
Abstract
The piriform cortex (PC) is highly susceptible to chemical and electrical seizure induction. Epileptiform activity is associated with an acid shift in extracellular pH, suggesting that acid-sensing ion channels (ASICs) expressed by PC neurons may contribute to this enhanced epileptogenic potential. In epileptic rats and surgical samples from patients with medial temporal lobe epilepsy (TLE), PC layer II ASIC1a-immunopositive neurons appeared swollen with dendritic elongation, and there was loss of ASIC1a-positive neurons in layer III, consistent with enhanced vulnerability to TLE-induced plasticity and cell death. In rats, pilocarpine-induced seizures led to transient downregulation of ASIC1a and concomitant upregulation of ASIC2a in the first few days post-seizure. These changes in expression may be due to seizure-induced oxidative stress as a similar reciprocal change in ASIC1a, and ASIC2a expression was observed in PC12 cells following H2O2 application. The proportion of ASIC1a/ASIC2a heteromers was reduced in the acute phase following status epilepticus (SE) but increased during the latent phase when rats developed spontaneous seizures. Knockdown of ASIC2a by RNAi reduced dendritic length and spine density in primary neurons, suggesting that seizure-induced upregulation of ASIC2a contributes to dendritic lengthening in PC layer II in rats. Administration of the ASIC inhibitor amiloride before pilocarpine reduced the proportion of rats reaching Racine level IV seizures, protected layer II and III neurons, and prolonged survival in the acute phase following SE. Our findings suggest that ASICs may enhance susceptibility to epileptogenesis in the PC. Inhibition of ASICs, particularly ASIC2a, may suppress seizures originating in the PC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Temporal lobe epilepsy (TLE) is the most common partial seizure syndrome and can involve many limbic structures including the piriform cortex (PC). PC is the largest area of the mammalian olfactory cortex and is extensively interconnected with the other major limbic regions, the amygdaloid nucleus, entorhinal cortex, and hippocampus. The PC has been implicated in olfaction and memory processing and also in the spread of excitatory waves during ictogenesis. Pioneering investigations using the kindling protocol confirmed the amygdala and PC as major epileptogenic areas [1], but the specific circuit and electrophysiological features that confer susceptibility to seizure induction are still not completely clear. During and following seizures, large quantities of lactic acid and glutamic acid are released into the extracellular space, resulting in an acid environment that activates acid-sensing ion channels (ASICs) [2, 3]. We speculated that the epileptic susceptibility of the PC may be associated with activation of excitatory ASICs.
ASICs are multimeric proton-gated channels belonging to the superfamily of degenerins/epithelial sodium channels (DEG/ENaC). Three channel subunits have been cloned in the mammalian central nervous system (CNS), ASIC1a, ASIC2a, and ASIC2b, while ASIC1b and ASIC3 subunits are almost exclusively localized or enriched in the peripheral nervous system [4, 5]. These channels are proposed to function at multiple subcellular locations, including dendritic spines and synapses [6, 7]. Central neurons express both homotrimeric and heterotrimeric ASICs. In vitro, ASIC trimers are composed of ASIC1a homotrimers, ASIC2a homotrimers, and 2:1 or 1:2 ASIC1a/ASIC2a heteromers [8]. The assembly ratio depends on the cRNA injection ratio in vitro, indicating random aggregation. The ASIC1a subunit has a major influence on current amplitude, while ASIC2a influences kinetics. Homotrimeric ASIC2a and ASIC1a/2a heterotrimeric channels recover more rapidly than ASIC1a homomultimers following a shift to pH 5 and so may induce repetitive depolarization [9, 10]. The ASIC2a subunit also influences the ASIC response to FRRFamide (Phe-Met-Arg-Phe amide) and zinc and targets ASIC1a to the synapse via an association with PSD-95 [11, 12].
ASICs may participate in the pathogenesis of cerebral ischemia and amyotrophic lateral sclerosis (ALS). Blockade or knockout of ASIC1a protected the brain from ischemic injury [13, 14], while ALS model mice with ASIC1a deficiency exhibited delayed onset and slower progression of motor dysfunction [15]. The expression and function of ASICs has also being linked to seizures and epileptogenesis. Channels with ASIC1a or ASIC2a generate excitatory currents in response to decreasing pH in vitro. In vivo, production of lactic acid during intense neural activity and seizures decreases extracellular pH which activates ASIC1a [2, 3, 16]. Mossy fiber sprouting from dentate gyrus (DG) granule neurons, a neuroplastic response to temporal lobe seizures involved in progression to recurrent spontaneous seizures, increases the release of Zn2+ within the DG and hippocampus, which could enhance ASIC2a-containing channel currents [17, 18]. Moreover, the frequency of the ASIC1 gene rs844347-A allele is higher in patients with TLE than in healthy controls [19]. ASIC blocker amiloride delayed the onset of pilocarpine-induced seizures and reduced both seizure frequency and myoclonic jerks [20, 21]. Conversely, activation of ASIC1a channels expressed on inhibitory interneurons decreased seizure severity and duration [22].
We speculate that epileptogenesis in the PC is associated with the expression levels and subunit combinations of ASIC1a and ASIC2a. To data, there are no published studies on the ASICs related to the susceptivity of PC. In this study, we found that ASIC1a-expressing PC layer II/III neurons in human TLE patients and rats were particularly vulnerable to damage. We also demonstrated that the expression levels of ASIC1a and ASIC2a, as well as the combination pattern, were changed in the rat PC after seizures. Furthermore, blockade of ASICs reduced the loss of PC neurons and pilocarpine-induced seizures in rats, suggesting that targeted inhibition of PC ASICs, particularly ASIC2a, may suppress TLE.
Materials and Methods
Ethics Statement
The present study was performed in accordance with the Guidelines for Animal Experimentation of the Fourth Military Medical University and the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) revised in 1996. The protocol and animal use for this study was approved by the Ethics Committee for Animal Experimentation of the Fourth Military Medical University. All efforts were made to minimize the number of animals and their suffering.
