
Abstract
The existing data about whether acid sensing ion channels (ASICs) are proconvulsant or anticonvulsant are controversial. Particularly, acid sensing ion channel 3 (ASIC3) is the most sensitive to extracellular pH and has the characteristic ability to generate a biphasic current, but few studies have focused on the role of ASIC3 in seizure. Here we found ASIC3 expression was increased in the hippocampus of pilocarpine induced seizure rats, as well as in hippocampal neuronal cultures undergoing epileptiform discharge elicited by Mg2+-free media. Furthermore, ASIC3 blockade by the selective inhibitor APETx2 shortened seizure onset latency and increased seizure severity compared with the control in the pilocarpine induced seizure model. Incubation with APETx2 enhanced the excitability of primary cultured hippocampal neurons in Mg2+-free media. Notably, the aggravated seizure was associated with upregulation of the N-methyl-d-aspartate subtype of glutamate receptors (NMDARs), increased NMDAR mediated excitatory neurotransmission and subsequent activation of the Ca2+/calmodulin-dependent protein kinase II (CaMKII) and cAMP-response element binding protein (CREB) signaling pathway. Moreover, co-immunoprecipitation confirmed the interaction between ASIC3 and NMDAR subunits, and NMDARs blockade prevented the aggravated seizure caused by ASIC3 inhibition. Taken together, our findings suggest that ASIC3 inhibition aggravates seizure and potentiates seizure induced hyperexcitability at least partly by the NMDAR/CaMKII/CREB signaling pathway, which implies that ASIC3 agonists may be a promising approach for seizure treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epilepsy, a common and disabling brain disorder, is characterized by recurrent unprovoked seizures and affects approximately 50 million people worldwide [1]. However, the mechanism of seizure regulation has been only partially unraveled, and 20–30% of patients remain refractory to treatment [2]. Therefore, elucidation of the essential mechanism underlying seizure and search for new antiepileptics to treat intractable epilepsy are justified.
The fact that seizures can reduce brain pH through CO2 accumulation, lactic acid production and other mechanisms has drawn much attention to ASICs [3, 4]. The ASICs, which belong to the degenerin/epithelial sodium channel superfamily, are proton-gated, cation-selective, amiloride-sensitive channels [5]. Of the six ASIC subunits cloned so far, ASIC1a, ASIC2a, and ASIC2b are widely expressed in the central nervous system, whereas ASIC1b and ASIC3 are traditionally considered to be mainly expressed in peripheral sensory neurons. However, recent studies have also demonstrated the existence of ASIC3 transcripts and immunoreactivity in neurons of some brain regions such as hypothalamus, suprachiasmatic nucleus and brainstem [6].
Previous research on the role of ASICs in epileptic seizures is puzzling and controversial. Some evidence demonstrates distinct changing patterns of ASIC subunits in animal seizure models or in specimens from TLE patients. ASIC1a and ASIC2b mRNA levels are decreased in the hippocampus of pilocarpine-treated rats, while ASIC1a mRNA and protein levels undergo a reduction and ASIC2b presents an increase in rat piriform cortex following pilocarpine injection [7, 8]. Inhibition of ASICs by amiloride has anticonvulsant effects in pentylenetetrazole and pilocarpine models of seizure [9,10,11,12]. Additionally, the selective ASIC1a blocker PcTX1 inhibits neuronal firing in cell culture and hippocampal slice models of epilepsy [13, 14]. These results suggest that the activation of ASICs, especially ASIC1a, may be proconvulsant. In contrast, other studies raise the opposite possibility that ASICs might terminate epileptic seizures. For example, seizures are more severe in ASIC1a knockout mice than in their wild-type counterparts, and ASIC1a overexpression mitigates seizure severity [15]. Thus, it is necessary to gain a better understanding of these ambiguities.
Another factor deserving attention is the characteristic kinetics and activation profile of ASIC3. ASIC3 is the most H+ sensitive of the six isoforms and has a pH value for half-maximal activation of approximately 6.4, which implies that it may be the first to respond to the acidosis in seizure [16]. Besides, ASIC3 has the particularity to generate a biphasic current—a rapidly inactivating followed by a sustained current [17]. This means it does not fully inactivate as the extracellular fluid remains acid, therefore having the possibility to modify neuronal excitability. So far, only one study has highlighted the pathophysiological significance of ASIC3 in epilepsy and suggested an antiepileptic role for ASIC3 [18]. However, data are still scarce about how ASIC3 influences neuronal excitability and seizure patterns.
Herein we examined the changes in ASIC3 expression in a pilocarpine-induced seizure model and an in vitro Mg2+-free epileptiform discharge model. Behavioral studies, measurements of epilepsy associated proteins and signaling pathways, and electrophysiological assessments of neuronal excitability and NMDAR mediated excitatory neurotransmission were performed to investigate the role of ASIC3 in seizure.
Materials and Methods
Animal Seizure Models and Behavioral Tests
All animal experimental procedures were approved by the Commission of Wuhan University for the Ethics of Experiments on Animals. Adult male rats weighing 220–250 g from the Experimental Animal Center of Wuhan University were maintained under standard conditions (22–24 °C, a 12/12-h light/dark cycle, free access to food and water). The rats were randomly divided into six groups: a control group and five groups examined at different time points after seizure induction. The pilocarpine model of seizure was established by intraperitoneal injection of lithium chloride (127 mg/kg, Sigma, St. Louis, MO, USA) and administration of pilocarpine (35 mg/kg, i.p., Sigma) 19 h later. The rats were pretreated with atropine sulfate (1 mg/ml, i.p.) 30 min before pilocarpine injection to reduce peripheral cholinomimetic effects. Seizures were evaluated by Racine’s scale. Generalized tonic-clonic seizures (GTCS) were considered stage 6. Only animals that reached stage 4 (rearing), stage 5 (rearing plus imbalance and falling) or stage 6 were regarded as successful models and used for subsequent experiments. Once seizure activity reached stage 4, the time was noted as status epilepticus (SE) onset. One hour after the onset of SE, the rats were injected with diazepam (10 mg/kg) to terminate seizures. The control animals were given equal volumes of saline instead of lithium chloride and pilocarpine.
