
Abstract
Traumatic brain injury (TBI) results in neuronal apoptosis, autophagic cell death and necroptosis. Necroptosis is a newly discovered caspases-independent programmed necrosis pathway which can be triggered by activation of death receptor. Previous works identified that necrostatin-1 (NEC-1), a specific necroptosis inhibitor, could reduce tissue damage and functional impairment through inhibiting of necroptosis process following TBI. However, the role of NEC-1 on apoptosis and autophagy after TBI is still not very clear. In this study, the amount of TBI-induced neural cell deaths were counted by PI labeling method as previously described. The expression of autophagic pathway associated proteins (Beclin-1, LC3-II, and P62) and apoptotic pathway associated proteins (Bcl-2 and caspase-3) were also respectively assessed by immunoblotting. The data showed that mice pretreated with NEC-1 reduced the amount of PI-positive cells from 12 to 48 h after TBI. Immunoblotting results showed that NEC-1 suppressed TBI-induced Beclin-1 and LC3-II activation which maintained p62 at high level. NEC-1 pretreatment also reversed TBI-induced Bcl-2 expression and caspase-3 activation, as well as the ratio of Beclin-1/Bcl-2. Both 3-MA and NEC-1 suppressed TBI-induced caspase-3 activation and LC3-II formation, Z-VAD only inhibited caspase-3 activation but increased LC3-II expression at 24 h post-TBI. All these results revealed that multiple cell death pathways participated in the development of TBI, and NEC-1 inhibited apoptosis and autophagy simultaneously. These coactions may further explain how can NEC-1 reduce TBI-induced tissue damage and functional deficits and reflect the interrelationship among necrosis, apoptosis and autophagy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI), a leading cause of mortality in young adults, occurs when brain subjected to an external mechanical force [1], and then it causes secondary injuries with a complex performance of pathophysiologic processes such as ischemia reperfusion injury, cerebral hemorrhage and edema [2]. All these events lead to induction of mitochondrial dysfunction and amplification of biochemical cell death signal cascade, all of which cause neuronal cell death and functional deficits [3, 4]. Our previous studies had proved that both apoptosis and autophagy participated in neuronal cell death and functional loss after TBI [3, 5]. Recently, a caspases-independent programmed cell death pathway, necroptosis, showed to be involved in ischemic neurological injury [6–9] and TBI [10]. When a pan-caspases inhibitor, Z-VAD-FMK, was used to block “extrinsic” apoptosis, tumor necrosis factor-α (TNF-α)-induced necroptotic cell death which showed a necrotic morphologic feature could serve as a backup mechanism of cellular demise [11–18]. These meaningful findings implied multiple cell death modes contributed to TBI-induced neuronal cell death.
Being a specialty inhibitor of receptor-interacting protein-1 (RIP-1), necrostatin-1 (NEC-1) was found to potently depress necroptotic cell death [6, 19] and became a hot topic of potential therapeutic agentia in different experimental protocols. As it was expected, NEC-1 reduced the disrupture of brain tissue and improved functional outcomes after TBI [10]. Moreover, interactions between necroptosis and other programmed cell death modes had been observed recently. Rosenbaum et al. found that NEC-1 could inhibit the induction of microtubule-associated protein 1 light chain 3-II (LC3-II) after retinal ischemic [9] and Zhang et al. found both of LC3-II formation and caspase-3 activation are inhibited by NEC-1[20]. Aforementioned evidences suggest that necroptosis is closely related to autophagy and apoptosis at least in particular conditions, and thereby, inhibition of necroptosis by NEC-1 may interfere with apoptosis and autophagy. But, to our knowledge, the effects of NEC-1 on TBI-induced autophagy and apoptosis are still unclear. So we hypothesized that administration of NEC-1 could produce an effect on TBI-induced apoptotic or autophagic programmed neuronal cell death pathway. To test our hypothesis, we firstly counted PI-positive cells at different time point post-TBI to investigate the time course of cell death [3, 5], and then we examined the potential effects of NEC-1 on TBI-induced autophagy and apoptosis. Inhibitors of autophagy (3-MA) and apoptosis (Z-VAD) were also used to confirm the effects of NEC-1 on autophagic and apoptotic activity in this study.