All patients gave written informed consent prior to participation in this study. The cases examined in the study were treated surgically for intractable TLE by the Department of Neurosurgery, Tangdu Hospital, Fourth Military Medical University. Eight specimens were randomly selected from an epilepsy brain tissue bank. The criteria, informed consent, and brain tissue processing were referenced in our previous publications [23]. The individual patient details are listed in Supplement Tables 1 and 2.
Animals and Treatments
Adult male Sprague–Dawley rats (200–240 g body weight) were obtained from the Experimental Animal Center of the Fourth Military Medical University. They were housed under standard environmental conditions (temperature 22 ± 1 °C, humidity 50–60 %, and 12-h light/dark cycle). The experimental procedures were approved by the Committee of Animal Use for Research and Education of the Fourth Military Medical University. Lithium chloride (125 mg/kg, intraperitoneal injection (i.p.), Sigma, St. Louis, MO, USA) was injected 24 h prior to injection of pilocarpine (40 mg/kg, i.p., Sigma). All experimental animals received injections of 1 ml 0.9 % NaCl (i.p.) immediately following a seizure and twice on the day following a seizure to prevent dehydration. The control animals were treated with saline. Methyl scopolamine (1 mg/kg in 0.9 % saline) was intraperitoneally administered 30 min before pilocarpine administration to minimize the peripheral effects of pilocarpine. There were eight animals per group in this experimental. Control rats were treated identically except they were injected with normal saline instead of pilocarpine. All animals received special care until they were sacrificed. The evoked behavioral seizures were scored using Racine’s scale. We used Racine stages (I ~ V) as a quantifiable means to describe seizure intensities: stage I, mouth and facial movement; stage II, head nodding; stage III, forelimb clonus; stage IV, rearing with forelimb clonus; and stage V, rearing and falling with forelimb clonus. Racine stage IV was recognized as the statistic criterion in this experiment. Animals were treated by diazepam (10 mg/kg, i.p.) to end the status epilepticus (SE) after at least 60 min. In the oxidative stress experimental, the animals were treated by diazepam (10 mg/kg) to end the SE. At the end of the scheduled time intervals from pilocarpine injection, the part of the animals were sacrificed by decapitation, and brains were rapidly extracted for the Western blot analysis, another animals were perfused and fixed by the paraformaldehyde for the immunohistochemistry analysis.
Tissue Preparation
Human
Immediately upon resection in the operating room, the tissues from patients with intractable epilepsy and control were rinsed in PBS. Tissue blocks (0.5–1.0 cm3) for immunohistochemistry (IHC) were fixed in 4 % paraformaldehyde for 4 h and then embedded in paraffin following standard laboratory procedures. The paraffin-embedded brains were cut into 5 μm thick slices for IHC for immunochemical staining. Samples for Western blotting were flash frozen on powdered dry ice and stored at −70 °C.
Rats
Various time points following pilocarpine-evoked seizures, six rats from each group were sacrificed by decapitation after i.p. administration of a lethal dose of chloral hydrate. The six rat brains were dissected to collect both the piriform cortex, which were then flash frozen in liquid nitrogen and stored at −70 °C for qPCR and Western blot analysis.
Quantitative Reverse Transcription Polymerase Chain Reaction
The total RNA of tissue was extracted by TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. RNA was reverse transcribed by PrimeScript™ Double Strand cDNA Synthesis Kit (Takara, JP). The products were used as the template. Quantitative reverse transcription PCR (qPCR) was carried out with SYBR® Premix Ex Taq™ using the iQ5 multicolor real-time PCR detection system (Bio-Rad). The thermal profile was 50 °C for 30 min, 95 °C for 15 min, and then 40 cycles of 95 °C for 30 s, and 55 °C for 30 s. The primer sequences were as follows: ASIC1a forward, 5′-AAAGTGCCAGAAGGAGGCTAAG-3′; ASIC1a reverse, 5′-AGGTAGGATGTTGGCAGCGTA-3′; ASIC2a forward, 5′-ATCCTTGTGTTGTTTGTGACCG-3′; ASIC2a reverse, 5′-TGGGAAAAGAAGGAGGTTGGT-3′; β-actin forward, 5′-GATCCTGACCGAGCGTGGCTACA-3′; and β-actin reverse, 5′-ACGGATGTCAACGTCACACTTCA-3′.
Primary Neuron Culture
The pregnant rats from the Experimental Animal Center of the Fourth Military Medical University were sacrificed in a CO2 chamber according to current Fourth Military Medical University (Animal Use for Research and Education Committee) guidelines. The pregnant rats were sacrificed. Place the pups (E15) in the large culture dish. The brain was then released by using a microspatula. The cerebrum was separated from the cerebellum and brain stem, and the cerebral hemispheres were separated from each other by gently separating along the midline fissure. The meninges were gently peeled from the individual cortical lobes that were placed into a culture dish containing neurobasal media. After 0.125 % trypsin solution digestion, cortices were triturated gently with a 1-ml tip and dissociated into a cell suspension. Cells were plated in 60-mm cell culture dish at a concentration of 5 × 106 cells. Incubate the dish at 37 °C in a moist 5 % CO2, 95 % air atmosphere. The medium was changed every 72 h until the cells were ready to be used.
