
Abstract
Neuronal autophagy and inflammatory responses are important in the pathogenesis of traumatic brain injury (TBI), and toll-like receptor 4 (TLR4) may play an important role in the related molecular cascade. The present study investigated the protective effect of apocynin, an inhibitor of NADPH oxidase, in a TBI rat model and further examined neuronal autophagy and the TLR4-mediated pathway. Adult male Sprague-Dawley rats were subjected to controlled cortical impact injury and intraperitoneally injected with apocynin (50 mg/kg) immediately after the trauma. In addition to motor and behavioral studies, brain water content and histology analyses were performed. Expression of autophagy-related proteins as well as TLR4/NF-κB signaling and inflammatory mediators was analyzed. The apocynin treatment significantly attenuated TBI-induced motor and behavioral impairment, brain edema and neuronal damage in rats. Immunohistochemical and Western blot analyses revealed that apocynin treatment significantly reduced the expression of NOX2, LC3 and Beclin1 in the hippocampus at 12–48 h after injury. Double immunolabeling demonstrated that apocynin decreased the co-localization of LC3 or TLR4-positive cells with hippocampal neurons at 24 h following TBI. In addition, CD11b (microglial marker) and GFAP (astrocyte marker)-immunopositive cells were also clearly decreased in hippocampal tissues. Meanwhile, protein levels of TLR4, NF-κB p65, TNF-α and IL-1β were found to be significantly downregulated by Western blot analysis. In conclusion, our findings indicate that the protective effects of apocynin may be related to modulation of neuronal autophagy and the TLR4/NF-κB signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI) is a major cause of death and disability in young individuals and is a critical public health problem in current society [1]. TBI begins with primary injury and gradually develops into secondary brain injury, resulting from a series of complex molecular cascade events that eventually lead to brain edema, neuron death and impairment of movement and cognitive function. Current experimental and clinical studies are focused on the neuroprotection following TBI, with the purpose of preventing secondary injury and improving functional outcomes [2]. However, the development of a safe and efficient treatment for TBI is greatly complicated by the existence of a highly complex injury-induced environment. Therefore, despite various strategies that have been employed to understand the molecular basis of TBI, no fully effective treatments for TBI are available to date.
Substantial evidence indicates that neuronal autophagy and inflammation play an important role in the secondary injury after TBI [3,4,5]. Autophagy is a regulated cellular pathway for the turnover of organelles and proteins by lysosomal-dependent processing. The autophagy pathway is involved in the pathophysiological process of brain injury, although controversy remains as to whether this process is protective or detrimental after injury [6]. Our previous study demonstrated that TBI activates autophagy and exacerbates neuronal damage, and suppression of neuronal autophagy by ceftriaxone and resveratrol can contribute to neuroprotection in the hippocampus of the rat brain [4, 7]. On the other hand, neuroinflammation is largely described in the acute phase after brain injury, frequently associated with blood brain barrier (BBB) dysfunction and followed by cerebral edema. Toll-like receptors (TLRs) play a vital role in the induction of inflammatory reaction and production of inflammatory mediators [8]. In particular, TLR4 and the downstream nuclear factor-kappa B (NF-κB) signaling pathway, which mediate the inflammatory reaction in brain injury or stroke, were demonstrated to be involved in the extension of cerebral infarction and the aggravation of traumatic brain damage [9, 10]. Interestingly, inhibiting the expression of inflammatory mediators by the TLR4/NF-κB pathway has been demonstrated to provide neuroprotection against TBI [5, 7].
Nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) is a multiunit enzyme composed of several subunits that include several isoforms of NOX1–5 [11]. A previous study demonstrated that NOX2, a catalytic subunit of NOX, is localized to the cerebral cortex and hippocampal CA1 subregion [12]. Experimental evidence suggests that over-activated NOX2 significantly contributes to oxidative damage to neurons and other cell types in ischemic and traumatic conditions [13, 14]. Apocynin, a natural organic compound isolated from the root extract of Picrohiza kurroa, has been used as an efficient inhibitor of the NADPH-oxidase complex [15]. Recently, apocynin was reported to protect against oxidative stress (ROS) production, microglial activation, BBB disruption and neuronal death after TBI in rats [16]. Moreover, treatment with apocynin also attenuates the expression and activation of the NOX2 protein, reducing brain edema and spatial learning deficits in TBI rats [14]. However, its role in the pathophysiology of TBI is not well understood. Specifically, whether the neuroprotection of apocynin is involved in neuronal autophagy via suppression of the inflammatory cascade after TBI remains underexplored to date.
Here, we generated a rat model of TBI and performed intraperitoneal (i.p.) injections of apocynin (50 mg/kg) immediately after trauma. The present study was designed to assess the protective effect of apocynin on post-TBI motor function, neurological impairment and brain edema in a rat model of TBI. We further examined whether apocynin could attenuate brain damage via suppression of neuronal autophagy and downregulation of the inflammatory cascade (TLR4/NF-κB signaling pathway) in the hippocampus following TBI in rats.