Surgical Procedures and Drug Injection
The rats were anesthetized with 3.5% chloral hydrate and placed in a stereotaxic instrument. The skull was exposed and 0.5 mm holes were drilled. Right-lateral guide cannulas were implanted aimed at the lateral ventricle (anterior–posterior 0.8 mm, medial–lateral 1.5 mm, dorsal–ventral 4 mm). The guide cannulas were fixed by dental cement, and dummy cannulas were inserted to prevent clogging. The rats were allowed 5 days to recover prior to the next experiment. APETx2 (5 µl at 20 or 30 µM dissolved in PBS, Smartox), a selective antagonist of ASIC3, is a 42 amino acid peptide isolated from the venom of the sea anemone Anthopleura elegantissima. It was intracerebroventricularly microinjected into brain through a 10 µl micropipette connected to an injection tube 0.5 h before pilocarpine injection. The concentration range of APETx2 was mainly determined according to previous studies [18, 19]. AP5 (5 µl at 5 mM dissolved in PBS, Sigma), a selective antagonist of NMDAR, was injected 1 h before pilocarpine injection as above. The dose of AP5 was chosen based on previous studies [20, 21]. These two drugs were injected for more than 3 min and the needle was left in place for 2 min to allow diffusion.
Western Blot Analysis and Co-immunoprecipitation (Co-IP)
Total protein was extracted from cultured neurons or rat hippocampal tissue in cell lysis buffer (Beyotime Institute of Biotechnology) with 1 mM phenylmethanesulfonyl fluoride (Beyotime Institute of Biotechnology), protease inhibitor cocktail (CWBIO) and phosphatase inhibitor cocktail (CWBIO). The protein in the supernatant was separated after centrifugation at 14,000×g for 5 min at 4 °C. For detection of the surface levels of NMDARs, membrane protein was extracted using the ProteoExtract Native Membrane Protein Extraction Kit (M-PEK Kit, Millipore) as previously described [22]. Briefly, the hippocampal tissue pieces were washed twice in wash buffer and then homogenized in ice-cold extraction buffer 1. After incubation for 10 min at 4 °C with gentle agitation, the homogenate was centrifuged at 16,000×g at 4 °C for 15 min and the supernatant was discarded. The pellet was resuspended in extraction buffer 2, incubated for 30 min at 4 °C with gentle agitation and then centrifuged at 16,000×g at 4 °C for 15 min. Finally, the supernatant enriched in integral membrane protein was collected. The protein concentrations were measured by BCA assay (Pierce). Forty micrograms of protein was subjected to SDS-polyacrylamide gel electrophoresis (5% spacer gel; 10% separation gel) and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore). The PVDF membrane was blocked in 5% skim milk or BSA for 1.5 h at room temperature and then incubated at 4 °C overnight with primary antibodies: rabbit anti-ASIC3 (ASC-018) 1:350, Alomone; rabbit anti-NMDAR1 (D65B7) 1:1000, Cell Signaling Technology; rabbit anti-NMDAR2A (AB1555P) 1:500, Millipore; mouse anti-NMDAR2B (sc-365597) 1:500, Santa Cruz Biotechnology; rabbit anti-Glutamate Receptor 1 (ab109450) 1:2000, Abcam; rabbit anti-CaMKIIα (phosphor-T286) (ab32678) 1:1000, Abcam; rabbit anti-CaMKIIα (AF6493) 1:1000, Affinity; rabbit anti-CREB (phosphor-S133) (ab32096) 1:2000, Abcam; rabbit anti-CREB (12208-1-AP) 1:1000, Proteintech; mouse anti-β-actin (A3854) 1:60000, Sigma. After being washed with TBST three times, the membrane was incubated with appropriate secondary antibodies (1:5000, CWBIO) for 1.5 h at room temperature. The resulting bands were visualized with chemiluminescent horseradish peroxidase (HRP) substrate, and the film was developed in a dark room. Band density was quantified using the software ImageJ, and protein expression levels were normalized to the corresponding levels of β-actin.
Protein extracts (500 µg) from rat hippocampal tissues 1 day after seizure were pre-cleared with protein A/G PLUS-agarose (Santa Cruz Biotechnology). The pre-cleared lysates were subsequently incubated with 2 µg indicated primary antibodies or the same species IgG as control on a rocking platform at 4 °C overnight followed by incubation with 50 µl protein A/G PLUS-agarose at 4 °C for 2 h. The agarose beads were then pelleted, washed 4 times with lysis buffer, boiled with 2× loading buffer and subjected to Western blot with the appropriate primary and secondary antibodies.
Reverse Transcription-PCR (RT-PCR) Analysis
Total RNA from hippocampal tissues was isolated using TRIzol reagent (Invitrogen) and then reverse transcribed into cDNA using AMV-RT enzyme (Promega) and oligo (dT) primers. The following sense and antisense primers were used: GAPDH, TGTGAAGCTCATTTCCTGGTATG and AGGGCCTCTCTCTTGCTCTC; ASIC3, CTGGCAACGGACTGGAGATTA and TGTAGTAGCGCACGGGTTGG [23]. Thirty-five cycles of PCR were performed for ASIC3 (95 °C for 30 s, 61 °C for 30 s, and 72 °C for 30 s) and 24 cycles for GAPDH (95 °C for 20 s, 55 °C for 20 s, and 72 °C for 20 s). The PCR parameters were designed to avoid overamplification and allow a better comparison between groups. One-half of the PCR products were visualized on 2% agarose gels by ethidium bromide staining.
Immunohistochemistry (IHC)
Paraffin embedded sections were deparaffinized in xylene for 20 min, rehydrated in a graded ethanol series for 5 min at each grade, and then incubated in 0.3% H2O2 for 20 min. The sections were heated at 92–98 °C in 10 mmol/l sodium citrate buffer at pH 6.0 for 20 min to achieve antigen retrieval. Nonspecific binding was blocked in 10% goat serum for 20 min at room temperature. The sections were incubated with rabbit anti-ASIC3 antibody (1:200, Alomone) overnight at 4 °C followed by incubation with biotinylated goat anti-rabbit secondary antibody (1:100, Zhongshan Golden Bridge Inc., Beijing, China) for 30 min at 37 °C. After treatment with ABC solution at 37 °C for 30 min, the sections were incubated with DAB (Zhongshan Golden Bridge Inc., Beijing, China) for 5 min. Hematoxylin was used for counterstaining. Images of slides were captured with a Nikon microscope (Nikon, Japan). Five random visual field images at ×200 magnification for each sample were selected for measurement. The mean optical density (MOD) of each visual field was calculated by Image-Pro Plus 6.0 software (Media Cybemetrics Inc., USA). Visualization and quantification were performed by an experimenter blinded to the groups to avoid bias.