Materials and Methods
TBI Model and Drug Administration
Mature male CD1 (20–25 g) mice were purchased from the Experimental Animal Center of Soochow University (certificate No. 20020008, grade II). All experimental procedures complied with the NIH Guide for the Care and Use of Laboratory Animals. Mice were anesthetized with 4 % chloral hydrate (0.4 mg/g) and fixed in a Kopf stereotactic apparatus [21], and were subjected to TBI in left part of the brain using a weight-drop model as described previously [3, 22]. The reproducibility and consistency of this TBI model were ensured by the accurate location stabilized hit pressure, depth and hitting duration. The craniotomy, which did not significantly affect physiological parameters, was closed immediately after TBI [22]. For separate investigation on the effects of NEC-1, NEC-1 (2.6 μg dissolved in 1 μl DMSO; Sigma-Aldrich Inc., St. Louis, MO, USA, Catalog No.: N9037, Lot No.: 077K4619) or Vehicle (1 μl DMSO) were pretreated with a single intracerebroventricular (i.c.v.) injection in ipsilateral hemisphere 15 min before TBI. Mice were euthanized at 1, 6, 12, 24, or 48 h after TBI. In presences of 3-MA and Z-VAD, Vehicle (1 μl DMSO dissolved in 1 μl saline), 3-MA (0.06 mg dissolved in 2 μl Vehicle; Santa Cruz Biotechnology, sc-205596, 400 nmol), Z-VAD (100 ng dissolved in 2 μl Vehicle; Beyotime Institute of Biotechnology, C1202, 20 μl), or NEC-1 (2.6 μg dissolved in 2 μl Vehicle) were also pretreated with a single intracerebroventricular (i.c.v.) injection in ipsilateral hemisphere 15 min before TBI. Mice were euthanized at 24 h after TBI. Sham-operated mice, which received the craniotomy without the impactor injury, were also received drugs or Vehicle by i.c.v. injection and killed at 24 h after operation. No operation or agentia applied to naïve mice which sacrificed at the same time point as the sham group. Each injured cortical (2 mm × 2 mm × 2 mm tissue block including the impact site and surroundings) or hippocampal tissue (the entire ipsilateral hippocampus including the impact site and surroundings) from each mouse was dissected for several assays.
Propidium Iodide Labeling and Tissue Preparation
Mice (n = 6 for both Vehicle group and NEC-1-treated group) were deeply anesthetized using 4 % chloral hydrate and subjected to TBI or sham-operation. Loss of plasmalemma integrity after TBI was evaluated by intraperitoneal injection of 0.4 mg/ml PI (Sigma-Aldrich Inc., St. Louis, MO, USA) 1 h before sacrificing. 8 series of 12 μm sections (200 μm apart from each section) from anterior to posterior hippocampus (bregma −1.90 to −3.00) were produced from each mouse using cryostat microtome (LEICA, CM1850). All slides preserved in sealed dark cool condition. Each section was examined by fluorescence microscope (NIKON, ECLIPSE Ti). All cortical regions of brain were chosen from 200× cortical fields from within contused cortex. From each mouse brain, five to eight 200× cortical fields from five to eight brain sections separated by at least 150 μm were selected for analysis using a random number generator. One cortical field per brain section was analyzed. The mean number of PI-positive cells for a given time (12, 24 or 48 h) point was calculated by summing the cell count data from all of the counted brain sections [23].
Western Blot Analysis
Mice were deeply anesthetized with chloral hydrate and killed at different time points post-TBI (n = 3 for each time point). Injured cortical (extending 2 mm to the center of the wound) or hippocampal samples were homogenized in Western blot analysis buffer containing 10 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 % (v/v) Triton X-100, 1 % sodium deoxycholate, 0.1 % SDS, 5 mM EDTA, 1 mM PMSF, 0.28 kU/l aprotinin, 50 mg/l leupeptin, 1 mM benzamidine, and 7 mg/l pepstain A (all chemicals were purchased from Sigma-Aldrich Inc., St. Louis, MO, USA). The homogenate was then centrifuged at 12 000 rpm for 10 min at 4 °C and the supernatant was retained and preserved at −80 °C for later use. Protein concentration was determined using a BCA kit (Pierce Chemical, Rockford, IL, USA). Twenty micrograms of protein from each sample was subject to electrophoresis on 10 % SDS-PAGE gel using a constant current [24]. Proteins were transferred to polyvinylidene fluoride membranes on a semidry electrotransferring unit (Bio-Rad) and incubated with antibodies against Beclin-1 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), LC3 (1:3 000; Abcam, ab81785), p62 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), caspase-3 (1:500; Bioword Technology, Minneapolis, MN, USA) and Bcl-2 (1:1 000; Bioword Technology, Minneapolis, MN, USA) in Tris–buffered saline containing 0.1 % Tween-20 (TBST) and 5 % nonfat dry milk overnight at 4 °C. After the overnight incubation with the primary antibodies, membrane were washed and incubated with horseradish peroxidase-conjugated second antibody in TBST for 2 h. Immunoreactivity was detected with enhanced chemoluminescent autoradiography (Pierce Chemical, Rockford, IL, USA), according to the manufacturer’s instructions. The membranes were reprobed with GAPDH (1:5 000; Bioword Technology, Minneapolis, MN, USA) after striping. The signal intensity of primary antibody binding was quantitatively analyzed with Sigma Scan Pro 5 and was normalized to a loading control, GAPDH.