Materials and Methods
Animals and TBI Model
Adult male Sprague-Dawley rats (age 10–12 weeks; weight 300–330 g; Shijiazhuang, China) were used in this study. All experimental procedures were approved by the Animal Care and Use Committee of Hebei Medical University, and they conformed to the guidelines of the Chinese Council on Animal Protection. The rats were housed under environmentally controlled conditions in a 12 h light/dark cycle at 25 °C and were provided with food and water ad libitum. A previously described TBI model was utilized [17]. Briefly, after inducing anesthesia with an i.p. injection of 10% chloral hydrate (3 ml/kg), the head of the animal was fixed on a stereotactic frame. Aseptic techniques were used throughout the surgery. A midline scalp incision was performed to expose the skull. A 6-mm craniotomy was performed over the right parietal cortex, centered on the coronal suture and 2.5 mm lateral to the sagittal suture (velocity = 5 m/s, depth = 2.5 mm and dwell time = 100 ms). The bone flap was immediately replaced and sealed, and the scalp was sutured closed. The rectal temperature was maintained at 37 °C with heating pads and lamps. The animals were returned to the feeding room after recovery from anesthesia. Sham-operated rats underwent procedures identical to those of the TBI animals, including anesthesia and surgery, but without TBI. Efforts were made to reduce animal suffering and minimize the number of animals used for these experiments. The severity of the controlled cortical impact injury model and the region affected was depicted in Fig. 1.

The severity of the controlled cortical impact injury model and the region affected. Scale bar 200/20 μm. (n = 5, per group)
Groups and Drug Administration
The 105 adult rats were each randomly assigned to one of three groups (n = 35): sham, TBI or TBI + apocynin group. Apocynin (Sigma-Aldrich) was dissolved using 1% dimethyl sulfoxide (DMSO) and physiological saline, and then it was i.p. injected (50 mg/kg) immediately after TBI as described previously [14]. Both sham and TBI groups received equal volumes of DMSO/saline by i.p. injection. Each sub-group was composed of five rats, and the rats were killed at 12, 24, and 48 h following TBI. In addition, neurobehavioral tests and assessments of brain edema were performed on the remaining rats between days 1–21. All investigations were blind, and the animal codes were revealed only at the end of the behavioral and histological analyses.
Rotarod Task
Animals were pre-trained for 3 days for the rotarod. The rotarod was conducted prior to and at 1, 4, 7, 14 and 21 days after TBI as previously described [18]. For each trial, the rat was placed on a rotating barrel, and the speed was slowly increased from 4 to 40 rpm within 5 min. The time during which the animals remained on the rotarod was recorded. Pre-training occurred on the 3 days preceding injury.
Beam Walking Test
The beam walking test was utilized to evaluate fine motor coordination and function by measuring the ability of the animals to traverse an elevated narrow beam as described previously [19]. Animals were pre-trained for 3 days for beam walking tests. The beam walking test was conducted prior to and at 1, 4, 7, 14 and 21 days after TBI. The time for the rat to cross the beam was recorded (not to exceed 60 s). Three measurements per trial were recorded 1 h before TBI (baseline) and at each tested time-point after TBI.
Modified Neurological Severity Score (mNSS)
The mNSS is a composite of motor, sensory, reflex and balance tests [20]. One point was given for the inability to perform each test or for the absence of a reflex; thus, the higher the score, the more severe the injury. Neurological function was graded on a scale of 0–18, where a normal score was 0, and a maximal deficit score was 18.
Histological Analysis
The brain tissues were fixed in 4% paraformaldehyde solution for 24 h, washed with running water for 4 h, then dehydrated with graded alcohol and embedded in paraffin following standard histological procedures. The tissues were serially sectioned at a thickness of 5 µm. All sections were mounted on glass slides and then stained with hematoxylin and eosin (H&E). The brain sections of the hippocampus area were at roughly 1.9 mm posterior to the bregma from each animal.
Brain Water Measurement
Brain edema was evaluated by analysis of brain water content as previously described [4]. Rat brains were harvested and weighed immediately on an electric analytic balance to obtain the wet weight (WW) and then dried at 100 °C for 24 h to obtain the dry weight (DW). Brain water content was calculated using the following formula: % brain water = [(WW − DW)/WW] × 100.
Immunohistochemistry
Formalin-fixed paraffin-embedded sections (4 mm) were prepared for IHC staining. Briefly, sections were blocked with 3% H2O2 for 20 min, followed by incubation with blocking 5% goat serum for 1 h at room temperature. The sections were then incubated with the primary antibodies (NOX2, LC3, Beclin1, CD11b, GFAP) overnight at 4 °C, followed by incubation with secondary biotinylated antibodies (1:500, Sigma) for 1 h. Color was developed with DAB reagent for 2–10 min, and counterstaining was performed with hematoxylin. Primary antibodies were replaced with PBS in the negative control. Images were captured using an AxioVision4Ac microscope system (Carl Zeiss, Germany).