Hippocampal Neuronal Culture and Induction of Epileptiform Discharge by Magnesium-Free Medium
Hippocampal neurons were prepared from 1-day-old Sprague-Dawley rats (Experimental Animal Center of Wuhan University). Briefly, hippocampal tissue was dissected and rinsed in ice cold PBS. Blood vessels and meninges were removed under a microscope. The tissue was minced and incubated with 0.25% trypsin for 10 min at 37 °C. Single-cell suspension was obtained by pipetting gently and repeatedly in high-glucose DMEM (Gibco) supplemented with 10% FBS (Gibco), 1 mM l-glutamine (Gibco), and 1% penicillin/streptomycin. Cells were plated at a density of 105 cells/cm2 onto 24-well (for electrophysiology) or 6-well (for Western blot) plates pre-coated with poly-d-lysine (0.1 mg/ml, Sigma) and were maintained at 37 °C in a 5% CO2 humidified atmosphere. The medium was replaced with serum-free Neurobasal (Gibco) containing 2% B27 (Gibco) plus 1 mM l-glutamine the next day. Treatment with 10 µM cytarabine (Sigma) was used on the third day to block division of glial cells. Half of the medium was changed every 3 days.
Epileptiform discharge in hippocampal neuronal culture was induced as described previously [24, 25]. Briefly, after neurons were maintained in culture for 12–14 days, Neurobasal medium was replaced with physiological basal recording solution (pBRS, 145 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose, 0.002 mM glycine, pH adjusted to 7.3 with NaOH, and osmolarity adjusted to 325 mOsm with sucrose), pBRS with 63 nM APETx2, pBRS without MgCl2 (referred to hereafter as Mg2+-free) or pBRS without MgCl2 plus 63 nM APETx2 (referred to hereafter as Mg2+-free plus APTEx2). The dose of APETX2 was chosen because it inhibits the rat ASIC3 peak current with an IC50 of 63 nM [26]. The neurons were then incubated at 37 °C in a 5% CO2 humidified atmosphere for 1 h before immediately being utilized for electrophysiology experiments. To evaluate ASIC3 protein expression after epileptiform discharge, we incubated the neurons in pBRS or pBRS without MgCl2 for 3 h and restored them to Neurobasal medium for at least 6 h before subjecting them to Western blot.
Electrophysiology
To measure excitability of cultured hippocampal neurons, whole-cell current clamp recordings were performed as described before [24, 25]. Briefly, the recording chamber was perfused with pBRS or pBRS without MgCl2. Patch electrodes with a resistance of 3–5 MΩ were pulled using a vertical puller (PC-10, Narishige International, London, UK), fire-polished and filled with pipette solution (140 mM K+ gluconate, 1.1 mM EGTA, 1 mM MgCl2, 10 mM HEPES, 4 mM Na2ATP, pH adjusted to 7.2 with KOH, and osmolarity adjusted to 290 mOsm with sucrose). Phase-bright pyramidal-shaped neurons with a resting membrane potential (RMP) more negative than − 50 mV were selected for recording. The neuronal membrane potential was held at − 60 mV, and recordings were terminated if the series resistance exceeded 20 MΩ. To record spontaneous discharge of action potential, we performed the recordings in current clamp mode at I = 0 setting. To record evoked discharge of action potential, we injected the neurons with 500-ms depolarizing currents from 20 to 120 pA in 20-pA incremental steps.
To prepare brain slices, the rats were anesthetized with 3.5% chloral hydrate 1 day after seizure. After cardiac perfusion with 4 °C slice solution (88 mM NaCl, 2.5 mM KCl, 7 mM MgCl2, 0.5 mM CaCl2, 1.25 mM NaH2PO4, 25 mM NaHCO3, 75 mM sucrose, saturated with 95% O2 and 5% CO2), the brain was rapidly removed from the skull and placed in 4 °C slice solution for 2 min. Coronal 300 µm brain slices containing hippocampus were cut on a vibratome (VT-1200S, Leica, Wetzlar, Germany) and recovered in artificial cerebrospinal fluid at 32 °C (ACSF, 126 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 2 mM MgSO4, 2 mM CaCl2, 25 mM NaHCO3, 10 mM glucose, saturated with 95% O2 and 5% CO2) for at least 1 h before clamp patch recording.
For NMDAR mediated mEPSC recording, the internal pipette solution contained 133 mM K+ gluconate, 8 mM NaCl, 0.6 mM EGTA, 2 mM MgATP, 0.3 mM Na3GTP and 10 mM HEPES (pH adjusted to 7.4 with KOH, osmolarity adjusted to 290 mOsm with sucrose). ACSF without MgSO4 (to relieve the Mg2+ block of NMDAR) was used as external solution in the presence of glycine (0.002 mM, as coagonist to activate NMDAR). Tetrodotoxin (TTX, 1 µM), bicuculline (10 µM) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 µM) were added in the recording ACSF to block action potentials, inhibitory synaptic transmission and AMPA receptor mediated mEPSC, respectively. Hippocampal CA1 pyramidal neurons were held at − 70 mV under a voltage-clamp to record NMDAR mediated mEPSC.
To calculate the ratio of evoked NMADR mediated EPSC to APMAR mediated EPSC (NMDA/AMPA ratio), the internal pipette solution contained 135 mM cesium methanesulfonate, 2 mM MgCl2, 1 mM EGTA, 2 mM MgATP, 0.3 mM Na3GTP, 10 mM HEPES, 8 mM Na2-phosphocreatine (pH adjusted to 7.2 with CsOH, osmolarity adjusted to 290 mOsm with sucrose). Brain slices were bathed in ACSF containing bicuculline (10 µM) to block inhibitory postsynaptic currents. A concentric stimulating electrode (FHC, Bowdoin, USA) was positioned 50–100 µm from the neuron being recorded. Evoked currents were generated using a 0.1 Hz pulse (duration, 100 µs) delivered by a stimulation isolation unit (SS-201J, Nihon Kohden). Membrane potential was held at − 70 mV to record AMPAR-mediated current. Stimulus intensity was adjusted to produce a single-peaked response with an average amplitude of approximately 60 pA. Membrane potential was held at + 40 mV to record NMDAR-mediated current. Amplitude of NMDAR current was quantified at 50 ms after stimulus, when the contribution of the AMPAR component was minimal. Typically, 30 responses were averaged to generate an average response. NMAD/AMPA ratios were calculated by dividing the amplitude of the NMDAR current by the amplitude of the AMPAR current (peak current, at − 70 mV), and were used as a way to assay relative NMDAR function as described previously [27,28,29].
