+ in astrocytes resembles the Crabtree effect, where a primary stimulation of glycolysis leads to a secondary inhibition of respiration (Fig. 1c). Whereas the effects of K+ and glutamate on astrocytic metabolism do not interact linearly [50], afferent stimulation in hippocampal slices provoked an increase in astrocytic ATP [18], showing that the Crabtree effect dominates over the Warburg effect, at least in the short term.
Additional intercellular signals involved in the control of astrocytic glycolysis by neuronal activity are nitric oxide (NO) and ammonium (NH4+). Astrocytes are devoid of NO synthase but are surrounded by the highest NO synthase activity of the body, located in endothelial cells and in neurons [65]. The initial observation that NO stimulates glycolysis and lactate production in astrocytes but not in neurons through inhibition of mitochondrial cytochrome oxidase [66] was recently followed by the demonstration that the modulation can be detected within seconds at nanomolar NO, levels that are deemed to be within the physiological range [67]. Neurons may not produce enough NO to reach astrocytes [68] but endothelium is a stronger NO source, activated by shear stress during local reactive hyperemia or by neuronal signals [69,70,71]. NH4+ is another candidate for the acute regulation of astrocytic metabolism. Most of the glutamate released during excitatory neurotransmission is returned to neurons in the form of glutamine. Within neurons, glutamine is reconverted to glutamate with the generation of one NH4+ [72]. It is not clear how much of this NH4+ is returned to astrocytes as such, or as amino acids [73], but activity-dependent local NH4+ surges have been recorded in several animal models [74,75,76,77]. NH4+ is efficiently captured by astrocytes via channels and transporters [78, 79]. Physiological ammonium levels in brain tissue have been estimated at 0.2–0.45 mM [79]. Intravenous infusion of NH4+ leading to an increase of 0.7 mM, caused a rapid reversible rise in brain tissue lactate and cerebral blood flow [80]. At 0.2 mM, NH4+ provoked an acute inhibition of mitochondrial pyruvate consumption in astrocytes resulting in deviation of the glycolytic flux towards lactate production and release, but glycolysis was not stimulated [81]. This lack of response is another example of the relative autonomy of glycolysis in these specialized cells. Given these metabolic effects of NH4+ it is unfortunate that so little is known about the speed and mechanism of its release by neurons. If stored in synaptic vesicles to be co-released with glutamate [82], its metabolic effects would be fast. The primary target of both NO and NH4+ (at low physiological levels) is the mitochondria, so both signals can be said to induce aerobic glycolysis of the Warburg type.
Mechanisms of the Pasteur, Warburg and Crabtree Effects
According to classic biochemistry, the second-to-second conversation between glycolysis and respiration is conducted via adenine nucleotides. Glycolysis responds to ATP and AMP (which amplifies ADP changes through adenylate kinase) and respiration responds to ADP. Thus, the Pasteur effect is mediated by the mitochondria sustaining high cytosolic ATP and low cytosolic AMP, resulting in glycolysis inhibition at PFK1. The Warburg effect is therefore seen as a suppression/failure of these inhibitory mechanisms, either because not enough ATP is produced or because the glycolytic machinery becomes insensitive to ATP or AMP. The Crabtree effect develops when a primary stimulation of glycolysis (e.g. by glucose or by K+) increases ATP and decreases ADP, leading to inhibition of mitochondrial respiration. All this sounds quite logical according to the test-tube properties of isolated enzymes and organelles, but there is no evidence that adenine nucleotides mediate these effects in intact cells under physiological conditions. For example, the strong NBCe1-dependent activation of glycolysis in astrocytes that occurs despite increased cytosolic ATP [18] demonstrates that alternative mechanisms may override the influence of adenine nucleotides. Conversely, in astrocytes exposed to glutamate, glycolysis remained unstimulated during several minutes despite severe ATP depletion [18, 46, 51], so there must be another, stronger influence interfering with the stimulatory effect of the nucleotides. In neurons, adenine nucleotides do not seem to be paramount either, because these cells are capable of increasing their rates of glycolysis and mitochondrial pyruvate consumption by several-fold in the absence of detectable changes in cytosolic ATP and ADP [36]. Adenine nucleotide-mediated control may well dominate under pathological conditions like ischemia. For normal workloads however, it is perhaps time to consider alternatives, for example glycolytic intermediates [15] or the conspicuous mitochondrial attachment of hexokinase to mitochondria [83, 84].