Immunofluorescence Analyses
Immunofluorescence analysis was performed as described previously by our laboratory [7]. Briefly, coronal brain sections (15 μm) were cut on a microtome after perfusion and cryoprotection. Staining was performed using a mouse anti-NeuN monoclonal antibody (dilution, 1:100; Millipore) following the manufacturer’s instructions. For double-labeling staining, sections were incubated with a mixture of rabbit anti-LC3 polyclonal antibody (dilution, 1:100; MBL) or goat anti-TLR4 polyclonal antibody (diluted, 1:100; Santa Cruz Biotechnology) and mouse anti-NeuN monoclonal antibody (dilution, 1:100; Millipore) overnight at 4 °C. On the following day, the sections were incubated with a mixture of fluorescein-conjugated donkey anti-rabbit or donkey anti-goat Alexa-Fluor594 and donkey anti-mouse Alexa-Fluor 488 (Santa Cruz Biotechnology; diluted 1:200) for 2 h at 37 °C in the dark. The sections were then stained with 4,6-diamidino-2-phenylindole (DAPI) and observed with a fluorescence microscope (Olympus Fluoview™ FV1000). Primary antibodies were replaced with PBS in the negative control group.
Western Blot Analysis
Western blotting was performed as described in detail by our laboratory [7]. Briefly, total proteins were extracted from the hippocampal tissues, and the protein concentration was determined by the BCA reagent (Solarbio, Beijing, China) method. The equivalent of 30 mg of protein was separated by gel electrophoresis, transferred to PVDF membranes and immunoblotted with an anti-LC3 (MBL, diluted 1:1000), anti-NOX2 (Invitrogen, diluted 1:1000), anti-Beclin1 (MBL, diluted 1:1000), anti-TLR4 (Santa Cruz Biotechnology; diluted 1:500), anti-NF-κB p65 (Santa Cruz Biotechnology; diluted 1:500), anti-TNF-α (Affinity, diluted 1:500) or anti-IL-1β (Affinity, diluted 1:500) antibody. The membranes were incubated with secondary biotinylated antibodies for 2 h at 37 °C, separately. The immunoblotted proteins on the membrane were visualized following development with an enhanced chemiluminescence (ECL) detection system, and the densitometric signals were quantified by using Image J software (Image Lab 4.1; BioRad).
Statistical Analysis
All experiments were repeated three times, and similar results were obtained. All data are presented as the mean ± standard deviation (SD) and analyzed using SPSS 16.0. Statistical analysis was performed using ANOVA, followed by Student-Newman-Keuls post hoc tests. P values of less than 0.05 were considered statistically significant.
Results
Apocynin Improved Behavioral Function and Reduced Brain Edema After TBI
To explore the neuroprotective potential of apocynin for TBI, we first evaluated motor and neurological functions, which are important indexes for assessing TBI severity. TBI induced a significant impairment in performances in both the rotarod and beam walk tests at all time points in vehicle-treated rats (Fig. 2a, b). Treatment with apocynin significantly increased the rotarod running time at 1, 4 and 7 days post-injury as compared to the vehicle group (*P < 0.05 vs. vehicle group). Similarly, apocynin-treated rats exhibited better beam walk performances with significantly reduced latency times when crossing the beam from 1 to 14 days after TBI (*P < 0.05 vs. vehicle group). Moreover, we found that the mNSS of rats in the TBI group was significantly increased in comparison with that of the sham group from 1 to 21 days, and apocynin treatment significantly reduced the mNSS compared with the TBI group (Fig. 2c). Finally, the dry-wet weight method was used to determine the effect of apocynin on brain edema from 1 to 21 days after TBI. As shown in Fig. 2d, TBI induced a significant increase in brain edema at 1–14 days compared with the sham group, and apocynin significantly reduced the formation of brain edema in rats with TBI. These results suggest that a single injection of apocynin is sufficient to improve behavioral functional outcomes after TBI.

Post-injury apocynin treatment improved long-term neurobehavioral functions and reduced brain edema. a Apocynin significantly increased the rotarod running time compared with vehicle-treated rat on days 1, 4, and 7 after TBI. b Beam walk latencies were significantly shorter for the apocynin group than the vehicle group at 1, 4, 7, and 14 days post-TBI. c The mNSSs were significantly lower in the apocynin group than the vehicle group at all time points analyzed after injury. d Treatment of apocynin significantly decreased brain edema compared with vehicle group on days 1, 4, 7, and 14 after TBI, as reflected by a decrease in brain water content. Values are presented as mean ± SD; #P < 0.05 versus sham group; *P < 0.05 versus vehicle group; (n = 5, per group)
Apocynin Attenuates Neurons Damage in the Cortex and Hippocampus
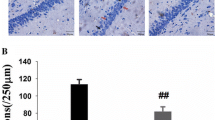
We next examined the neuroprotective effect of apocynin in the cortex and hippocampal region by immunostaining with the neuronal marker NeuN following TBI. The number of surviving neurons was quantified by counting NeuN-positive cells per 1 mm length in the hippocampal area of all animals. As shown in Fig. 3a, sham animals showed robust NeuN immunostaining in the cortex and hippocampal CA1 and CA3 regions, indicating clear and moderate-sized normal neurons in the non-injured state. By contrast, TBI resulted in a profound decrease in the number of surviving neurons in the cortex and hippocampal region at 24 h after TBI, as compared to the sham control (Fig. 3a, b). Intriguingly, in the apocynin group, NeuN immunostaining and the number of surviving neurons were markedly enhanced, suggesting a strong neuroprotective effect was exerted against TBI-induced neuronal cell loss.