Antibodies and Reagents
The tissues from patients with TLE or epileptic rats were lysised by the lysis buffer containing EDTA-free protease inhibitor cocktail (Roche, CH). Immunoprecipitation was performed overnight at 4 °C using anti-ASIC1 (sc-13905, Santa Cruz, CA) and anti-ASIC2 (sc-22333, Santa Cruz, CA). The complex was captured with 20 μl of 25 % slurry of Protein A/G plus agarose beads (sc-2003, Santa Cruz, CA) for 4 h at 4 °C. Proteins were separated by 10 % SDS-PAGE and transferred onto PVDF (Millipore, USA) at 350 mA at 90 min. Nonspecific binding sites were blocked by immersing the PVDF membrane in 1 % blocking solution (TBST solution, 1 % BSA power). Proteins were probed with antibodies in 5 % nonfat milk: anti-ASIC1 (ab87514, Abcam, UK), anti-ASIC2a (ab169768 and ab77384, Abcam, UK), anti-ASIC1a (LS-C155923, LSBio, USA), anti-ASIC2a (LS-B4731, LSBio, USA), anti-beta-actin (04-1116, Millipore, USA), NeuN (ABN78, Millipore, USA), and DAPI (28718-90-3, Millipore, USA). The secondary antibodies included goat anti-rabbit IgG H&L (Alexa Fluor® 647) (ab150079, Abcam, UK), goat anti-mouse IgG H&L (Alexa Fluor® 488) (ab150113, Abcam, UK), goat anti-rabbit IgG conjugated to HRP (ab6721, Abcam, UK), and goat anti-mouse IgG conjugated to HRP (ab97023, Abcom, UK). The dendritic spines were labeled by DiI (468495, Sigma, USA).
Immunocytochemistry and Immunofluorescence
The rats were sacrificed and perfused with physiological saline and then fixed by 4 % paraformaldehyde. Fifteen micrometers thick sections were cut by freezing microtome. The sections were processed for immunostaining. The endogenous peroxidase activity was blocked by 1 % H2O2 in the PBS for 5 min. Nonspecific immunoglobulin binding of the tissue was blocked by 1 % bovine serum albumin in PBS. The sections were incubated with the primary antibodies overnight at 4 °C: ASIC1 (Abcam, UK) and ASIC2 (Abcam, UK). The sections were washed with PBS (3 × 10 min) and incubated with the goat anti-rat IgG HRP secondary antibody. DAB Color Developing Reagent Kit (Millipore, USA) was used. The images were acquired with a Nikon eclipse Ti microcopy.
The primary neurons were separated from the E15 rats and cultured in the polylysine coating sterile glasses for 8 days. The cells were washed with PBS (3 × 10 min) and fixed with 4 % paraformaldehyde. Before immunostaining, the cells were washed with PBS (3 × 10 min) and stained by ASIC2 (Abcam, UK) and NeuN (Millipore, USA) overnight at 4 °C. Then, the cells were washed with PBS (3 × 15 min) and incubated with Goat Anti-Rabbit IgG H&L (Alexa Fluor® 647) and Goat Anti-Mouse IgG H&L (Alexa Fluor® 488) (Abcam, USA) for 4 h at room temperature. Confocal images were acquired with a Nikon C2 confocalmicrocopy (Nikon, JP).
The primary neurons were cultured for 2 weeks and then rinsed in PBS. Cells were fixed in 4 % paraformaldehyde for 30 min and then washed with PBS (3 × 10 min). The cells were stained with DiI overnight at 4 °C and then washed with PBS (3 × 15 min). Confocal images were acquired with a Nikon C2 confocalmicrocopy (Nikon, JP).
Western Blotting
Total proteins from tissues or cell extracts (50 μg/gel lane) were separated by SDS-PAGE, blotted, and probed with anti-ASIC1a, anti-ASIC2a, and anti-β-actin, and then probed with secondary antibodies (goat anti-mouse IgG conjugated to HRP and goat anti-rabbit IgG conjugated to HRP). The images were collected by the ChemiDoc XRS+ (Bio-Rad, USA).
Co-immunoprecipitation
The tissue of pyriform cortex was obtained from the pilocarpine-treated rats and control rats in RIPA buffer (150 mM NaCl, 20 mM Tris–HCl, pH 7.4, 5 mM EDTA, 1 % NP-40, 1 % Na-deoxycholate, 0.1 % SDS, 1 mM PMSF, 20 μg/ml leupeptin, 20 μg/ml aprotinin, and 3 μg/ml pepstatin A). Lysates were incubated overnight with ASIC1a and ASIC2a antibody before being absorbed with protein A/G PLUS-agarose beads. Precipitated immunocomplexes were released by boiling with 2× SDS electrophoresis sample buffer and prepared for Western blot analysis.
Behavior Analysis After the Oral Administration of Amiloride
Amiloride was chosen as ASICs inhibitor against LiCl-pilocarpine-induced seizures. Amiloride was dissolved in water. The rats were randomly divided into two groups (control group and treated group). Each group had 20 rats. The treated group was injected by amiloride (30 mg/kg) 90 min before injection of pilocarpine (40 mg/kg). The control group was only administered by physiologic saline before the injection of pilocarpine. The behaviors of seizures were observed. The time from the end of pilocarpine injection to appearance of Racine stages IV was recorded. Data analysis of the time to Racine stages IV was performed using Student’s t test.
Statistical Analysis
All data are presented as the mean ± SEM (n = number of individual samples). Statistical analysis was performed using GraphPad Software. The statistical data were presented as follows. Paired t test was mainly applied in the analysis of the number of neurons in layers II and III in Fig. 1. Paired t test was also applied in the analysis of protein level of co-IP. In vitro experiments, the data came from the primary neuron culture experiments (n = 4). Paired t test was applied in the analysis of dendritic length and number of spines. One-way ANOVA (Dunnett’s multiple comparison test) was applied in the analysis of the differences of mRNA and protein expression. In the analysis of ratio of Racine IV of rats, chi-squared test was used (Fig. 7a). We defined statistical significance as P < 0.05.