In summary, based on experiments in animals, in vivo, ex vivo and in cultured cells, the main locus of acute activity-dependent aerobic glycolysis in brain tissue appears to be the astrocyte. A fast Crabtree effect triggered by K+ coincides with a fast Warburg effect mediated by NO, superimposed on a tonic, glutamate-dependent Warburg effect. The time course of the Warburg effect induced by NH4+ remains to be determined. The combined result of these modulations is that lactate and oxygen are made available to neurons, on demand. In the meantime neurons maintain a balance between glycolysis and respiration mediated by parallel upstream control of both pathways by the Na+ pump (Fig. 2). Technical developments are eagerly awaited to confirm these observations in humans.

Acute activity-dependent aerobic glycolysis in brain tissue. Excitatory neuronal activity triggers the release of multiple small molecules, which act as intercellular metabolic signals. K+ stimulates astrocytic glycolysis leading to inhibition of respiration, a Crabtree effect. Neuronal glutamate and NH4+, and endothelial NO, also inhibit astrocytic respiration, a Warburg effect. As a result, neurons are supplied with lactate and oxygen. Glycolysis and mitochondrial respiration in neurons are controlled by the Na+ pump, not by canonical mechanisms involving adenine nucleotides
References
Hinkle PC (2005) P/O ratios of mitochondrial oxidative phosphorylation. Biochim Biophys Acta 1706:1–11
Fox PT, Raichle ME, Mintun MA, Dence C (1988) Nonoxidative glucose consumption during focal physiologic neural activity. Science 241:462–464
Prichard J, Rothman D, Novotny E, Petroff O, Kuwabara T, Avison M, Howseman A, Hanstock C, Shulman R (1991) Lactate rise detected by 1H NMR in human visual cortex during physiologic stimulation. Proc Natl Acad Sci USA 88:5829–5831
Hu Y, Wilson GS (1997) A temporary local energy pool coupled to neuronal activity: fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J Neurochem 69:1484–1490
Warburg O (1925) The metabolism of carcinoma cells. J Cancer Res 9:148–163
Goyal MS, Hawrylycz M, Miller JA, Snyder AZ, Raichle ME (2014) Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab 19:49–57
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Pasteur L (1861) Expériences et vues nouvelles sur la nature des fermentations. Comptes Rendus 52:1260–1264
Warburg O (1926) Über die wirkung von blausaureäthylester (Athylcarbylamin) auf die pasteursche reaktion. Biochemische Zeitschrift 172:432–441
Warburg O, Posener K, Negelein E (1924) Über den stoffwechsel der carcinomzelle. Naturwissenschaften 12:1131–1137
Racker E (1972) Bioenergetics and the problem of tumor growth. Am Sci 60:56–63
Liberti MV, Locasale JW (2016) The warburg effect: how does it benefit cancer cells? Trends Biochem. Sci. 41(3):211–218
Crabtree HG (1929) Observations on the carbohydrate metabolism of tumours. Biochem J 23:536–545
Rosenthal O, Bowie MA, Wagoner G (1940) On the interdependence of respiration and glycolysis. Science 92:382–383
Diaz-Ruiz R, Averet N, Araiza D, Pinson B, Uribe-Carvajal S, Devin A, Rigoulet M (2008) Mitochondrial oxidative phosphorylation is regulated by fructose 1,6-bisphosphate. A possible role in crabtree effect induction? J Biol Chem 283:26948–26955
Thomson JM, Gaucher EA, Burgan MF, De Kee DW, Li T, Aris JP, Benner SA (2005) Resurrecting ancestral alcohol dehydrogenases from yeast. Nat Genet 37:630–635
Hagman A, Sall T, Compagno C, Piskur J (2013) Yeast "make-accumulate-consume" life strategy evolved as a multi-step process that predates the whole genome duplication. PLoS ONE 8:e68734
Fernandez-Moncada I, Ruminot I, Robles-Maldonado D, Alegria K, Deitmer JW, Barros LF (2018) Neuronal control of astrocytic respiration through a variant of the Crabtree effect. Proc Natl Acad Sci USA 115:1623–1628
Harris JJ, Jolivet R, Attwell D (2012) Synaptic energy use and supply. Neuron 75:762–777
Bouzier-Sore AK, Pellerin L (2013) Unraveling the complex metabolic nature of astrocytes. Front Cell Neurosci 7:179
Stobart JL, Anderson CM (2013) Multifunctional role of astrocytes as gatekeepers of neuronal energy supply. Front Cell Neurosci 7:38