All recordings were performed with a MultiClamp 700B amplifier and a Digidata 1550 (Molecular Devices). Data were filtered at 2 kHz, acquired at 10 kHz, and analyzed by Clampfit 10 software (Molecular Devices). The MiniAnalysis 6.0.3 software (Synaptosoft) was used to analyze synaptic activity.
Statistical Analysis
All data were expressed as the mean ± SEM. Student’s t test was applied for the comparison of two independent groups. More than two groups were compared by one-way analysis of variance (ANOVA) followed by post hoc LSD test where appropriate. Chi-squared test or Fisher’s exact test was used for categorical variables. Data in cumulative fraction of mEPSC amplitude and inter-event interval was analyzed by Kolmogorov–Smirnov test. Statistical analyses were performed using SPSS 20.0 and P < 0.05 was considered statistically significant.
Results
The Level of ASIC3 is Elevated in Pilocarpine-Induced Seizure Model and In Vitro Mg2+-Free Epileptiform Discharge Model
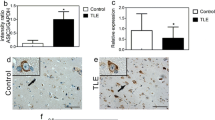
Rat ASIC3 protein presented as a band at approximately 60 kDa with β-actin at 42 kDa (Fig. 1a). ASIC3 protein expression began to increase 2 h after SE, reached a peak at 48-h and then slightly decreased at 72-h but still higher than the control. The hippocampal cDNA template yielded a unique PCR product band for ASIC3 at 506 bp and for GAPDH at 142 bp (Fig. 1c). The relative mRNA expression of ASIC3 in the seizure group was higher than in the control at all time points. IHC showed that the MOD value of ASIC3 in the hippocampus of the rats 24 h after seizure was higher than that in the control (Fig. 1e, f).

The level of ASIC3 is elevated in pilocarpine induced seizure models. Representative Western blot (a) and densitometric analysis (b) showed that ASIC3 protein expression was increased significantly in seizure rats at each time point compared with the control (n = 4 for each group). Representative RT-PCR images (c) and densitometric analysis (d) showed that ASIC3 mRNA expression was increased significantly in seizure rats at each time point compared with the control (n = 4 for each group). IHC showed faint ASIC3 immunoreactivity in the hippocampus of the control (upper panels of e) but relatively strong ASIC3 immunoreactivity in the rats 24 h after seizure (lower panels of e). Analysis of mean optical density value proved that the difference was significant (f) (n = 3 for each group). *P < 0.05, **P < 0.01 compared with the control group. Scale bars: 500 µm in left panels of e, 100 µm in the remaining panels of e
Age-matched control neurons displayed infrequent spontaneous action potentials, while neurons after a treatment with Mg2+-free solution for 3 h demonstrated characteristic epileptiform discharges (Fig. 2a). The ASIC3 protein level in Mg2+-free group was approximately 1.5 times higher than in the control group (Fig. 2b, c).

The level of ASIC3 is elevated in an in vitro Mg2+-free epileptiform discharge model. a Control neurons displayed occasional spontaneous action potentials, while neurons after 3 h exposure to Mg2+-free solution demonstrated high-frequency epileptiform discharges. Representative Western blot (b) and densitometric analysis (c) showed that the ASIC3 protein level in the Mg2+-free group was significantly increased compared with the control. Western blot images and data were from 3 independent experiments. *P < 0.05 compared with the control group
ASIC3 Blockade Increases Seizure Severity
We explored the impact of ASIC3 blockade on epileptic seizure behavior by intracerebroventricular injection of APETx2. We found that APETx2 significantly shortened the latency to seizure of level 4 and enhanced the maximum Racine score compared with the sham or PBS treatment (Fig. 3b, c). There was no distinct difference between the 20 and 30 µM APETx2 groups in terms of latency or maximum Racine score. Additionally, more than half the rats in the APETx2 groups developed GTCS, whereas those in the sham or PBS group were less likely to have GTCS (Fig. 3d). The APETx2-treated groups had more severe seizures than the sham or PBS group except at the 45-min time point (Fig. 3e).

ASIC3 blockade increases seizure severity. a Schematic overview of APETx2 administration and pilocarpine induced seizure model. Either 20 or 30 µM APETx2 significantly decreased the latency to seizure of level 4 (b) and increased the maximum Racine score of seizure (c) compared with the PBS group (n = 11 for each group). d The incidence of GTCS in 20 or 30 µM APETx2 group was higher than that of the PBS group (n = 18 for each group). e Racine score of seizure over time was higher in APETx2 treated groups except at 45 min time point compared with the PBS group (n = 11 for each group). Pilocarpine was injected at time zero. *P < 0.05, **P < 0.01, 20 µM APETx2 group compared with the PBS group. #P < 0.05, ##P < 0.01, 30 µM APETx2 group compared with the PBS group. NS not significant
ASIC3 Inhibition Enhances the Excitability of Cultured Hippocampal Neurons in Mg2+-Free Epileptiform Discharge Model
Meanwhile, we investigated the impact of ASIC3 inhibition on neuronal excitability in an in vitro Mg2+-free epileptiform discharge model. Control neurons exhibited occasional spontaneous action potentials in pBRS and in pBRS with APETx2. Incubation with Mg2+-free medium for 1 h triggered continuous high-frequency (approximately 5 Hz) epileptiform discharges throughout the recording period. Treatment with 63 nM APETx2 significantly raised the frequency of burst discharges induced by Mg2+-free medium (Fig. 4a, b). No significant differences in RMP or peak amplitude were observed among groups. Moreover, compared with neurons treated with Mg2+-free medium alone, neurons treated with Mg2+-free medium plus APETx2 generated more action potentials in response to each current injection (Fig. 4c, d).