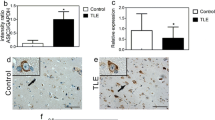
Abnormal morphology and reduced numbers of ASIC1a-positive neurons in epileptic tissues. a, b Sections from rats (n = 4). c, d Sections from the TLE patient (n = 4). All sections were immunostained for ASIC1a. The cortical layer II and III (II and III). Scale bar in a–d at 100 μm. Data in e–i are presented as mean ± standard error of mean (SEM). Student’s t test: *P < 0.05 compared to control
Results
Abnormal Morphology of ASIC1a-Positive Neurons in Tissue from Epileptic Rats and TLE Patients
The similar phenomena in piriform cortex from the epileptic rat and TLE patients included the decreased number of ASIC1a-positive cells in the layer III and the increased number of abnormal morphology in the SIC1a-positive cells (Fig. 1a–i). But, there were some differences between epileptic human and rats. In the cortical layer II from epileptic rats, the number of neurons with abnormally elongated dendrites increased (Fig. 1a, b, f). Compared with the rats, the neurons with abnormally elongated dendrites were not found in epileptic patients (Fig. 1d). The number of ASIC1a-positive neurons was higher in layer II of piriform cortex from TLE patients compared to controls (Fig. 1g), while individual layer II neurons exhibited altered morphology with swollen cell bodies (Fig. 1c, d, h). There were remaining neurons that exhibited aberrant dendritic structure and ill-defined somal outlines in layer III compared to controls (Fig. 1i).
Abnormal Morphology and Cell Loss in Rat Piriform Cortex After Status Epilepticus
To examine the relationship between ASICs expression and the epileptic susceptibility of piriform cortex, we measured ASIC1a expression levels in rats administered LiCl-pilocarpine, a well-established model of TLE. After intraperitoneal injection, rats experienced at least 1 h of status epilepticus (SE) and gradually developed spontaneous seizures after the acute and latent phases. We observed obvious tissue damage and redistribution of ASIC1a immunohistochemical staining in the PC at 2 weeks after SE (Fig. 2a). In PC layer II, the dendrites of semilunar and pyramidal cells appeared abnormally elongated, extending into layer I or III, while the dendrites of pyramidal cells and large multipolar cells in layer III shrank and even disappeared. Loss of layer III cells was greater than loss of layer II neurons (Fig. 2c). Unexpectedly, we observed no change in ASIC1a protein level in PC homogenate at 2 weeks after SE compared to control (Fig. 2b, d), although changes in the early phase following SE are still possible.

ASIC1a immunohistochemical staining in rat brain sections. a The left panel is a coronal section from a saline-treated (control) rat, and the right is a coronal section from a pilocarpine-treated rat at 2 weeks after status epilepticus, both immunostained for ASIC1a. In saline-treated control rats (n = 4), ASIC1a immunoreactivity did not differ between the piriform cortex and other areas. In pilocarpine-treated rats (n = 4), ASIC1a immunoreactivity was markedly stronger in the piriform cortex compared to other areas. b, d The protein level of ASIC1a did not differ significantly from control (Con) at 2 weeks (2 w) after pilocarpine treatment (n = 4). c Higher resolution photomicrograph of the demarcated region (right, region in black box) in a, right panel. Left is control. Layers of the piriform cortex are separated by black dashed curves. The dendrites of layer II neurons grew abnormally and extended into layer I. Karyopyknosis appeared in layer III. Scale bar in c–d: 200 μm. Data in d are presented as mean ± standard error of mean (SEM). Student’s t test: P > 0.05 compared to control
Downregulation of ASIC1a and Upregulation of ASIC2a in the Acute Phase
To determine whether ASIC expression changes in the acute phase after status epilepticus (before returning to control levels, Fig. 2b), we examined mRNA levels of ASIC1a and ASIC2a at 1, 6, 12, 24, 48, and 72 h post-SE. Expression of ASIC1a mRNA was downregulated at 24, 48, and 72 h after SE before gradually returning to control levels in latent phase (Fig. 3a and Supplemental Fig. 1a). In contrast, ASIC2a mRNA expression was upregulated 12, 24, 48, and 72 h after SE and then gradually declined to control levels in latent phase (Fig. 3b and Supplemental Fig. 1b). The protein expression patterns of ASIC2a were similar with the respective mRNA expression patterns (Fig. 3d and Supplemental Fig. 1d). Though mRNA and protein of ASIC1a gradually declined in the acute phase, the statistically significant differences of protein level were prior to mRNA level (Fig. 3c and Supplemental Fig. 1c).

The mRNA and protein expression levels of ASIC1a and ASIC2a in rat piriform cortex after pilocarpine-induced SE. a The expression of ASIC1a mRNA was 35 ± 6 % of control on 24 h post-SE, 44 ± 3 % on 48 h, and 45 ± 5 % on 72 h (n = 3 rats/group). b Expression of ASIC2a mRNA was 2.7 ± 0.5-fold higher than control on 12 h, 2.7 ± 0.3-fold on 24 h, 3.1 ± 0.5-fold on 48 h, and 2.5 ± 0.3-fold on 72 h (n = 3 rats/group). c Representative immunoblot and densitometric analysis of ASIC1a protein showing that expression was 39 ± 4 % of control on 24 h, 51 ± 8 % on 48 h, and 62 ± 3 % on 72 h (n = 3 rats/group). d Representative immunoblot and densitometric analysis of ASIC2a showing that protein expression was 1.7 ± 0.8-fold higher than control on 12 h, 1.8 ± 0.2-fold on 24 h, 1.9 ± 0.8-fold on 48 h, and 1.9 ± 0.3-fold on 72 h (n = 3 rats/group). e Representative immunoblot and densitometric analysis of ASIC1a protein showing that expression was 60 ± 7 % of control on 1 h and 52 ± 8 % on 2 h. f Representative immunoblot and densitometric analysis of ASIC2a protein showing that expression was 1.4 ± 0.1-fold higher than control on 1 h and 2.1 ± 0.1-fold on 2 h. Data in a–f are presented as mean ± standard error of mean (SEM). *P < 0.05 and **P < 0.01 by one-way ANOVA compared to the control
We also examined if the changes of ASIC1a and ASIC2a were associated with seizure duration by comparing expression in rats in which seizures were terminated by diazepam 1 or 2 h following pilocarpine-induced SE. After 24 h, ASIC1a protein expression decreased with SE duration in 2-h group compared with 1-h group (Fig. 3e). Conversely, the level of ASIC2a increased in 2-h group compared with 1-h group (Fig. 3f).