Statistics Analysis
All data shown here were expressed as the mean ± standard error (SE) for all mice in each group. Statistical analysis in Western blot and PI-positive cells data were carried out by one-way ANOVA with Dunnett t test. Statistical analysis was given using the related programs in SPSS 13.0. Differences were considered when P < 0.05.
Results
NEC-1 Reduced TBI-Induced Neural Cell Death
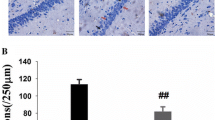
To determine whether NEC-1 can reduce cell death post-TBI in our protocol, PI labeling was used to detect the dead cells. Our previous work had proved that TBI could arouse a distinct increase in the number of PI-positive cells from 12 to 72 h after TBI and the amount of PI-positive cells peaked in the 48 h group [3]. So we selected 12, 24 and 48 h to test if NEC-1 could reduce PI-positive cells versus Vehicle-treated groups. Results revealed that pretreatment with a single intracerebroventricular (i.c.v.) injection of NEC-1 significantly decreased the amount of PI-positive cells versus the Vehicle-treated groups post-TBI (Fig. 1a, b; P < 0.05).

Pre-treatment with NEC-1 reduced the amount propidium iodide (PI)-positive cells from 12 to 48 h after TBI. a Representative photomicrographs show numbers of PI-positive cells in cortical region of brain after TBI in NEC-1 and Vehicle-treated mice (n = 6/group, magnification ×200). b Results of quantitation of PI-positive cells in injured cortex. *P < 0.05 versus. TBI Vehicle-treated mice at the same time point
NEC-1 Attenuated TBI-Induced Autophagic Activity
The levels of LC3-II, p62 and Beclin-1 were examined to determine the ability of NEC-1 in autophagy activation. Beclin-1/Bcl-2 ratio was also calculated here.
The results showed that:
-
1.
The level of LC3-II significantly increased versus sham group from 6 to 48 h after TBI (Fig. 2a; P < 0.05). Pretreatment with NEC-1 could effectively reduce LC3-II level from 6 to 48 h after TBI versus Vehicle-treated groups in the injured cortex and hippocampus, respectively (Fig. 2a; P < 0.05).
Fig. 2 
NEC-1 treatment inhibited TBI-induced LC3-II activation and maintained p62 level. Mice were sacrificed at various time points after TBI as indicated. Tissue extracts from injured cortex and hippocampus were separated on SDS-PAGE, and protein levels of LC3-II and p62 were detected by immunoblotting. a Time course of TBI-induced LC3-II up-regulation and reversal of TBI-induced LC3-II activation by NEC-1. b Time course of TBI-induced decline in p62 and NEC-1 pretreatment inhibited p62 decrease following TBI. Optical densities of the protein bands were quantitatively analyzed with Sigma Scan Pro 5 and normalized with loading control GAPDH. Data were expressed as means ± SEM (a, b; n = 6). Statistical comparisons were carried out with ANOVA followed by Dunnett t test. *P < 0.05, **P < 0.01 versus TBI Vehicle-treated mice at the same time point; # P < 0.05, ## P < 0.01 versus Vehicle-treated sham mice
-
2.
A time course related decline of p62 levels was observed in the injured cortex and hippocampus following TBI (Fig. 2b; P < 0.05). Pretreatment with NEC-1 significantly maintained p62 level versus the Vehicle-treated mice (Fig. 2b; P < 0.05).
-
3.