ASIC3 inhibition increases the excitability of cultured hippocampal neurons in Mg2+-free epileptiform discharge model. Representative traces (a) and quantification analysis (b) showed that APETx2 alone did not influence the action potential frequency of normal neuronal cultures. Mg2+ withdrawal for 1 h triggered high-frequency epileptiform discharges in cultured hippocampal neurons. Incubation with 63 nM APETx2 further raised the spontaneous action potential frequency of neurons in Mg2+-free medium (n = 10 for each group). c Representative traces of action potential firing generated by incremental depolarizing current injections in primary neurons of the control, APETx2, Mg2+-free and Mg2+-free plus APETx2 groups. d Comparison of number of action potentials in neurons from various groups in response to different current injections (n = 8 for each group). **P < 0.01, the Mg2+-free group compared with the control group. ##P < 0.01, the Mg2+-free plus APETx2 group compared with the Mg2+-free group. NS not significant, the APETx2 group compared with the control group
APETx2 Upregulates the Surface NMDAR Expression in the Hippocampus of Rat Seizure Models
To test whether the effect of ASIC3 blockade on seizure phenotype was because of changes in neuronal excitability, we examined the hippocampal membrane expression of NMDAR and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), two main types of ionotropic excitatory amino acid receptors. The membrane expression of NMDAR1 (NR1), NMDAR2A (NR2A) and NMDAR2B (NR2B) was significantly increased 2 h after SE onset, and the vehicle PBS had no effect on their expression. The membrane expression of these NMDARs was even higher in the APETx2 group than in the seizure or the PBS group (Fig. 5a, c). The expression profile of NMDAR subunits 24 h after SE onset was similar to that at the 2-h time point except that, in the APETx2 group, NR1 remained statistically unchanged compared with the seizure and the PBS group (Fig. 5e, g). For AMPAR, although a significant increase in the membrane expression of glutamate receptor 1 (GluR1) was present 2 h after SE onset, the protein level in the APETx2 treated seizure group did not differ from that in the seizure group (Fig. 5b, d). Likewise, there were no significant differences in the expression of GluR1 between the seizure group and the APETx2 treated seizure group 24 h after SE (Fig. 5f, h). Since ASIC3 inhibition correlated with increase of surface NMDAR expression, we further examined whether ASIC3 can interact with NMDAR in vivo by Co-IP experiments using hippocampal lysates. Our study confirmed that ASIC3 antibody effectively precipitated NR1, NR2A and NR2B. This interaction was validated again by reciprocal Co-IP using antibodies of NR1, NR2A and NR2B respectively (Fig. 6).

APETx2 increases the membrane expression level of NMDAR subunits in rat seizure models. Rats were sacrificed and hippocampal tissues were collected for Western blot 2 h after SE onset. Representative Western blot (a, b) and densitometric analysis (c, d) showed that the membrane expression of NMDAR subunits and GluR1 was significantly elevated in seizure rats. APETx2 pretreatment further increased the levels of surface NMDARs, but not GluR1. There was no significant difference in protein expression between the seizure group and the PBS group. e–h A similar pattern of change in NMDAR subunits and GluR1 was also observed 24 h after SE onset except that, in the APETx2 group, NR1 remained statistically unchanged compared with the seizure group and the PBS group. n = 4 for each group. *P < 0.05, **P < 0.01, compared with the pilocarpine or PBS plus pilocarpine group. NS not significant

Co-IP assay confirms the physical interaction between ASIC3 and NMDARs. a Protein extracts from hippocampal tissue was immunoprecipitated with NR1 antibody and then blotted with ASIC3 antibody. The Western blot showed an interaction of NR1 with ASIC3. This interaction was verified again by immunoprecipitating with ASIC3 antibody and then blotting with NR1 antibody. Rabbit IgG was used as negative control. Likewise, the interactions between ASIC3 and NR2A (b) or NR2B (c) were found respectively
ASIC3 Inhibition Strengthens NMDAR Mediated Excitatory Neurotransmission
To test whether upregulation of NMDAR protein level contributes to NMDAR mediated excitatory neurotransmission. We recorded NMDAR mediated mEPSC and NMDA/AMPA ratios in hippocampal slices. The amplitude and frequency of NMDAR mediated mEPSC were significantly increased in APETx2 treated seizure group compared with those observed in seizure group (Fig. 7a–c). Moreover, NMDA/AMPA ratios increased significantly in seizure group compared with control group. NMDA/AMPA ratios in APETx2 treated seizure group were higher than those in seizure group, which demonstrated a relatively increased NMDAR function when ASIC3 was inhibited (Fig. 7d, e).

ASIC3 inhibition strengthens NMDAR mediated excitatory neurotransmission. a Representative traces of NMDAR mediated mEPSC in slices from control group, pilocarpine induced seizure group and seizure group treated with APETx2. Cumulative fraction of mEPSC inter-event interval (b) and amplitude (c) showed that the amplitude and frequency of NMDAR mediated mEPSC were significantly increased in APETx2 treated seizure group compared with those observed in seizure group (n = 8 for each group). d For each pair, lower traces were recorded at − 70 mV (AMPA), and upper traces were recorded at + 40 mV (NMDA, calculated 50 ms post-stimulus). e Quantification analysis showed that NMDA/AMPA ratios increased significantly in seizure group compared with control group. NMDA/AMPA ratios in APETx2 treated seizure group were higher than those in seizure group (n = 8 for each group). *P < 0.05 compared with the control group, **P < 0.01 compared with pilocarpine induced seizure group
APETx2 Activates the CaMKII and CREB Signaling Pathway
Next, we measured the levels of total and phosphorylated CaMKIIα and CREB. Both p-CaMKIIα and total CaMKIIα were significantly upregulated 2 h after SE onset. APETx2 pretreatment further increased the level of p-CaMKIIα compared with the seizure group but had no effect on the level of total CaMKIIα (Fig. 8a, b). There was no significant difference in p-CaMKIIα or total CaMKIIα expression between the seizure group and the PBS group. Additionally, both p-CREB and total CREB were significantly increased 2 h after SE onset. There was a significant increment of p-CREB expression in the APETx2 group compared with the seizure group (Fig. 8a, c). The level of p-CaMKIIα and p-CREB were also significantly elevated in the APETx2 group compared with the seizure group 24 h after SE onset (Fig. 8d–f).