Apocynin increases neuronal density following TBI. Coronal brain sections containing cerebral cortex and hippocampal region were subjected to immunofluorescent staining with anti-NeuN antibodies (green). a Representative images indicate that both sham and post-treatment of apocynin markedly increased neuronal density at 24 h after TBI. Scale bar 50 μm. b Quantification of the number of viable neurons/mm length of hippocampal area in each group. (n = 5, per group; *P < 0.05 vs. sham group; #P < 0.05 vs. TBI group). (Color figure online)
Apocynin Reduces Upregulation of NOX2 Protein Expression
We next examined the role of the major NOX2 isoform of NADPH oxidase in TBI damage by IHC and Western blot. As shown in Fig. 4a, we could occasionally observe positive cells, and the positive cells were lightly stained in the sham group. Compared with the sham group, NOX2 positive cells were increased in the TBI group, staining with a deep color and indicating enhanced immune reactivity. However, the immunoreactivity of NOX2 declined in the apocynin group. Following TBI, NOX2 levels were markedly increased at 12, 24 and 48 h. As demonstrated by densitometry analysis in Fig. 4b, treatment with apocynin significantly reduced the relative protein expression of NOX2 at 12, 24 and 48 h in the hippocampus compared with the TBI group.

Apocynin reduces upregulation of NOX2 protein expression. a Representative immunohistochemical staining of NOX2 in the hippocampus from sham, TBI and apocynin treated group at 24 h. Scale bar 50 μm. b Western blot analysis of NOX2 bands in the hippocampus at 12, 24 and 48 h following TBI or sham surgery. c Densitometry analysis of NOX2 band corresponding to β-actin. Results demonstrated that NOX2 protein increased markedly at 12, 48 and 72 h following TBI induction (*P < 0.05 vs. sham group). Treatment with apocynin significantly decreased NOX2 protein expression (#P < 0.05 vs. TBI group). Data were expressed as mean ± SD (n = 5, per group)
Apocynin Suppresses Neuronal Autophagy in Hippocampus
To further clarify the roles of apocynin in the attenuation of TBI-induced brain damage and in neuroprotection, we first examined the autophagy marker proteins, LC3 and Beclin1, in the hippocampus at 24 h after TBI by immunohistochemistry. The results show robust increases in immunostaining intensity for LC3 and Beclin1 in the hippocampal CA1 region in the TBI group as compared to sham controls (Figs. 5a, 6a). Interestingly, treatment with the NADPH oxidase inhibitor apocynin markedly attenuated the LC3 and Beclin1 immunostaining in the hippocampal CA1 region as compared to TBI controls. Next, the co-localization of NeuN and LC3 was assessed by immunofluorescence staining at 24 h. As shown in Fig. 5b, few or no LC3-positive cells were detected at 24 h in the hippocampal tissue of sham control rats. However, autophagy was mainly induced following TBI, and LC3-positive cells co-localized with NeuN in the hippocampus CA1 region at 24 h. Moreover, the LC3 immunofluorescence intensity in NeuN-positive cells was also reduced after apocynin treatment. In order to confirm the ability of apocynin to inhibit autophagy, the protein levels of LC3II/I and Beclin1 were evaluated by Western blot at 12, 24 and 48 h in the hippocampus. The LC3II/I ratio and Beclin1 expression were significantly upregulated at 12, 24 and 48 h after TBI compared with the sham group. Interestingly, treatment with apocynin significantly decreased the protein expression of LC3II/I and Beclin1 in the injured hippocampus compared with the TBI groups. (Figs. 5c, 6b, c).

Evaluated of LC3 protein expression in the hippocampus. a Representative immunohistochemical staining of LC3 in the hippocampus from sham, TBI and apocynin treated group at 24 h. Scale bar 50 μm. b Co-localization of LC3 and NeuN at 24 h following TBI was determined by immunofluorescent staining. As shown, few LC3-positive NeuN cells were detected in the hippocampal in sham group. The majority of LC3-positive NeuN cells were found in the hippocampal in TBI group. Orange labeling is indicative of co-localization. Treatment with apocynin significantly inhibited the increase in LC3-positive NeuN post-trauma. Scale bar 50 μm. c Western blot analysis of LC3 bands in the hippocampus and densitometry analysis of LC3II/I bands corresponding to β-actin. Results demonstrated that LC3II/I protein expression was significantly elevated after TBI (*P < 0.01 vs. sham group). Treatment with apocynin significantly decreased the level of LC3II/I protein expression (#P < 0.05 vs. TBI group). Data were expressed as mean ± SD (n = 5, per group)

Evaluated of Beclin1 protein expression in the hippocampus. a Representative immunohistochemical staining of Beclin1 in the hippocampus from sham, TBI and apocynin treated group at 24 h. Scale bar 50 μm. b Western blot analysis of Beclin1 bands in the hippocampus at 12, 24 and 48 h following TBI or sham surgery. c Densitometry analysis of Beclin1 band corresponding to β-actin. Results demonstrated that Beclin1 protein increased markedly at 12, 48 and 72 h following TBI induction (*P < 0.05 vs. sham group). Treatment with apocynin significantly decreased Beclin1 protein expression (#P < 0.05 vs. TBI group). Data were expressed as mean ± SD (n = 5, per group)
Apocynin Suppresses TLR4 Expression in the Hippocampus
To determine whether TLR4 is involved in TBI-induced neuronal damage, we performed double immunofluorescence staining at 24 h after TBI. As clearly shown in Fig. 7a, few or no TLR4-positive cells co-labeled with NeuN were detected in the hippocampus tissue of sham control rats. However, the majority of TLR4-positive NeuN cells were found in the hippocampal CA1 region at 24 h in the TBI group. Interestingly, apocynin also significantly reduced the number of TLR4-positive cells co-labeled with NeuN after TBI. The protein expression of TLR4 also was subsequently analyzed by Western blot (Fig. 7b) and found to be at low levels in the hippocampus in the sham group and markedly increased at 12, 24 and 48 h after TBI. As demonstrated in Fig. 7c, apocynin significantly inhibited the upregulation of TLR4 protein levels compared with the TBI groups at 12–48 h.