Fernandez-Fernandez S, Almeida A, Bolanos JP (2012) Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem J 443:3–11
Barros LF, Weber B (2018) CrossTalk proposal: an important astrocyte-to-neuron lactate shuttle couples neuronal activity to glucose utilisation in the brain. J Physiol 596:347–350
Magistretti PJ, Allaman I (2018) Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci 19:235–249
Pellerin L, Magistretti PJ (1994) Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA 91:10625–10629
Volkenhoff A, Weiler A, Letzel M, Stehling M, Klambt C, Schirmeier S (2015) Glial glycolysis is essential for neuronal survival in drosophila. Cell Metab 22:437–447
Gonzalez-Gutierrez A, Ibacache A, Esparza A, Barros LF, Sierralta J (2019) Neuronal lactate levels depend on glia-derived lactate during high brain activity in Drosophila. Glia. https://doi.org/10.1002/glia.23772
Yellen G (2018) Fueling thought: Management of glycolysis and oxidative phosphorylation in neuronal metabolism. J Cell Biol 217:2235–2246
Bak LK, Walls AB (2018) Lack of evidence supporting an astrocyte-to-neuron lactate shuttle coupling neuronal activity to glucose utilisation in the brain. J Physiol 596:351–353
Dienel GA (2019) Brain glucose metabolism: integration of energetics with function. Physiol Rev 99:949–1045
Porras OH, Loaiza A, Barros LF (2004) Glutamate mediates acute glucose transport inhibition in hippocampal neurons. J Neurosci 24:9669–9673
Tescarollo F, Covolan L, Pellerin L (2014) Glutamate reduces glucose utilization while concomitantly enhancing AQP9 and MCT2 expression in cultured rat hippocampal neurons. Front Neurosci 8:246
Bak LK, Walls AB, Schousboe A, Ring A, Sonnewald U, Waagepetersen HS (2009) Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J Neurochem 109:87–93
Surin AM, Gorbacheva LR, Savinkova IG, Sharipov RR, Khodorov BI, Pinelis VG (2014) Study on ATP concentration changes in cytosol of individual cultured neurons during glutamate-induced deregulation of calcium homeostasis. Biochemistry 79(2):146–157
Lange SC, Winkler U, Andresen L, Byhro M, Waagepetersen HS, Hirrlinger J, Bak LK (2015) Dynamic changes in cytosolic ATP levels in cultured glutamatergic neurons during NMDA-induced synaptic activity supported by glucose or lactate. Neurochem Res 40:2517–2526
Baeza-Lehnert F, Saab AS, Gutierrez R, Larenas V, Diaz E, Horn M, Vargas M, Hosli L, Stobart J, Hirrlinger J, Weber B, Barros LF (2019) Non-canonical control of neuronal energy status by the Na(+) pump. Cell Metab 29:668–680
Hochachka PW, Buck LT, Doll CJ, Land SC (1996) Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci USA 93:9493–9498
Diaz-Garcia CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G (2017) Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab 26:361–374
Newman LA, Korol DL, Gold PE (2011) Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS ONE 6:e28427
Albensi BC, Oliver DR, Toupin J, Odero G (2007) Electrical stimulation protocols for hippocampal synaptic plasticity and neuronal hyper-excitability: are they effective or relevant? Exp Neurol 204:1–13
Rodriguez-Rodriguez P, Fernandez E, Almeida A, Bolanos JP (2012) Excitotoxic stimulus stabilizes PFKFB3 causing pentose-phosphate pathway to glycolysis switch and neurodegeneration. Cell Death Differ 19:1582–1589
Herrero-Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolanos JP (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 11:747–752
Hasel P, Dando O, Jiwaji Z, Baxter P, Todd AC, Heron S, Markus NM, McQueen J, Hampton DW, Torvell M, Tiwari SS, McKay S, Eraso-Pichot A, Zorzano A, Masgrau R, Galea E, Chandran S, Wyllie DJA, Simpson TI, Hardingham GE (2017) Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat Commun 8:15132
Sotelo-Hitschfeld T, Niemeyer MI, Machler P, Ruminot I, Lerchundi R, Wyss MT, Stobart J, Fernandez-Moncada I, Valdebenito R, Garrido-Gerter P, Contreras-Baeza Y, Schneider BL, Aebischer P, Lengacher S, San MA, Le DJ, Bonvento G, Magistretti PJ, Sepulveda FV, Weber B, Barros LF (2015) Channel-mediated lactate release by k+-stimulated astrocytes. J Neurosci 35:4168–4178