Up-regulation of Beclin-1 was detected from 6 to 48 h after TBI and peaked between 12 and 24 h (Fig. 3a; P < 0.05). Pretreatment with NEC-1 resulted in a significant decrease in Beclin-1 protein levels from 6 to 48 h after TBI in the injured cortex and hippocampus, versus Vehicle-treated groups respectively (Fig. 3a; P < 0.05).
Fig. 3 
NEC-1 pretreatment reversed TBI-induced up-regulation of Beclin-1 and Beclin-1/Bcl-2 ratio increase. Mice were euthanized at various times after TBI as described before. Tissue extracts from injured cortex and hippocampus were separated on SDS-PAGE, and protein levels of Beclin-1 were detected by immunoblotting. a TBI-induced up-regulation of Beclin-1 was inhibited by NEC-1. b The ratio of Beclin-1/Bcl-2 was increased from 12 to 48 h following TBI and NEC-1 pretreatment reversed the ratio. Optical densities of the protein bands were quantitatively analyzed with Sigma Scan Pro 5 and normalized with loading control GAPDH. Data were expressed as means ± SEM (a, b; n = 6). Statistical comparisons were carried out with ANOVA followed by Dunnett t test *P < 0.05, **P < 0.01 versus TBI Vehicle-treated mice at the same time point; # P < 0.05, ## P < 0.01 versus Vehicle-treated sham mice
-
4.
NEC-1 significantly reversed TBI-induced up-regulation of the Beclin-1/Bcl-2 ratio from 12 to 48 h respectively (Fig. 3b; P < 0.05).


NEC-1 Reversed TBI-Induced Caspase-3 Activation and Bcl-2 Down-Regulation
To determine whether NEC-1 could influence apoptosis, we examined the protein levels of Bcl-2 and activated caspase-3. Time course related down-regulation of anti-apoptotic protein, Bcl-2, was observed from 6 to 48 h post-TBI (Fig. 4a; P < 0.05). Mice pretreated with NEC-1 showed a reductive expression of Bcl-2 in the injured cortex and hippocampus (Fig. 4a; P < 0.05). Time course related increase of activated caspase-3, an executor protein of apoptosis, was observed from 1 to 48 h after TBI versus sham group and peaked at 24 h time point post TBI (Fig. 4b; P < 0.05). Pretreatment with a single injection of NEC-1 could significantly inhibit TBI-induced activation of caspase-3 versus Vehicle-treated group from 6 to 48 h after TBI, respectively (Fig. 4b; P < 0.05).

TBI-induced Bcl-2 decline and activated caspase-3 increase was reversed by NEC-1. Mice were euthanized at various times after TBI as described. Tissue extracts from the TBI and sham-operated cortex and hippocampus were separated on SDS-PAGE, and protein levels of activated caspase-3 and Bcl-2 were detected by immunoblotting. a Time course of TBI-induced down-regulation of Bcl-2 was reversed by NEC-1. b Time course of TBI-induced up-regulation of caspase-3 activaty was inhibited by NEC-1. Optical densities of the protein bands were quantitatively analyzed with Sigma Scan Pro 5 and normalized with loading control GAPDH. Data were expressed as means ± SEM (a, b; n = 6). Statistical comparisons were carried out with ANOVA followed by Dunnett t test.*P < 0.05, **P < 0.01 versus TBI Vehicle-treated mice at the same time point; # P < 0.05, ## P < 0.01 versus Vehicle-treated sham group
The Different Effects of NEC-1, 3-MA and Z-VAD on Activation of Autophagy and Apoptosis at 24 h Post-TBI
To confirm the inhibitory effects of NEC-1 on autophagy and apoptosis, expression levels of LC3-II and caspase-3 were examined by immunoblotting in the presence or absence of 3-MA, Z-VAD, or NEC-1. Pretreatment with either NEC-1 or 3-MA reversed TBI-induced LC3-II up-regulation and caspase-3 activation in the injured cortex and hippocampus versus Vehicle-treated group at 24 h post-TBI (Fig. 5; P < 0.05). Pretreatment with Z-VAD inhibited TBI-induced caspase-3 activation potently in the injured cortex and hippocampus at 24 h post-TBI. However, pretreatment with Z-VAD increased LC3-II level at 24 h post-TBI (Fig. 5; P < 0.05).