Apocynin reduces protein expression of TLR4. a Identification of TLR4-positive cells 24 h post-TBI in the hippocampus using immunofluorescence. TLR4 immunoreactivity (red) was present in NeuN-positive cells (green) at 24 h following TBI, and cell nuclei were counterstained by DAPI (blue). Orange labeling indicates co-localization. Scale bar 50 μm. b Western blot analysis of TLR4 bands in the hippocampus at 12, 24 and 48 h following TBI or sham surgery. c Densitometry analysis of TLR4 band corresponding to β-actin. Results demonstrated that TLR4 protein increased markedly at 12, 48 and 72 h following TBI induction (*P < 0.05 vs. sham group). Treatment with apocynin significantly decreased TLR4 protein expression (#P < 0.05 vs. TBI group). Data were expressed as mean ± SD (n = 5, per group). (Color figure online)
Apocynin Downregulates the Expression of Inflammatory Mediators
Results of the immunohistochemical analysis showed little CD11b and GFAP immunoreactivity in sham-operated rats, but it was obviously increased in the hippocampal region 24 h after TBI (Fig. 8a). Apocynin administration obviously reduced the number of CD11b and GFAP-immunopositive cells in the hippocampal region as compared to TBI controls (Fig. 8b, c). In order to further investigate the mechanism of action of apocynin, Western blot analysis was performed to examine the expression of inflammation-related molecules in the hippocampus, including NF-κB p65, TNF-α and IL-1β. Our results showed that the protein expression levels of NF-κB p65, TNF-α and IL-1β were significantly upregulated in the hippocampus tissues of rats in the TBI group compared with the sham group at 12–48 h following TBI. In addition, their protein content reached a maximum level at the same time, 24 h following injury (Fig. 9a–c). However, treatment with apocynin significantly inhibited the expression levels of all three of these proteins in the hippocampus tissues of rats at the same time points (Fig. 9d–f).

Apocynin reduces expression of CD11b and GFAP. a Representative immunohistochemical staining of CD11b and GFAP in the hippocampus from sham, TBI and apocynin treated group at 24 h. Scale bar 50 μm. b Quantification showing that apocynin treated rats had significantly fewer CD11b or GFAP-positive cells in the hippocampal area than the TBI group. The total numbers of CD11b or GFAP-positive cells were expressed as the mean number per 100 lm length of hippocampal area in each group. (n = 5, per group; *P < 0.01 vs. sham group, #P < 0.05 vs. TBI group)

a Western blot analysis demonstrates levels of NF-κB p65, TNF-α, and IL-1β in the hippocampus at 12, 24 and 48 h following TBI or sham surgery. b Densitometry of the NF-κB p65, TNF-α, and IL-1β band correlated to the β-actin band. The results demonstrated that the expression of NF-κB p65, TNF-α and IL-1β was significantly elevated at 12–48 h following TBI (*P < 0.05 vs. sham group). Administration of apocynin significantly decreased the levels of NF-κB p65, TNF-α and IL-1β (#P < 0.05 vs. TBI group). Data were expressed as mean ± SD (n = 5, per group)
Discussion
This study utilized the rat TBI injury model to investigate neuroprotective effects of apocynin, as well as changes in neuronal autophagy and the TLR4/NF-κB signaling pathway following TBI injury. For the first time, a single injection of apocynin post-TBI was shown to improve behavioral function and reduce neuronal damage and brain edema in rats. Apocynin can inhibit the “activity” of NADPH oxidase. And it has been demonstrated that treatment with apocynin also attenuates the expression and activation of the NOX2 protein [14]. In present study, we focused on the expression of NOX2 in the rat TBI model. We found that NOX2 protein expression was attenuated after apocynin treatment. This phenomenon reveals that NOX2-depedent NADPH oxidase activity was inhibited by apocynin. These protective effects also might be associated with suppression of neuronal autophagy and the TLR4/NF-κB signaling pathway.