APETx2 increases p-CaMKIIα and p-CREB activity. Representative Western blot (a) and densitometric analysis (b, c) showed that both p-CaMKIIα and total CaMKIIα were significantly upregulated 2 h after SE onset. APETx2 pretreatment further increased the level of p-CaMKIIα compared with the seizure group but had no effect on the level of total CaMKIIα. Additionally, both p-CREB and total CREB were significantly increased 2 h after SE onset. There was a significant increment of p-CREB but not total CREB expression in the APETx2 group. d–f Consistent with the results at the 2-h time point, p-CaMKIIα and p-CREB were significantly elevated in the APETx2 group compared with the seizure group 24 h after SE onset. n = 4 for each group. *P < 0.05, **P < 0.01, compared with the pilocarpine or PBS plus pilocarpine group. NS not significant
AP5, a Selective NMDAR Antagonist, Abolishes the Aggravated Seizures Caused by ASIC3 Inhibition
To determine whether the effect of ASIC3 inhibition on seizure susceptibility require the participation of NMDAR, AP5 was utilized 1 h before pilocarpine administration to block NMDAR activity. In the presence of AP5, We found no significant differences in the latency to seizure of level 4 (Fig. 9a) and maximum Racine score (Fig. 9b) among the sham, the vehicle and the APETx2 groups.

AP5 abolishes the aggravated epileptic seizures caused by ASIC3 inhibition. When AP5 was applied to block NMDAR activity, no significant differences were seen in the latency to seizure of level 4 (a) or the maximum Racine score (b) among the sham, the vehicle and the APETx2 groups (n = 9 for each group). NS not significant
Discussion
The principal findings of this study are as follows. First, ASIC3 was increased in the hippocampus of pilocarpine-induced seizure model and in cultured hippocampal neurons undergoing epileptiform discharge after Mg2+-free induction. Second, the ASIC3 inhibition exacerbated seizure severity and potentiated epileptiform discharges in hippocampal neurons treated with Mg2+-free solution. Third, the above phenomenon was probably mediated by elevated NMDAR function and subsequent activation of the CaMKII/CREB signaling pathway. These findings support a proconvulsant role for inhibition of ASIC3.
A previous study has reported elevated ASIC3 protein expression in the temporal cortex and hippocampus of pilocarpine-induced seizure rats [18], which was confirmed by our data. However, contrary to the decreased ASIC3 mRNA level in TLE patients [18], we found an increase in ASIC3 mRNA level in the hippocampus of seizure rats. Our results are supported by another study, which reported that pilocarpine treatment enhanced ASIC3 mRNA expression in a time-dependent manner within the first 2 h of seizure [30]. This inconsistency may be ascribed to different stages of disease, namely, acute seizure and subsequent chronic epilepsy. Additionally, a disparity in regulatory mechanisms due to species differences cannot be ignored, as the homology between human and rat orthologs of ASIC3 is 83%, while it is close to 100% for other ASIC subunits [31].
Utilizing Mg2+-free media to elicit SE-like activity in hippocampal neuronal culture is a commonly used model to evaluate the effects of SE on neuronal molecular and physiological changes [24, 25, 32]. Despite lack of true anatomical connections, this model exhibits several features similar to the development of acquired epilepsy in animal models and clinical cases. Consistent with the variation tendency of ASIC3 expression at the animal level, our results showed, for the first time, that continuous high-frequency burst discharges elicited by Mg2+-free solution upregulated ASIC3 protein levels in cultured neurons, further suggesting that ASIC3 is involved in seizure.
Pharmacological inhibition of ASIC3 by APETx2 in our study aggravates seizure, which implies that ASIC3 may play a part in inhibiting seizure. This phenomenon could be explained by the previous conclusion that acidosis inhibits seizures [33, 34]. Extracellular acidosis generated by seizures, in turn, could activate ASIC3 and then terminate seizure; that is to say, acidosis may exert antiepileptic effects through elevated ASIC3. In this way, it is possible that any treatment inactivating ASIC3 will bring about more serious epileptic seizures. Our work was supported by that of Cao et al. [18], which ascribed the antiepileptic ability of ASIC3 to increased activity of inhibitory interneurons. Interestingly, ASIC1 was also reported to mediate the effects of decreased pH to end seizures [15]. On the other hand, a number of studies advocated a proconvulsant role for ASICs as described before [9,10,11,12,13,14]. Regarding this controversy, we believe there are three aspects that need to be addressed. First, aside from ASIC, amiloride inhibits other channels such as Na+/Ca2+ and Na+/H+ exchangers; therefore, blockade experiments cannot conclude whether and which ASICs are involved in epilepsy, let alone whether ASICs are proconvulsant or anticonvulsant. Second, different models of epilepsy were employed, which may influence the consistency of outcomes. Finally, each ASIC may exert a different function rather than the same, in which case it would not be surprising that some contribute to development of seizure while the others offer protection.
In accord with the abovementioned behavioral assays, APETx2 increased neuronal excitability as evidenced by more spontaneous and evoked action potentials spikes in Mg2+-free epileptiform discharge model. The frequent discharges of neurons induced by inhibition of ASIC3 could thus contribute to seizure onset and may eventually develop into epileptogenesis. These in vitro results further consolidate the role for ASIC3 in regulating neuronal excitability and seizure phenotype.
Our research reveals a previously unrecognized regulatory effect whereby ASIC3 inhibition could exacerbate seizure by promoting NMDAR function followed by activation of the CaMKII/CREB signaling pathway. Overstimulation of glutamatergic synaptic transmission and activation of glutamate receptors play a pivotal role in the etiology of epileptic seizure [35]. Among ionotropic glutamate receptors, NMDAR and AMPAR are crucial regulators of glutamatergic synaptic transmission. NMDARs consist of the obligatory subunit, NR1, and at least two main modulatory subunits, NR2A and NR2B. Increased expression of cell surface NMDARs is implicated in prolonged seizure and consequent neuronal damage, and NMDAR blockade can protect against SE and excitotoxicity [36,37,38,39]. In the present study, a significant increase in NR1, NR2A and NR2B membrane expression was observed in the pilocarpine group compared with the control. ASIC3 inhibition by APETx2 further increased the membrane expression level of NMDAR subunits. Meanwhile, upregulation of NMDAR protein level by ASIC3 inhibition contributes to NMDAR mediated excitatory neurotransmission. In contrast, membrane expression level of GluR1 did not change after APETx2 treatment. This selective change in NMDAR level suggests that ASIC3 likely influences the level of NMDAR subunits on the cell membrane. ASIC3 inhibition may accelerate NMDAR subunits trafficking from the intracellular pool to the surface and thus increase their surface expression, which could increase seizure susceptibility. Additionally, reciprocal Co-IP confirmed the interaction between ASIC3 and NMDAR subunits, and this may be the basis of regulation of NMDARs by ASIC3. Moreover, NMDARs blockade by AP5 prevented the aggravated epileptic behaviors caused by ASIC3 inhibition, which further demonstrated that the NMDARs activity was necessary for ASIC3 regulation of epilepsy. In other words, NMDARs mediated the worsening seizures induced by decreased function of ASIC3.