Oxidative Stress Regulates the Expression of ASIC1a and ASIC2a
Enhanced metabolism and other events associated with the brief or prolonged seizures, such as NO release, induce oxidative stress in epileptic tissue [24]. We examined if oxidative stress induces a similar reciprocal change in ASIC1a and ASIC2a expression by treating PC12 cells with different doses of H2O2 (50, 100, and 400 μM) and measuring ASIC1a and ASIC2a protein expression levels by Western blotting after 24 h. ASIC1a expression was progressively downregulated with H2O2 dose (Fig. 4a), while expression of ASIC2a was upregulated at the lowest dose (50 μM) but was downregulated by higher doses (100 and 200 μM) (Fig. 4b). Thus, at least, moderate oxidative stress induces these changes similar to those observed in piriform cortex 1 day after SE.

Changes in ASIC1a and ASIC2a protein expression in PC12 cells under oxidative stress. a Representative immunoblot and densitometric analysis showing that ASIC1a protein expression decreased progressively with H2O2 dose (64 ± 14 % of control in 50 μM, 53 ± 12 % in 100 μM, 43 ± 4 % in 400 μM, n = 3). b Representative immunoblot and densitometric analysis of ASIC2a expression showing that protein levels increased to 1.5 ± 0.06-fold in 50 μM but did not change significantly at higher doses (n = 3). c In pilocarpine-treated rats, ASIC2a expression increased with seizure duration. Protein level of ASIC2a increased to 1.4 ± 0.04-fold in 1-h group and 1.9 ± 0.26-fold in 2-h group compared with control groups (n = 3 rats/group). Data in a–c are presented as mean ± standard error of mean (SEM). *P < 0.05 and **P < 0.01 by one-way ANOVA compared to the control
Changes in ASIC Subunit Combinations After SE
The kinetic properties of ASICs depend on subunit composition (ASIC1a homotrimer, ASIC2a homotrimer, or ASIC1a/2a heterotrimer). To investigate whether seizures affect the subunit composition of ASICs, we conducted co-immunoprecipitation experiments. Expression of the ASIC1a−ASIC2a complex was lower 1 day after pilocarpine-induced SE (Fig. 5a, c) but higher in the latent phase 2 weeks post-SE (Fig. 5b, d).

Changes in expression of the ASIC1a−ASIC2a complex in piriform cortex of rats after SE. a Expression of the ASIC1a−ASIC2a complex was lower than control in the acute phase (1 day) following SE (n = 3 rats/group). b In the latent phase, expression of the ASIC1a−ASIC2a complex was higher than control (n = 3 rats/group). c Representative immunoblot and densitometric analysis of ASIC1a protein showing that expression was 24 ± 5 % of control. d Representative immunoblot and densitometric analysis of ASIC2a protein showing that expression was 4.1 ± 0.5-fold higher than control. Data in c and d are presented as mean ± standard error of mean (SEM). Student’s t test: *P < 0.05 compared to control
Knockdown of ASIC2a Interferes with Development of Dendrites and Dendritic Spines
ASIC1a and ASIC2a participate in synaptic plasticity [6]. ASIC2a targets ASIC1a to synapse via an association with PSD-95 [11]. To investigate whether upregulation of ASIC2a is involved in the abnormal dendritic morphology of layer II neurons, we interfered with ASIC2a expression in primary cortical neurons. Mean length of dendrites decreased about 20 % in the siRNA group compared to the control group (Fig. 6a, b). The mean number of dendritic spines per 10-μm length of dendrite was reduced about 59 % in the knockdown group (Fig. 6c, d).

Knockdown of ASIC2a interferes with the development of dendrites and dendritic spines in primary cortical neurons. a ASIC2a/NeuN double-labeling immunofluorescence in primary neurons. Higher resolution photomicrographs of the single cell were inserted (the demarcated region in white box) in a. b ASIC2a knockdown reduced mean dendritic length to 80 ± 2 % of control (n = 4). c, d ASIC2a knockdown reduced mean number of spines per 10 μm of dendritic length to 41 ± 7 % of control (n = 4). Scale bar in a at 100 μm. Scale bar in c at 10 μm. Data in b, d are presented as mean ± standard error of mean (SEM). *P < 0.05 by Student’s t test compared to control
Amiloride Prevents Neuronal Loss in PC Layer III after Seizures
To further investigate whether the abnormal expression of ASIC subunits in PC neurons confers vulnerability to seizures, we examined the effects of the ASCI inhibitor amiloride on seizure semiology and outcome following pilocarpine. Racine IV stage (rearing with forelimb clonus) was easy to discern and so was used as an index of seizure severity. The fraction of rats reaching Racine stage IV and the time to Racine IV were lower in amiloride-treated rats compared to the control pilocarpine group (Fig. 7a, b). Survival rate was also higher in the early phase (12–48 h) following pilocarpine in the group pretreated with amiloride (Fig. 7c). Finally, Nissl staining and immunohistochemistry revealed decreased loss of PC neurons and greater preservation of morphology in the amiloride-treated group (Fig. 7d, e).