TBI-induced LC3-II formation and caspase-3 activation tested in the presence or absence of 3-MA, Z-VAD or NEC-1 at 24 h post-TBI. Mice were euthanized at various times after TBI as described before. Tissue extracts from injured cortex and hippocampus were separated on SDS-PAGE, and protein levels of LC3-II and activated caspase-3 were detected by immunoblotting. Both 3-MA and NEC-1 suppressed TBI-induced caspase-3 activation and LC3-II formation at 24 h post-TBI. Z-VAD inhibited caspase-3 activation, but enhanced LC3-II expression at 24 h post-TBI. Optical densities of the protein bands were quantitatively analyzed with Sigma Scan Pro 5 and normalized with loading control GAPDH. Data were expressed as means ± SEM (n = 3). Statistical comparisons were carried out with ANOVA followed by Dunnett t test *P < 0.05 versus TBI Vehicle-treated mice at the same time point; # P < 0.05, ## P < 0.01 versus Vehicle-treated sham mice
Discussion
Previous studies identified that TBI initiates physiopathologic cascades of cell death signals and induces multiple cell death modes [3, 5, 25, 26]. Beyond the classical programmed cell death pathways, apoptosis (type1) and autophagy (type2), necroptosis termed a newfound caspase-independent programmed necrotic mode. It has been demonstrated that the occurrence of these cell death modes participate in neural injury in TBI and ischemia models [3, 5, 7–10].
NEC-1, identified as a specific inhibitor of receptor-interacting protein-1 kinase, effectively inhibits necroptosis [6, 19]. NEC-1 is also found to reduce amount of injured cell and tissue damage, even improve function outcome after TBI [10]. The literatures definitely elucidated that necroptosis occurred after TBI and NEC-1 achieved the therapeutic action to reduce TBI-induced neural cell lose and neurological dysfunction. However, several lines of evidences indicate that necroptosis has indeterminate relationships with the other two types of programmed cell death [9, 20, 27]. Considered these previous findings, we contemplated that NEC-1 could change TBI-induced activity of autophagy and apoptosis.
Consistent with the study before [10], we found that NEC-1 pretreatment could reduce the amount of PI-positive cells following TBI, suggesting that our weight-drop TBI model was successfully established for exploring the role of NEC-1 on autophagy and apoptosis following TBI.
Evidences implied that autophagy is a probable downstream consequence of necroptosis other than a contributing factor to necroptosis and is activated as a clean-up mechanism for cell death [6, 28–30]. As an echo, treatment with autophagy inhibitors also inhibits Z-VAD-FMK-induced cell death, and knockdown of autophagy related genes such as beclin-1 and atg7 were shown to inhibit necroptosis in L929 cells [31]. These significant results reflect an interacted mechanism between necroptosis and autophagy. Being autophagic biomarker, Beclin-1, the mammalian orthologue of yeast Atg6, is a crucial importance protein found to participate in the regulation of autophagy with several co-actors [32, 33]. LC3, mammalian ortholog of yeast Atg8, synthesized as a pro-LC3 which is cleaved by ATG4 protease and converted to LC3-I. When autophagy is activated, LC3-I is conjugated with PE (lipidated) to form LC3-II [34]. Detection of transformation from LC3-I to LC3-II after TBI is regarded as a biochemical evidence of autophagosomal vacuole formation [5]. Accumulation of LC3-II reflects the autophagic flux [35]. The immunoblotting results in this study showed a dramatic up-regulation of Beclin-1 expression and LC3-II levels post-TBI in the injured cortex and hippocampus, pretreatment with NEC-1 partially decreased TBI-induced up-regulation of Beclin-1 and LC3-II levels. In addition, p62 is identified as a specific substrates captured by LC3 which is selectively transported into the autophagosome [36] and accumulation of p62 inversely correlate with autophagic activity [37]. Previous work proved that the amount of p62 could assist in assessing the decrease of autophagic activity [5]. Our data revealed that NEC-1 pretreatment maintained a steady status of p62 levels post-TBI, suggesting degradation of endogenous autophagic substrates was attenuated by administration of NEC-1. Accordingly, down-regulation of TBI-induced autophagy activation by NEC-1 pretreatment should attribute to the less substrate in NEC-1 pretreated group for autophagy to clean-up. Recently, a breakthrough has been made in the understanding of the role of Bcl-2 in controlling autophagy via its interaction with Beclin-1. Bcl-2 interacts with Beclin-1 via BH3 domain and the ratio of Beclin-1/Bcl-2 levels has been demonstrated to be a significant measurement of autophagy regulation [38–41]. Pretreatment with NEC-1 resulted in a significant decrease of Beclin-1/Bcl-2 ratio in our experimental model. The decrease may be partially illuminated that the addition to Bcl-2 led to the reduction of free Beclin-1 and then to weaken the activation of autophagy [5]. All these new observation showed the anti-autophagic role of NEC-1 in our TBI model, and this observation is supported by the result in a retinal ischemia-reperfusion injury model [9].