In the study, we demonstrated that rats treated with apocynin (50 mg/kg, i.p.) exhibited reduced motor deficits at 1–7 and 1–14 days by rotarod and beam walk tests, respectively, and also showed reduced mNSS at 1–21 days and attenuated brain edema formation at 1–14 days. These results are consistent with previous studies suggesting that apocynin treatment affords neuroprotection against a variety of neurological disorders, including reduced motor and cognitive impairment [21, 22] and attenuation of brain edema [23, 24]. It is interesting to note that other previous studies did not verify any protective effect of apocynin on brain water content or against neuromotor impairment induced by different brain injury models [25, 26]. The absence of beneficial results with apocynin treatment in brain edema could indicate that NOX2-depedent NADPH oxidase activity is not involved in the development of brain edema induced by TBI. One possible reason for these discrepant results is the diversity of treatment protocols (different doses, time or routes of administration) and different experimental models. Therefore, further studies are necessary to clarify the involvement of NOX2-depedent NADPH oxidase activity in the development of brain edema after TBI.
NOX2 has been shown to be highly expressed in the cortex and hippocampus and contributes significantly to neuronal cell death and functional impairments after TBI [14, 27]. Therefore, we evaluated neuronal damage in the cortex and hippocampus using immunostaining for NeuN. The results showed marked morphological changes and neuronal loss in the cortex and hippocampal CA1 and CA3 neurons of rats in the TBI group. Our study also revealed that NOX2 was highly expressed in hippocampal neurons at 24 h after TBI. NOX2 protein expression was enhanced at 12 to 48 h following TBI induction. Treatment with apocynin not only reduced protein expression of NOX2, but also markedly attenuated the morphological changes and increased neuronal survival. These results indicate that following TBI, NOX2 activation is pivotal in the additional aggravation of secondary brain injury. Our findings are similar to those of Zhang et al., who demonstrated that treatment with apocynin also selectively protected vulnerable neurons in the cortex and hippocampus and downregulated NOX2 activity following TBI [24]. Previous studies using NOX2 mutant knockout mice or the specific NOX2 inhibitor, gp91ds-tat, found that TBI damage to the brain was likewise significantly attenuated in NOX2 knockout mice or the gp91ds-tat group [25, 28].
Previous studies have indicated that neuronal autophagy and NOX expression commonly aggravate secondary brain damage after TBI [4, 6, 7, 14, 24]. Teng et al. found that autophagy may contribute to impaired angiogenesis in persistent pulmonary hypertension-pulmonary artery endothelial cells (PPHN-PAEC) through increasing NOX expression [29]. Those results suggest that, in PPHN-PAEC, a positive feedback relationship between autophagy and NOX expression may regulate angiogenesis. Lu et al. also showed that the pharmacological inhibition of NADPH oxidase using apocynin or gp91ds-tat could decrease autophagy in hippocampal slice cultures and the rat brain, respectively [30]. However, whether the autophagy signal transduction pathway is mediated by NADPH oxidase after TBI is not yet clear. We hypothesized that the mechanism underlying the neuroprotective effects of apocynin on TBI is mediated through the prevention of autophagic neuronal death. Results of our immunohistochemistry and Western blot analyses demonstrated that apocynin inhibited neuronal autophagy and decreased the expression of LC3 d Beclin1 in rat hippocampal tissues at 12–48 h after TBI. Recently, autophagy flux was assessed in several models of TBI based on levels of the autophagic substrate protein sequestosome 1 (SQSTM1)/p62 [31]. Lipinski et al. [32] demonstrated that augmentation and/or restoration of autophagy flux may provide a potential therapeutic target for treatment of TBI and spine cord injury. Because recent studies and our previous research have shown the accumulation of autophagosomes based on electron microscopy studies and/or accumulation of the autophagosome marker proteins LC3-II and Beclin1 [3, 4, 6, 7], we did not address the issue of flux in the current study. It is therefore reasonable to hypothesize that the neuroprotective effect of apocynin on TBI is associated with the attenuation of neuronal autophagy, which is a contributing factor to neuronal damage.