Pharmacological inhibition of ASICs reduced seizure severity, protected piriform cortex neurons, and enhanced survival in the early period after pilocarpine treatment. a, b Amiloride (30 mg/kg; i.p.) was administered 90 min before pilocarpine (40 mg/kg; i.p.). a The proportion of rats with Racine IV motor seizures after pilocarpine treatment was lower in the amiloride-treated group (41.10 ± 6.75 %) than the control pilocarpine group (80.57 ± 3.38 %) (n = 3). Chi-square test: P value = 0.0012, chi-square = 10.52, df = 1. b The time interval from pilocarpine injection to Racine IV seizures was longer in the amiloride-treated group (32 ± 4 min) compared with control group (21 ± 1 min) (n = 7 rats/group). Student’s t test: *P = 0.024, t = 2.58, df = 12. c Amiloride enhanced survival in the acute phase. d Nissl staining showed that injury of the piriform cortex after seizures was attenuated in the amiloride-treated group (n = 3 rats/group). Scale bar in d at 50 μm. e In amiloride-treated rats, ASIC1a immunoreactivity was reduced in the piriform cortex compared to pilocarpine controls (n = 4 rats/group). Higher resolution photomicrographs of single cell were inserted (the demarcated region in black box) in e. Scale bar in e at 100 μm
Discussion
Our study provides evidence that ASICs contribute to seizure-induced damage of PC neurons and to the epileptogenic susceptibility of piriform cortex. This conclusion is based on the following lines of evidence. Piriform cortex neurons expressing ASIC2a appeared more sensitive to neuroplastic and destructive changes in human TLE patients. In a rat model of TLE, the patterns of ASIC1a and ASIC2a expression were altered by seizures, possibly due to seizure-associated oxidative stress. Among these changes was an increase in ASCI2a-containing channels in the latent phase, and ASCI2a RNAi knockdown experiments suggested that this might account for the abnormal dendritic morphology of PC layer II neurons during the latent phase when rats develop spontaneous seizures. Finally, the ASIC antagonist amiloride suppressed epileptogenesis, preserved PC neuronal morphology and viability, and enhanced survival in rats.
Piriform cortex is a highly epilepsy-prone region. Moreover, the PC projects directly to amygdala, lateral entorhinal cortex, and subiculum [25, 26], thereby allowing the PC to particulate in spread of seizure activity. HSP90 immunoreactivity is decreased in PC neurons at 12 h–4 weeks after SE [27], and HSP90 degradation is related to neuronal vulnerability to SE insult, which indicates that a loss of synaptic input from the dying neurons may be a signal to induce axonal sprouting and synaptic-circuit reorganization [28, 29]. Indeed, the PC is involved in the epileptogenic network of patients with focal epilepsies [30, 31]. Under pathophysiological condition, interneuronal plasticity results in a change in PC firing behavior. Cezar et al. reported loss of interneuron functional diversity in the PC after induction of experimental epilepsy [32], which could lead to hyperexcitability. In the present study, ASIC1a expression was reduced and ASIC2a expression elevated in the acute phase of TLE modeling by pilocarpine (1–3 days post-SE), accompanied by changes in dendritic architecture (elongation in layer II and shrinkage in layer III) and loss of layer III neurons. Layer II consists of semilunar cells and pyramidal cells thought to be glutamatergic, whereas layer III consists of pyramidal cells and large presumably GABAergic multipolar cells. Channels composed of ASIC2a only or containing at least one ASIC2a subunit recover more rapidly than ASIC1a homomultimers following a shift to pH 5 in vitro [9, 10]. We speculate that excitatory ASICs may recurrently open, inducing excitotoxicity in layer III and hyperexcitability and neuroplasticity in layer II. Loss of layer III neurons and elongation of layer II dendrites into other layers may disturb intrinsic inhibitory loops, resulting in epileptogenesis.
Seizures increase the frequency of future seizures, suggesting that neural processes or factors generated by seizures may facilitate epileptogenesis. Oxidative stress is one possible mechanism for “seizures begetting seizures.” Oxidative stress may damage key inhibitory interneurons. In addition, oxidative stress leads to protein degradation [33, 34]. In the kainic acid model of epilepsy, oxidative damage to proteins increased in the piriform cortex and hippocampus at 8 h post-treatment and returned to control levels by 48 h [35]. Reactive oxygen species (ROS) damage protein structure and increase proteolytic susceptibility. We show that the protein levels of ASIC1a and ASIC2a in PC12 cells were both reduced at higher doses of H2O2. Unexpectedly, however, ASIC2a protein was higher in PC12 cells under lower dose of H2O2, suggesting that ASIC2a expression may be upregulated by milder oxidative stress associated with seizures. Indeed, seizures activate certain stress-responsive transcription factors, such as NF-kappa B, HIF-1, and Sp-1 [36–38], the latter of which can bind to the ASIC2 promoter. The increase in Sp-1 activity is associated with long-term changes in hippocampal functional plasticity after KA-induced seizures, possibly resulting in ASIC2a upregulation. The increase in ASIC2 expression and ensuing increase in ASIC channels containing ASIC2a could lead to hyperexcitability and epileptogenesis in the acute phase, in turn triggering cell loss and changes in microcircuitry in the latent phase (such as death of layer III interneurons and ectopic projections of layer II dendrites) that confer spontaneous epileptogenic potential on the PC.
Inhibition of ASIC1a appears neuroprotective under certain pathological conditions [6, 10, 14]. The acidosis-induced death of motorneurons was reduced by inhibiting ASIC1a in an ALS model [15], and inhibition of ASIC1a endocytosis exacerbated injury caused by acidosis [39]. In a previous study [40], ASIC1a mRNA was reduced to approximately 43 % of baseline 24 h after pilocarpine treatment as evidenced by in situ hybridization, consistent with our findings using quantitative PCR. These results suggest that downregulation of ASIC1a following pilocarpine-induced SE may be a self-protective mechanism to prevent further seizures in the acute phase.
In contrast to ASIC1a, ASIC2a has not been linked directly to epilepsy. Unlike ASIC1a, the presence of ASIC2a has little effect on ASIC current amplitude but does influence desensitization, recovery from desensitization, pH sensitivity, and the response to modulatory agents such as zinc [10]. ASIC1a and ASIC2a subunits are randomly mixed and yield ASIC1a/ASIC2a heterotrimers together with ASIC1a and ASIC2a homotrimers in vitro [8]. Reducing ASIC1a expression increased the severity of chemoconvulsant-induced seizures [22], strongly suggesting that circuits with predominantly homotrimeric ASIC2a channels are more seizure-prone. Our study showed that channels containing ASIC2a increased following seizures. The higher proportion of ASIC2a-containing channels may promote neuronal loss in the PC. ASIC1a/ASIC2a heteromers recover more quickly from depolarization [10] and so may induce repeated depolarization, leading to excitotoxicity (in layer III) or dendritic and synaptic plasticity (in layer II) [41, 42], resulting in the formation of epilepsy-prone neural circuits.