A point deserving attention is that the significant difference in NR1 disappeared at 24-h time point of SE, while other differences remained significant. A likely reason for this is that NR1 is not the direct protein that ASIC3 regulates. The change in NR1 is possibly subsequent to ASIC3 mediated regulation of other NMDAR subunits such as NR2A and NR2B, so the increase in NR1 is not sufficiently profound to maintain significance at 24-h. With regard to the positive result of interaction between ASIC3 and NR1 in the CO-IP experiment, we should be aware that both direct and indirect protein interaction can be detected by CO-IP. The identity of the subunit directly regulated by ASIC3 remains to be determined in our future studies. Alternatively, another possible explanation involves the exact function and population of NR1 that are detected. In the immunoblot analysis, the total number of membrane NR1 molecules is measured, regardless of their functional state or cellular location. However, the NR1 molecules that truly play a role in epileptic seizure are those that are on synapses and functional. Therefore, although the level of NR1 measured by immunoblot is not significantly different between the seizure group and the APETx2 treated seizure group at 24-h time point of SE, its function and subcellular location are not known. Importantly, despite the lack of a significant difference in NR1 at this time point, the overall biochemical and electrophysiological results are basically consistent, both showing an enhancing effect of ASIC3 inhibition on NMDARs.
Activation of NMDA receptors increases the intracellular calcium level, which can activate many calcium-dependent enzymes including CaMKII [40]. Once activated, CaMKII undergoes autophosphorylation and phosphorylates various substrates such as CREB [41, 42]. CREB is a transcription factor, and its phosphorylation gives rise to induction of immediate early genes. CREB plays an important role in epilepsy, whose activation occurs in rodent epilepsy models and in the seizure onset zone of patients with neocortical epilepsy [43, 44]. Suppressing CREB activity shortens the duration of SE and reduces the number of spontaneous seizures in the pilocarpine model of epilepsy [45, 46]. In our study, APETx2 administration resulted in enhanced CaMKII and CREB phosphorylation in seizure rats, which correlated with an increase in NMDAR function. These findings suggest for the first time the involvement of the NMDAR/CaMKII/CREB signaling pathway in the increased seizure susceptibility caused by ASIC3 inhibition.
This study has certain limitations. Owing to the lack of a selective ASIC3 agonist, we cannot observe the direct influence of that protein on seizure but merely infer from the effects of the antagonist. Thus, it is necessary to develop a selective ASIC3 agonist or use ASIC3 transgenic animals to obtain a more comprehensive understanding of the function of ASIC3 in seizure and even epilepsy.
In conclusion, our work provides new evidence that ASIC3 inhibition aggravates seizure by upregulating NMDAR function and subsequent CaMKII/CREB signaling. This effect implies that ASIC3 functions as part of an endogenous anticonvulsant system and may emerge as a new target for prevention and control of seizure.
References
McNamara JO (1999) Emerging insights into the genesis of epilepsy. Nature 399:A15–A22
French JA (2007) Refractory epilepsy: clinical overview. Epilepsia 48(Suppl 1):3–7
Somjen GG (1984) Acidification of interstitial fluid in hippocampal formation caused by seizures and by spreading depression. Brain Res 311:186–188
Wang RI, Sonnenschein RR (1955) PH of cerebral cortex during induced convulsions. J Neurophysiol 18:130–137
Boscardin E, Alijevic O, Hummler E, Frateschi S, Kellenberger S (2016) The function and regulation of acid-sensing ion channels (ASICs) and the epithelial Na(+) channel (ENaC): IUPHAR review 19. Br J Pharmacol 173:2671–2701
Wu W, Cheng C, Sun W, Wong C, Chen C (2012) Targeting ASIC3 for pain, anxiety, and insulin resistance. Pharmacol Ther 134:127–138
Biagini G, Babinski K, Avoli M, Marcinkiewicz M, Seguela P (2001) Regional and subunit-specific downregulation of acid-sensing ion channels in the pilocarpine model of epilepsy. Neurobiol Dis 8:45–58
Wu H, Wang C, Liu B, Li H, Zhang Y, Dong S, Gao G, Zhang H (2015) Altered expression pattern of acid-sensing ion channel isoforms in piriform cortex after seizures. Mol Neurobiol 53:1782–1793
Ali A, Pillai KP, Ahmad FJ, Dua Y, Vohora D (2006) Anticonvulsant effect of amiloride in pentetrazole-induced status epilepticus in mice. Pharmacol Rep 58:242–245
N’Gouemo P (2008) Amiloride delays the onset of pilocarpine-induced seizures in rats. Brain Res 1222:230–232
Ali A, Ahmad FJ, Pillai KK, Vohora D (2004) Evidence of the antiepileptic potential of amiloride with neuropharmacological benefits in rodent models of epilepsy and behavior. Epilepsy Behav 5:322–328
Ali A, Ahmad FJ, Pillai KK, Vohora D (2005) Amiloride protects against pentylenetetrazole-induced kindling in mice. Br J Pharmacol 145:880–884
Ievglevskyi O, Isaev D, Netsyk O, Romanov A, Fedoriuk M, Maximyuk O, Isaeva E, Akaike N, Krishtal O (2016) Acid-sensing ion channels regulate spontaneous inhibitory activity in the hippocampus: possible implications for epilepsy. Philos Trans R Soc Lond B Biol Sci 371:20150431
Xiong ZG, Pignataro G, Li M, Chang SY, Simon RP (2008) Acid-sensing ion channels (ASICs) as pharmacological targets for neurodegenerative diseases. Curr Opin Pharmacol 8:25–32
Ziemann AE, Schnizler MK, Albert GW, Severson MA, Howard MR, Welsh MJ, Wemmie JA (2008) Seizure termination by acidosis depends on ASIC1a. Nat Neurosci 11:816–822
Qadri YJ, Rooj AK, Fuller CM (2012) ENaCs and ASICs as therapeutic targets. Am J Physiol Cell Physiol 302:C943–C965