TLR4 acts as a proinflammatory cytokine that mediates inflammation following insults such as sepsis and acute cerebral ischemia or trauma during the acute phase [9]. It serves as a sensor for autophagy associated with innate immunity through MyD88-dependent and TRIF-dependent signaling [33]. Accumulating evidence also supports that TLR4 is expressed in diverse cell types of the brain, including neurons, microglia, astrocytes and oligodendrocytes [34], and plays an important role in neuronal death in animal models. Thus, it may be an important therapeutic target following TBI. In our study, TLR4 was determined to have a significant role in TBI, as dual immunohistochemistry studies revealed that it was highly co-localized in hippocampal neurons at 24 h after TBI. Furthermore, the protein expression of TLR4 was significantly increased at 12 h after TBI and remained high until 48 h compared with those levels observed in the sham group. Our results also showed that apocynin could significantly reduce the expression of TLR4 in the hippocampal tissues after TBI. Other studies similarly showed that apocynin treatment may exert a protective effect in myocardial ischemia-reperfusion (MIR) injury by affecting TLR4, MPO and ADMA levels [35]. Activation of TLR2/4 was previously shown to generally lead to nuclear translocation of NF-κB, a key regulator involved in the inducible expression of inflammatory mediators leading to inflammatory reactions [36]. Microglia, the principal cells involved in the innate immune response in the CNS, have been implicated in mediating the inflammation that occurs after TBI and contributing to neuronal damage through release of inflammatory cytokines [37, 38]. Thus, the attenuation of the NF-κB signaling cascade and microglial activation following NADPH oxidase inhibition can potentially reduce inflammatory reactions and facilitate neuronal survival following TBI. On the other hand, Huang et al. revealed that NOX2 activity and the generated ROS are key signals that induce TLR-activated autophagy of phagosomes [39]. Therefore, we also hypothesize that the autophagic pathway may be influenced by apocynin via downregulation of inflammatory cytokines in the hippocampus of the rat brain. As shown in our results, apocynin clearly reduced the CD11b and GFAP-immunopositive cells in the hippocampus as compared to TBI controls. In addition, we demonstrated that apocynin also decreased the protein expression of NF-κB p65 and attenuated the release of inflammatory cytokines (TNF-α and IL-1β) in the hippocampal tissue of rats.
It should be noted that all the protein and staining data were collected up to 48 h after the injury. However, the behavior data was analyzed up to 21 days. This is because oxidative stress, autophagy, and inflammatory reactions occur within a relatively short time-course after TBI. All factors can play a role in secondary brain damage, and thereby affect the recovery of long-term neurological deficits caused by TBI. Otherwise, few studies have focused on the molecular changes of autophagy, oxidative stress, and inflammatory response after a relatively long period of post-traumatic brain injury. This topic is very interesting and requires further investigations.
In summary, our study showed that administration of apocynin alleviated neurobehavioral deficits, brain edema and neuronal damage following TBI. These effects may be generated through down-regulating NOX2 activity, suppression of neuronal autophagy and the TLR4/NF-κB signaling pathway (Fig. 10). Apocynin may be promising as a protective intervention after TBI, and more study is warranted for its clinical testing in the future.

A simplified schematic diagram representing autophagy and TLR4 signaling in the TBI, and we hypothesized the mechanisms of apocynin’s action
References
Georgiou AP, Manara AR (2013) Role of therapeutic hypothermia in improving outcome after traumatic brain injury: a systematic review. Br J Anaesth 110:357–367
Luo T, Wu J, Kabadi SV, Sabirzhanov B, Guanciale K, Hanscom M et al (2013) Propofol limits microglial activation after experimental brain trauma through inhibition of nicotinamide adenine dinucleotide phosphate oxidase. Anesthesiology 119:1370–1388
Wang YQ, Wang L, Zhang MY, Wang T, Bao HJ, Liu WL et al (2012) Necrostatin-1 suppresses autophagy and apoptosis in mice traumatic brain injury model. Neurochem Res 37:1849–1858
Cui CM, Cui Y, Gao JL, Sun L, Wang Y, Wang K et al (2014) Neuroprotective effect of ceftriaxone in a rat model of traumatic brain injury. Neurol Sci 35:695–700
Zhu HT, Bian C, Yuan JC, Chu WH, Xiang X, Chen F et al (2014) Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-κB signaling pathway in experimental traumatic brain injury. J Neuroinflammation 11:59
Luo CL, Li BX, Chen XP, Sun YX, Bao HJ, Dai DK et al (2011) Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience 184:54–63
Feng Y, Cui Y, Gao JL, Li MH, Li R, Jiang XH et al (2016) Resveratrol attenuates neuronal autophagy and inflammatoryinjury by inhibiting the TLR4/NF-κB signaling pathwayin experimental traumatic brain injury. Int J Mol Med 37(4):921–930
Hoffmann O, Braun JS, Becker D, Halle A, Freyer D, Dagand E et al (2007) TLR2 mediates neuroinflammation and neuronal damage. J Immunol 178:6476–6481
Ahmad A, Crupi R, Campolo M, Genovese T, Esposito E, Cuzzocrea S (2013) Absence of TLR4 reduces neurovascular unit and secondary inflammatory process after traumatic brain injury in mice. PLoS ONE 8:e57208
Fang H, Wang PF, Zhou Y, Wang YC, Yang QW (2013) Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. J Neuroinflammation 10:27