Aside from autophagy, apoptosis is another important type of programmed cell death post TBI [42]. As a caspase-independent cell death mode, necroptosis exists as an alternative form of programmed cell death when caspase-dependant apoptosis is blocked [30]. However, definite interactions between necroptosis and apoptosis are far from obvious. Caspase-3, defined as an ‘executioner’ caspase, can be activated via amplification of extrinsic or intrinsic apoptotic signals [43, 44]. On the contrary, Bcl-2 is referred to as an anti-apoptotic member of B-cell lymphoma-2 (Bcl-2) family of proteins, plays an important role in regulation of both caspase-dependent and caspase-independent apoptosis [45]. To date, the effects of NEC-1 on apoptosis are still controversial. NEC-1 could not change the activation of caspase-3 or number of TUNEL-positive cell in the ischemic brain [6]. Moreover, NEC-1 reversed shikonin-induced necroptosis to apoptosis [46]. Discrepantly, elevated activation of caspase-3 induced by 11′-deoxyverticillin-A in human colon carcinoma cell death was also partially inhibited by NEC-1 [20]. The causes of these differences are probably associated with the different cell types or different strategies in particular protocols. Here, in our TBI protocol, we validated an anti-apoptotic role of NEC-1 through inhibiting Bcl-2 decline and caspase-3 activation after TBI.
Moreover, our above findings were also confirmed by utilizing of autophagy or apoptosis inhibitor in this experimental protocol. In our previous study, we found that the specific autophagic inhibitor 3-MA inhibited TBI-induced caspase-3 activation and LC3-II formation [5]. Application of pan-caspase inhibitor Z-VAD potently inhibited C42-induced apoptotic activation with a decrease in activation of caspase-3, but increased necroptotic cell death and autophagic process [20]. Consistent with previous findings, changes of LC3-II and caspase-3 protein level in the presence or absence of 3-MA, Z-VAD or NEC-1 indicated that both 3-MA and NEC-1 could inhibit TBI-induced casapase-3 activation and LC3-II conversion, and administration of Z-VAD decreased TBI-induced caspase-3 activation but enhanced formation of LC3-II after TBI. Thus, inhibition of apoptotic activation may enhance TBI-induced necroptotic and autophagic activation by a feedback mechanism. Additionally, application of specific autophagic inhibitor may potentially suppress both autophagic and apoptotic activation. So, we assumed that NEC-1 inhibits necroptotic activation may leads to suppression of autophagy. This process may leads to attenuation of TBI-induced apoptotic activation via mechanism of crosstalk between autophagy and apoptosis. In addition, other mechanisms that NEC-1 interfere the three cell death modes should be considered. Recently, studies found that NEC-1 could change the activities of other types of cell death associated factors. e.g.: (1) NEC-1 prevented glutamate-induced nuclear translocation of apoptosis inducing factor (AIF), production of reactive oxygen species (ROS) and poly ADP-ribose polymerase (PARP) activation [27, 49]; (2) NEC-1 efficiently reduced arachidonic acid (AA)-induced cell death via blocking reactive oxygen species (ROS) production and c-Jun N-terminal kinases (JNKs) activation [50]; (3) NEC-1 inhibited Z-VAD-FMK-induced phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) in mouse model of Huntington’s disease [51]. These findings implied that several possibilities that NEC-1 could be able to alter other cell death modes except necroptosis, at least in certain conditions. These accessory effects of NEC-1 coincide with the potential role of receptor-interacting protein-1 (RIP-1), a multifunctional protein, which is involved in different pathways of cell death and survival [47, 48].
To sum up, we confirmed that multiple programmed cell death modes could be involved in TBI-induced cell damage. For the first time, we found that NEC-1 suppressed TBI-induced autophagy and apoptosis in vivo. Both 3-MA and NEC-1 suppressed TBI-induced caspase-3 activation and LC3-II formation. Z-VAD inhibited TBI-induced caspase-3 activation. However, Z-VAD increased LC3-II expression following TBI. Our finding implied a complex crosstalk among different type of cell death after TBI and made a further understanding to NEC-1 as a potential therapeutic agent for TBI treatment.