Waldmann R, Bassilana F, de Weille J, Champigny G, Heurteaux C, Lazdunski M (1997) Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J Biol Chem 272:20975–20978
Cao Q, Wang W, Gu J, Jiang G, Wang K, Xu Z, Li J, Chen G, Wang X (2014) Elevated expression of acid-sensing ion channel 3 inhibits epilepsy via activation of interneurons. Mol Neurobiol 53:485–498
Karczewski J, Spencer RH, Garsky VM, Liang A, Leitl MD, Cato MJ, Cook SP, Kane S, Urban MO (2010) Reversal of acid-induced and inflammatory pain by the selective ASIC3 inhibitor, APETx2. Br J Pharmacol 161:950–960
Kawai N, Sakai N, Okuro M, Karakawa S, Tsuneyoshi Y, Kawasaki N, Takeda T, Bannai M, Nishino S (2015) The sleep-promoting and hypothermic effects of glycine are mediated by NMDA receptors in the suprachiasmatic nucleus. Neuropsychopharmacology 40:1405–1416
Lei LG, Sun S, Gao YJ, Zhao ZQ, Zhang YQ (2004) NMDA receptors in the anterior cingulate cortex mediate pain-related aversion. Exp Neurol 189:413–421
Zhang Y, Li Z, Gu J, Zhang Y, Wang W, Shen H, Chen G, Wang X (2015) Plic-1, a new target in repressing epileptic seizure by regulation of GABAAR function in patients and a rat model of epilepsy. Clin Sci 129:1207–1223
Deval E, Friend V, Thirant C, Salinas M, Jodar M, Lazdunski M, Lingueglia E (2006) Regulation of sensory neuron-specific acid-sensing ion channel 3 by the adaptor protein Na+/H+ exchanger regulatory factor-1. J Biol Chem 281:1796–1807
Nagarkatti N, Deshpande LS, Carter DS, DeLorenzo RJ (2010) Dantrolene inhibits the calcium plateau and prevents the development of spontaneous recurrent epileptiform discharges following in vitro status epilepticus. Eur J Neurosci 32:80–88
Deshpande LS, Nagarkatti N, Ziobro JM, Sombati S, DeLorenzo RJ (2008) Carisbamate prevents the development and expression of spontaneous recurrent epileptiform discharges and is neuroprotective in cultured hippocampal neurons. Epilepsia 49:1795–1802
Diochot S, Baron A, Rash LD, Deval E, Escoubas P, Scarzello S, Salinas M, Lazdunski M (2004) A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO J 23:1516–1525
Zhu Y, Wienecke CFR, Nachtrab G, Chen X (2016) A thalamic input to the nucleus accumbens mediates opiate dependence. Nature 530:219–222
Parker PRL, Lalive AL, Kreitzer AC (2016) Pathway-specific remodeling of thalamostriatal synapses in parkinsonian mice. Neuron 89:734–740
Lewitus GM, Konefal SC, Greenhalgh AD, Pribiag H, Augereau K, Stellwagen D (2016) Microglial TNF-α suppresses cocaine-induced plasticity and behavioral sensitization. Neuron 90:483–491
Liang JJ, Huang LF, Chen XM, Pan SQ, Lu ZN, Xiao ZM (2015) Amiloride suppresses pilocarpine-induced seizures via ASICs other than NHE in rats. Int J Clin Exp Pathol 8:14507–14513
Kellenberger S, Schild L (2002) Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev 82:735–767
Sombati S, Delorenzo RJ (1995) Recurrent spontaneous seizure activity in hippocampal neuronal networks in culture. J Neurophysiol 73:1706–1711
Velisek L, Dreier JP, Stanton PK, Heinemann U, Moshe SL (1994) Lowering of extracellular pH suppresses low-Mg(2+)-induces seizures in combined entorhinal cortex-hippocampal slices. Exp Brain Res 101:44–52
Mitchell WG, Grubbs RC (1956) Inhibition of audiogenic seizures by carbon dioxide. Science 123:223–224
Barker-Haliski M, White HS (2015) Glutamatergic mechanisms associated with seizures and epilepsy. Cold Spring Harb Perspect Med 5:a22863
Naylor DE, Liu H, Niquet J, Wasterlain CG (2013) Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol Dis 54:225–238
Ghasemi M, Schachter SC (2011) The NMDA receptor complex as a therapeutic target in epilepsy: a review. Epilepsy Behav 22:617–640
Rice AC, DeLorenzo RJ (1998) NMDA receptor activation during status epilepticus is required for the development of epilepsy. Brain Res 782:240–247
Di Maio R, Mastroberardino PG, Hu X, Montero L, Greenamyre JT (2011) Pilocapine alters NMDA receptor expression and function in hippocampal neurons: NADPH oxidase and ERK1/2 mechanisms. Neurobiol Dis 42:482–495
Wang H, Peng RY (2016) Basic roles of key molecules connected with NMDAR signaling pathway on regulating learning and memory and synaptic plasticity. Mil Med Res 3:26
Steven A, Seliger B (2016) Control of CREB expression in tumors: from molecular mechanisms and signal transduction pathways to therapeutic target. Oncotarget 7:35454–35465
Johannessen M, Delghandi MP, Moens U (2004) What turns CREB on? Cell Signal 16:1211–1227
Lund IV, Hu Y, Raol YH, Benham RS, Faris R, Russek SJ, Brooks-Kayal AR (2008) BDNF selectively regulates GABAA receptor transcription by activation of the JAK/STAT pathway. Sci Signal 1:a9
Rakhade SN, Yao B, Ahmed S, Asano E, Beaumont TL, Shah AK, Draghici S, Krauss R, Chugani HT, Sood S, Loeb JA (2005) A common pattern of persistent gene activation in human neocortical epileptic foci. Ann Neurol 58:736–747
Zhu X, Dubey D, Bermudez C, Porter BE (2015) Suppressing cAMP response element-binding protein transcription shortens the duration of status epilepticus and decreases the number of spontaneous seizures in the pilocarpine model of epilepsy. Epilepsia 56:1870–1878
Zhu X, Han X, Blendy JA, Porter BE (2012) Decreased CREB levels suppress epilepsy. Neurobiol Dis 45:253–263
Acknowledgements
This work was funded by National Natural Science Foundation of China (81471133), Natural Science Foundation of Hubei Province (2014CFB734), Health and Family Planning Commission of Hubei Province scientific research project (WJ2015MA007), and Wuhan Science and Technology Bureau scientific research project (2015060101010047).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Cao, Q., Xiao, ZM., Wang, X. et al. Inhibition of Acid Sensing Ion Channel 3 Aggravates Seizures by Regulating NMDAR Function. Neurochem Res 43, 1227–1241 (2018). https://doi.org/10.1007/s11064-018-2540-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-018-2540-9