In conclusion, our study implicates ASICs in the high epileptic susceptibility of the piriform cortex. Seizure-induced oxidative stress may alter ASCI subunit expression, resulting in enhanced expression of ASCI2a-containing channels that contribute to hyperexcitability, excitotoxicity, ectopic dendrite formation, and ultimately in spontaneous seizures. Inhibition of ASICs was neuroprotective in the acute phase after seizures. Thus, ASIC2a inhibitors are a potential therapeutic target for epilepsy. Further studies are needed to thoroughly unravel the role of ASICs in epilepsy.
References
Kelly ME, McIntyre DC (1996) Perirhinal cortex involvement in limbic kindled seizures. Epilepsy Res 26(1):233–243
Chen X, Kalbacher H, Grunder S (2005) The tarantula toxin psalmotoxin 1 inhibits acid-sensing ion channel (ASIC) 1a by increasing its apparent H+ affinity. J Gen Physiol 126(1):71–79. doi:10.1085/jgp.200509303
Ekholm A, Kristian T, Siesjo BK (1995) Influence of hyperglycemia and of hypercapnia on cellular calcium transients during reversible brain ischemia. Exp Brain Res 104(3):462–466
Chen CC, England S, Akopian AN, Wood JN (1998) A sensory neuron-specific, proton-gated ion channel. Proc Natl Acad Sci U S A 95(17):10240–10245
Waldmann R, Bassilana F, de Weille J, Champigny G, Heurteaux C, Lazdunski M (1997) Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J Biol Chem 272(34):20975–20978
Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E et al (2002) The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron 34(3):463–477
Alvarez de la Rosa D, Krueger SR, Kolar A, Shao D, Fitzsimonds RM, Canessa CM (2003) Distribution, subcellular localization and ontogeny of ASIC1 in the mammalian central nervous system. J Physiol 546(Pt 1):77–87
Bartoi T, Augustinowski K, Polleichtner G, Grunder S, Ulbrich MH (2014) Acid-sensing ion channel (ASIC) 1a/2a heteromers have a flexible 2:1/1:2 stoichiometry. Proc Natl Acad Sci U S A 111(22):8281–8286. doi:10.1073/pnas.1324060111
Benson CJ, Xie J, Wemmie JA, Price MP, Henss JM, Welsh MJ, Snyder PM (2002) Heteromultimers of DEG/ENaC subunits form H+-gated channels in mouse sensory neurons. Proc Natl Acad Sci U S A 99(4):2338–2343. doi:10.1073/pnas.032678399
Askwith CC, Wemmie JA, Price MP, Rokhlina T, Welsh MJ (2004) Acid-sensing ion channel 2 (ASIC2) modulates ASIC1 H+-activated currents in hippocampal neurons. J Biol Chem 279(18):18296–18305. doi:10.1074/jbc.M312145200
Zha XM, Costa V, Harding AM, Reznikov L, Benson CJ, Welsh MJ (2009) ASIC2 subunits target acid-sensing ion channels to the synapse via an association with PSD-95. J Neurosci Off J Soc Neurosci 29(26):8438–8446. doi:10.1523/JNEUROSCI.1284-09.2009
Askwith CC, Cheng C, Ikuma M, Benson C, Price MP, Welsh MJ (2000) Neuropeptide FF and FMRFamide potentiate acid-evoked currents from sensory neurons and proton-gated DEG/ENaC channels. Neuron 26(1):133–141
Pignataro G, Simon RP, Xiong ZG (2007) Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain J Neurol 130(Pt 1):151–158. doi:10.1093/brain/awl325
Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA et al (2004) Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 118(6):687–698. doi:10.1016/j.cell.2004.08.026
Behan AT, Breen B, Hogg M, Woods I, Coughlan K, Mitchem M, Prehn JH (2013) Acidotoxicity and acid-sensing ion channels contribute to motoneuron degeneration. Cell Death Differ 20(4):589–598. doi:10.1038/cdd.2012.158
Siesjo BK, von Hanwehr R, Nergelius G, Nevander G, Ingvar M (1985) Extra- and intracellular pH in the brain during seizures and in the recovery period following the arrest of seizure activity. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 5(1):47–57. doi:10.1038/jcbfm.1985.7
Baron A, Waldmann R, Lazdunski M (2002) ASIC-like, proton-activated currents in rat hippocampal neurons. J Physiol 539(Pt 2):485–494
Baron A, Schaefer L, Lingueglia E, Champigny G, Lazdunski M (2001) Zn2+ and H+ are coactivators of acid-sensing ion channels. J Biol Chem 276(38):35361–35367. doi:10.1074/jbc.M105208200
Lv RJ, He JS, Fu YH, Zhang YQ, Shao XQ, Wu LW, Lu Q, Jin LR et al (2011) ASIC1a polymorphism is associated with temporal lobe epilepsy. Epilepsy Res 96(1–2):74–80. doi:10.1016/j.eplepsyres.2011.05.002
N'Gouemo P (2008) Amiloride delays the onset of pilocarpine-induced seizures in rats. Brain Res 1222:230–232. doi:10.1016/j.brainres.2008.05.010
Tai KK, Truong DD (2013) Amiloride but not memantine reduces neurodegeneration, seizures and myoclonic jerks in rats with cardiac arrest-induced global cerebral hypoxia and reperfusion. PLoS One 8(4):e60309. doi:10.1371/journal.pone.0060309
Ziemann AE, Schnizler MK, Albert GW, Severson MA, Howard MA 3rd, Welsh MJ, Wemmie JA (2008) Seizure termination by acidosis depends on ASIC1a. Nat Neurosci 11(7):816–822. doi:10.1038/nn.2132
Liu B, Niu L, Shen MZ, Gao L, Wang C, Li J, Song LJ, Tao Y et al (2014) Decreased astroglial monocarboxylate transporter 4 expression in temporal lobe epilepsy. Mol Neurobiol 50(2):327–338. doi:10.1007/s12035-013-8619-z
Waldbaum S, Patel M (2010) Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res 88(1):23–45. doi:10.1016/j.eplepsyres.2009.09.020