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313
Serrano F, Kolluri NS, Wientjes FB, Card JP, Klann E (2003) NADPH oxidase immunoreactivity in the mouse brain. Brain Res 24:193–198
Ano Y, Sakudo A, Kimata T, Uraki R, Sugiura K, Onodera T (2010) Oxidative damage to neurons caused by the induction of microglial NADPH oxidase in encephalomyocarditis virus infection. Neurosci Lett 469:39–43
Song SX, Gao JL, Wang KJ, Li R, Tian YX, Wei JQ et al (2013) Attenuation of brain edema and spatial learning de fi cits by the inhibition of NADPH oxidase activity using apocynin following diffuse traumatic brain injury in rats. Mol Med Rep 7(1):327–331
Hayashi T, Juliet PA, Kano-Hayashi H, Tsunekawa T, Dingqunfang D, Sumi D et al (2005) NADPH oxidase inhibitor, apocynin, restores the impaired endothelial-dependent and-independent responses and scavenges superoxide anion in rats with type 2 diabetes complicated by NO dysfunction. Diabetes Obes Metab 7:334–343
Choi BY, Jang BG, Kim JH, Lee BE, Sohn M, Song HK et al (2012) Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain Res 1481:49–58
Mahmood A, Lu D, Chopp M (2004) Marrow stromal cell transplantation after traumatic brain injury promotes cellular proliferation within the brain. Neurosurgery 55:1185–1193
Vonder Haar C, Emery MA, Hoane MR (2012) Chronic folic acid administration confers no treatment effects in either a high or low dose following unilateral controlled cortical impact injury in the rat. Restor Neurol Neurosci 30:291–302
Chen CC, Hung TH, Wang YH, Lin CW, Wang PY, Lee CY et al (2012) Wogonin improves histological and functional outcomes, and reduces activation of TLR4/NF-kappaB signaling after experimental traumatic brain injury. PLoS ONE 7:e30294
Chen Y, Constantini S, Trembovler V, Weinstock M, Shohami E (1996) An experimental model of closed head injury in mice: pathophysiology, histopathology, and cognitive deficits. J Neurotrauma 13:557–568
Loane DJ, Stoica BA, Byrnes KR, Jeong W, Faden AI (2013) Activation of mGluR5 and inhibition of NADPH oxidase improves functional recovery after traumatic brain injury. J Neurotrauma 30:403–412
Lu XY, Wang HD, Xu JG, Ding K, Li T (2014) NADPH oxidase inhibition improves neurological outcome in experimental traumatic brain injury. Neurochem Int 69:14–19
Jinnouchi Y, Yamagishi S, Matsui T, Takenaka K, Yoshida Y, Nakamura K et al (2007) Administration of pigment epithelium-derived factor (PEDF) inhibits cold injury-induced brain edema in mice. Brain Res 1167:92–100
Zhang QG, Laird MD, Han D, Nguyen K, Scott E, Dong Y et al (2012) Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS ONE 7:e34504
Jackman KA, Miller AA, De Silva TM, Crack PJ, Drummond GR, Sobey CG (2009) Reduction of cerebral infarct volume by apocynin requires pretreatment and is absent in Nox2-deficient mice. Br J Pharmacol 156:680–688
Ferreira AP, Rodrigues FS, Della-Pace ID, Mota BC, Oliveira SM, Veiho Gewehr Cde C et al (2013) The effect of NADPH-oxidase inhibitor apocynin on cognitive impairment induced by moderate lateral fluid percussion injury: Role of inflammatory and oxidative brain damage. Neurochem Int 63:583–593
Ansari MA, Roberts KN, Scheff SW (2008) Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med 45:443–452
Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K et al (2010) Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation 7:41
Teng RJ, Du J, Welak S, Guan T, Eis A, Shi Y et al (2012) Cross talk between NADPH oxidase and autophagy in pulmonary artery endothelial cells with intrauterine persistent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 302(7):L651–L663
Lu Q, Harris VA, Kumar S, Mansour HM, Black SM (2015) Autophagy in neonatal hypoxia ischemic brain is associated with oxidative stress. Redox Biol 6:516–523
Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K et al (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8:445–544
Lipinski MM, Wu J, Faden AI, Sarkar C (2015) Function and mechanisms of autophagy in brain and spinal cord trauma. Antioxid Redox Signal 23(6):565–577
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT (2007) Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27:135–144
Carty M, Bowie AG (2011) Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem Pharmacol 81:825–837
Uysal A, Sahna E, Ozguler IM, Burma O, Ilhan N (2015) Effects of apocynin, an NADPH oxidase inhibitor, on levels of ADMA, MPO, iNOS and TLR4 induced by myocardial ischemia reperfusion. Perfusion 30(6):472–477
Tu XK, Yang WZ, Shi SS, Wang CH, Zhang GL, Ni TR et al (2010) Spatio-temporal distribution of inflammatory reaction and expression of TLR2/4 signaling pathway in rat brain following permanent focal cerebral ischemia. Neurochem Res 35:1147–1155
Kelley BJ, Lifshitz J, Povlishock JT (2007) Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol 66:989–1001
Venkatesan C, Chrzaszcz M, Choi N, Wainwright MS (2010) Chronic upregulation of activated microglia immunoreactive for galectin-3/Mac-2 and nerve growth factor following diffuse axonal injury. J Neuroinflammation 7:32
Huang J, Brumell JH (2009) NADPH oxidases contribute to autophagy regulation. Autophagy 5(6):887–889
Acknowledgements
This work was supported by the Natural Science Foundation of Hebei Province (Grant No. H2014105079).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Feng, Y., Cui, C., Liu, X. et al. Protective Role of Apocynin via Suppression of Neuronal Autophagy and TLR4/NF-κB Signaling Pathway in a Rat Model of Traumatic Brain Injury. Neurochem Res 42, 3296–3309 (2017). https://doi.org/10.1007/s11064-017-2372-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-017-2372-z