Karagiannis A, Sylantyev S, Hadjihambi A, Hosford PS, Kasparov S, Gourine AV (2016) Hemichannel-mediated release of lactate. J Cereb Blood Flow Metab 36:1202–1211
Bittner CX, Valdebenito R, Ruminot I, Loaiza A, Larenas V, Sotelo-Hitschfeld T, Moldenhauer H, San Martín A, Gutiérrez R, Zambrano M, Barros LF (2011) Fast and reversible stimulation of astrocytic glycolysis by K+ and a delayed and persistent effect of glutamate. J Neurosci 31:4709–4713
Loaiza A, Porras OH, Barros LF (2003) Glutamate triggers rapid glucose transport stimulation in astrocytes as evidenced by real-time confocal microscopy. J Neurosci 23:7337–7342
Porras OH, Ruminot I, Loaiza A, Barros LF (2008) Na(+)-Ca(2+) cosignaling in the stimulation of the glucose transporter GLUT1 in cultured astrocytes. Glia 56:59–68
McKenna MC, Stridh MH, McNair LF, Sonnewald U, Waagepetersen HS, Schousboe A (2016) Glutamate oxidation in astrocytes: roles of glutamate dehydrogenase and aminotransferases. J Neurosci Res 94:1561–1571
Rimmele TS, de Castro AH, Wellbourne-Wood J, Lengacher S, Chatton JY (2018) Extracellular potassium and glutamate interact to modulate mitochondria in astrocytes. ACS Chem Neurosci 9:2009–2015
Juaristi I, Llorente-Folch I, Satrustegui J, del Arco A (2019) Extracellular ATP and glutamate drive pyruvate production and energy demand to regulate mitochondrial respiration in astrocytes. Glia 67(4):759–774
Saab AS, Tzvetavona ID, Trevisiol A, Baltan S, Dibaj P, Kusch K, Mobius W, Goetze B, Jahn HM, Huang W, Steffens H, Schomburg ED, Perez-Samartin A, Perez-Cerda F, Bakhtiari D, Matute C, Lowel S, Griesinger C, Hirrlinger J, Kirchhoff F, Nave KA (2016) Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 91:119–132
Magistretti PJ, Chatton JY (2005) Relationship between l-glutamate-regulated intracellular Na+ dynamics and ATP hydrolysis in astrocytes. J Neural Transm 112:77–85
Ding F, O'Donnell J, Xu Q, Kang N, Goldman N, Nedergaard M (2016) Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science 352:550–555
Heinemann U, Schaible HG, Schmidt RF (1990) Changes in extracellular potassium concentration in cat spinal cord in response to innocuous and noxious stimulation of legs with healthy and inflamed knee joints. Exp Brain Res 79:283–292
Frohlich F, Bazhenov M, Iragui-Madoz V, Sejnowski TJ (2008) Potassium dynamics in the epileptic cortex: new insights on an old topic. Neurosci 14:422–433
Peng L, Zhang X, Hertz L (1994) High extracellular potassium concentrations stimulate oxidative metabolism in a glutamatergic neuronal culture and glycolysis in cultured astrocytes but have no stimulatory effect in a GABAergic neuronal culture. Brain Res 663:168–172
Takahashi S, Driscoll BF, Law MJ, Sokoloff L (1995) Role of sodium and potassium ions in regulation of glucose metabolism in cultured astroglia. Proc Natl Acad Sci USA 92:4616–4620
Sokoloff L, Takahashi S, Gotoh J, Driscoll BF, Law MJ (1996) Contribution of astroglia to functionally activated energy metabolism. Dev Neurosci 18:344–352
Ruminot I, Gutiérrez R, Peña-Munzenmeyer G, Añazco C, Sotelo-Hitschfeld T, Lerchundi R, Niemeyer MI, Shull GE, Barros LF (2011) NBCe1 mediates the acute stimulation of astrocytic glycolysis by extracellular K+. J Neurosci 31:14264–14271
Ruminot I, Schmalzle J, Leyton B, Barros LF, Deitmer JW (2017) Tight coupling of astrocyte energy metabolism to synaptic activity revealed by genetically encoded FRET nanosensors in hippocampal tissue. J Cereb Blood Flow Metab 39:513–523
Kohler S, Winkler U, Sicker M, Hirrlinger J (2018) NBCe1 mediates the regulation of the NADH/NAD(+) redox state in cortical astrocytes by neuronal signals. Glia 66:2233–2245
Choi HB, Gordon GR, Zhou N, Tai C, Rungta RL, Martinez J, Milner TA, Ryu JK, McLarnon JG, Tresguerres M, Levin LR, Buck J, MacVicar BA (2012) Metabolic communication between astrocytes and neurons via bicarbonate-responsive soluble adenylyl cyclase. Neuron 75:1094–1104
Sotelo-Hitschfeld T, Fernández-Moncada I, Barros LF (2012) Acute feedback control of astrocytic glycolysis by lactate. Glia 60:674–680
Garthwaite J, Boulton CL (1995) Nitric oxide signaling in the central nervous system. Annu Rev Physiol 57:683–706