Panuccio G, Sanchez G, Levesque M, Salami P, de Curtis M, Avoli M (2012) On the ictogenic properties of the piriform cortex in vitro. Epilepsia 53(3):459–468. doi:10.1111/j.1528-1167.2012.03408.x
Loscher W, Ebert U (1996) The role of the piriform cortex in kindling. Prog Neurobiol 50(5–6):427–481
Kim YJ, Kim JY, Ko AR, Kang TC (2013) Reduction in heat shock protein 90 correlates to neuronal vulnerability in the rat piriform cortex following status epilepticus. Neuroscience 255:265–277. doi:10.1016/j.neuroscience.2013.09.050
Dudek FE, Staley KJ (2011) The time course of acquired epilepsy: implications for therapeutic intervention to suppress epileptogenesis. Neurosci Lett 497(3):240–246. doi:10.1016/j.neulet.2011.03.071
Dingledine R, Varvel NH, Dudek FE (2014) When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv Exp Med Biol 813:109–122. doi:10.1007/978-94-017-8914-1_9
Centeno M, Vollmar C, Stretton J, Symms MR, Thompson PJ, Richardson MP, O'Muircheartaigh J, Duncan JS et al (2014) Structural changes in the temporal lobe and piriform cortex in frontal lobe epilepsy. Epilepsy Res 108(5):978–981. doi:10.1016/j.eplepsyres.2014.03.001
Demir R, Haberly LB, Jackson MB (2001) Epileptiform discharges with in-vivo-like features in slices of rat piriform cortex with longitudinal association fibers. J Neurophysiol 86(5):2445–2460
Gavrilovici C, Pollock E, Everest M, Poulter MO (2012) The loss of interneuron functional diversity in the piriform cortex after induction of experimental epilepsy. Neurobiol Dis 48(3):317–328. doi:10.1016/j.nbd.2012.07.002
Muller W, Bittner K (2002) Differential oxidative modulation of voltage-dependent K+ currents in rat hippocampal neurons. J Neurophysiol 87(6):2990–2995
Simon F, Varela D, Cabello-Verrugio C (2013) Oxidative stress-modulated TRPM ion channels in cell dysfunction and pathological conditions in humans. Cell Signal 25(7):1614–1624. doi:10.1016/j.cellsig.2013.03.023
Bruce AJ, Baudry M (1995) Oxygen free radicals in rat limbic structures after kainate-induced seizures. Free Radic Biol Med 18(6):993–1002
Schreck R, Albermann K, Baeuerle PA (1992) Nuclear factor kappa B: an oxidative stress-responsive transcription factor of eukaryotic cells (a review). Free Radic Res Commun 17(4):221–237
Rong Y, Baudry M (1996) Seizure activity results in a rapid induction of nuclear factor-kappa B in adult but not juvenile rat limbic structures. J Neurochem 67(2):662–668
Li J, Jiang G, Chen Y, Chen L, Li Z, Wang Z, Wang X (2014) Altered expression of hypoxia-Inducible factor-1alpha participates in the epileptogenesis in animal models. Synapse 68(9):402–409. doi:10.1002/syn.21752
Zeng WZ, Liu DS, Duan B, Song XL, Wang X, Wei D, Jiang W, Zhu MX et al (2013) Molecular mechanism of constitutive endocytosis of Acid-sensing ion channel 1a and its protective function in acidosis-induced neuronal death. J Neurosci Off J Soc Neurosci 33(16):7066–7078. doi:10.1523/JNEUROSCI.5206-12.2013
Biagini G, Babinski K, Avoli M, Marcinkiewicz M, Seguela P (2001) Regional and subunit-specific downregulation of acid-sensing ion channels in the pilocarpine model of epilepsy. Neurobiol Dis 8(1):45–58. doi:10.1006/nbdi.2000.0331
Hao J, Oertner TG (2012) Depolarization gates spine calcium transients and spike-timing-dependent potentiation. Curr Opin Neurobiol 22(3):509–515. doi:10.1016/j.conb.2011.10.004
Feldman DE (2012) The spike-timing dependence of plasticity. Neuron 75(4):556–571. doi:10.1016/j.neuron.2012.08.001
Acknowledgments
We sincerely thank the patients and their families for their participation and support in this study. This work was supported by the National Natural Science Foundation of China (grant numbers are 81271433 and 81471322).
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Hao Wu and Chao Wang contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplement Table 1
(DOCX 13 kb)
Supplement Table 2
(DOCX 13 kb)
Supplemental Fig. 1
The mRNA and protein expression levels of ASIC1a and ASIC2a in rat piriform cortex from acute phase to latent phase. A The expression of ASIC1a mRNA was 64±7% of control on day 1 post-SE and 67±5% on day 3 (n=4 rats/group). B Expression of ASIC2a mRNA was 3.6±0.7-fold higher than control on day 1 and 2.8±0.5-fold higher on day 3 (n=4 rats/group). C Representative immunoblot and densitometric analysis of ASIC1a protein showing that expression was 50±7% of control on day 1 and 66±7% of control on day 3 (n=3 rats/group). D Representative immunoblot and densitometric analysis of ASIC2a showing that protein expression was 1.9±0.1-fold higher than control on day 1 and 1.9±0.2-fold higher on day 3 (n=3 rats/group). Data in A-D are presented as mean ± standard error of mean (SEM). *P<0.05 and **P<0.01 by one-way ANOVA compared to the control. (GIF 40 kb)
Rights and permissions
About this article

Cite this article
Wu, H., Wang, C., Liu, B. et al. Altered Expression Pattern of Acid-Sensing Ion Channel Isoforms in Piriform Cortex After Seizures. Mol Neurobiol 53, 1782–1793 (2016). https://doi.org/10.1007/s12035-015-9130-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9130-5