Almeida A, Almeida J, Bolanos JP, Moncada S (2001) Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci USA 98:15294–15299
San Martín A, Arce-Molina R, Galaz A, Perez-Guerra G, Barros LF (2017) Nanomolar nitric oxide concentrations quickly and reversibly modulate astrocytic energy metabolism. J Biol Chem 292:9432–9438
Garthwaite J (2016) From synaptically localized to volume transmission by nitric oxide. J Physiol 594:9–18
Garthwaite G, Bartus K, Malcolm D, Goodwin D, Kollb-Sielecka M, Dooldeniya C, Garthwaite J (2006) Signaling from blood vessels to CNS axons through nitric oxide. J Neurosci 26:7730–7740
LeMaistre JL, Sanders SA, Stobart MJ, Lu L, Knox JD, Anderson HD, Anderson CM (2012) Coactivation of NMDA receptors by glutamate and d-serine induces dilation of isolated middle cerebral arteries. J Cereb Blood Flow Metab 32:537–547
Stobart JL, Lu L, Anderson HD, Mori H, Anderson CM (2013) Astrocyte-induced cortical vasodilation is mediated by d-serine and endothelial nitric oxide synthase. Proc Natl Acad Sci USA 110:3149–3154
Schousboe A, Westergaard N, Waagepetersen HS, Larsson OM, Bakken IJ, Sonnewald U (1997) Trafficking between glia and neurons of TCA cycle intermediates and related metabolites. Glia 21:99–105
Rothman DL, De Feyter HM, Maciejewski PK, Behar KL (2012) Is there in vivo evidence for amino acid shuttles carrying ammonia from neurons to astrocytes? Neurochem Res 37:2597–2612
Tashiro S (1922) Studies on alkaligenesis in tissues. Am J Physiol 60:519–543
Richter D, Dawson RM (1948) The ammonia and glutamine content of the brain. J Biol Chem 176:1199–1210
Tsukada Y, Takagaki G, Sugimoto S, Hirano S (1958) Changes in the ammonia and glutamine content of the rat brain induced by electric shock. J Neurochem 2:295–303
Coles JA, Marcaggi P, Vega C, Cotillon N (1996) Effects of photoreceptor metabolism on interstitial and glial cell pH in bee retina: evidence of a role for NH4+. J Physiol 495(Pt 2):305–318
Kelly T, Rose CR (2010) Ammonium influx pathways into astrocytes and neurones of hippocampal slices. J Neurochem 115:1123–1136
Thrane VR, Thrane AS, Wang F, Cotrina ML, Smith NA, Chen M, Xu Q, Kang N, Fujita T, Nagelhus EA, Nedergaard M (2013) Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat Med 19(12):1643–1648
Provent P, Kickler N, Barbier EL, Bergerot A, Farion R, Goury S, Marcaggi P, Segebarth C, Coles JA (2007) The ammonium-induced increase in rat brain lactate concentration is rapid and reversible and is compatible with trafficking and signaling roles for ammonium. J Cereb Blood Flow Metab. 27:1830–1840
Lerchundi R, Fernandez-Moncada I, Contreras-Baeza Y, Sotelo-Hitschfeld T, Machler P, Wyss MT, Stobart J, Baeza-Lehnert F, Alegria K, Weber B, Barros LF (2015) NH4+ triggers the release of astrocytic lactate via mitochondrial pyruvate shunting. Proc Natl Acad Sci USA 112:11090–11095
Marcaggi P (2006) An ammonium flux from neurons to glial cells. Proc Phys Soc 3:SA16
Wilson JE (2003) Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol 206:2049–2057
Jackson JG, O'Donnell JC, Krizman E, Robinson MB (2015) Displacing hexokinase from mitochondrial voltage-dependent anion channel impairs GLT-1-mediated glutamate uptake but does not disrupt interactions between GLT-1 and mitochondrial proteins. J Neurosci Res 93:999–1008
Acknowledgements
We thank all members of the Barros Lab for their contributions and discussions. We also thank Karen Everett for critical reading of the manuscript. This work was partially funded by CONICYT-BMBF Grant 180045. The Centro de Estudios Científicos (CECs) is funded by the Chilean Government through the Centers of Excellence Basal Financing Program of CONICYT.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Special Issue: In Honor of Professor Juan Bolaños.
Rights and permissions
About this article

Cite this article
Barros, L.F., Ruminot, I., San Martín, A. et al. Aerobic Glycolysis in the Brain: Warburg and Crabtree Contra Pasteur. Neurochem Res 46, 15–22 (2021). https://doi.org/10.1007/s11064-020-02964-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-020-02964-w