Abbreviations
- TBI:
-
Traumatic brain injury
- NEC-1:
-
Necrostatin-1
- i.c.v:
-
Intracerebroventricle
- PI:
-
Propidium iodide
- LC3:
-
Microtubule-associated protein- 1A/1B light chain 3
- 3-MA:
-
3-Methyladenine
- Z-VAD:
-
Pan-caspase inhibitor Z-VAD-FMK
- p62/SQSTM1:
-
Sequestosome 1 protein
References
Maas AI, Stocchetti N, Bullock R (2008) Moderate and severe traumatic brain injury in adults. Lancet Neurol 7(8):728–741
Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT (2008) Classification of traumatic brain injury for targeted therapies. J Neurotrauma 25(7):719–738
Luo CL, Chen XP, Yang R, Sun YX, Li QQ, Bao HJ, Cao QQ, Ni H, Qin ZH, Tao LY (2010) Cathepsin B contributes to traumatic brain injury-induced cell death through a mitochondria-mediated apoptotic pathway. J Neurosci Res 88(13):2847–2858
Xiong Y, Gu Q, Peterson PL, Muizelaar JP, Lee CP (1997) Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J Neurotrauma 14(1):23–34
Luo CL, Li BX, Li QQ, Chen XP, Sun YX, Bao HJ, Dai DK, Shen YW, Xu HF, Ni H, Wan L, Qin ZH, Tao LY, Zhao ZQ (2011) Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience 184:54–63
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1(2):112–119
Meloni BP, Meade AJ, Kitikomolsuk D, Knuckey NW (2011) Characterisation of neuronal cell death in acute and delayed in vitro ischemia (oxygen-glucose deprivation) models. J Neurosci Methods 195(1):67–74
Xu X, Chua KW, Chua CC, Liu CF, Hamdy RC, Chua BH (2010) Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain Res 1355:189–194
Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI (2010) Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res 88(7):1569–1576
You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ (2008) Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab 28(9):1564–1573
Denecker G, Vercammen D, Declercq W, Vandenabeele P (2001) Apoptotic and necrotic cell death induced by death domain receptors. Cell Mol Life Sci 58(3):356–370
Denecker G, Vercammen D, Steemans M, Vanden Berghe T, Brouckaert G, Van Loo G, Zhivotovsky B, Fiers W, Grooten J, Declercq W, Vandenabeele P (2001) Death receptor-induced apoptotic and necrotic cell death: differential role of caspases and mitochondria. Cell Death Differ 8(8):829–840
Vanden Berghe T, Denecker G, Brouckaert G, Vadimovisch Krysko D, D’Herde K, Vandenabeele P (2004) More than one way to die: methods to determine TNF-induced apoptosis and necrosis. Methods Mol Med 98:101–126
Vanden Berghe T, Kalai M, van Loo G, Declercq W, Vandenabeele P (2003) Disruption of HSP90 function reverts tumor necrosis factor-induced necrosis to apoptosis. J Biol Chem 278(8):5622–5629
Vanden Berghe T, van Loo G, Saelens X, Van Gurp M, Brouckaert G, Kalai M, Declercq W, Vandenabeele P (2004) Differential signaling to apoptotic and necrotic cell death by Fas-associated death domain protein FADD. J Biol Chem 279(9):7925–7933
Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P (1998) Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 187(9):1477–1485
Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, Vandenabeele P (1998) Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med 188(5):919–930
Vercammen D, Vandenabeele P, Beyaert R, Declercq W, Fiers W (1997) Tumour necrosis factor-induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine 9(11):801–808
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4(5):313–321
Zhang N, Chen Y, Jiang R, Li E, Chen X, Xi Z, Guo Y, Liu X, Zhou Y, Che Y, Jiang X (2011) PARP and RIP 1 are required for autophagy induced by 11′-deoxyverticillin A, which precedes caspase-dependent apoptosis. Autophagy 7(6):598–612
Qin ZH, Chen RW, Wang Y, Nakai M, Chuang DM, Chase TN (1999) Nuclear factor kappaB nuclear translocation upregulates c-Myc and p53 expression during NMDA receptor-mediated apoptosis in rat striatum. J Neurosci 19(10):4023–4033
Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG (1981) Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res 211(1):67–77
Whalen MJ, Dalkara T, You Z, Qiu J, Bermpohl D, Mehta N, Suter B, Bhide PG, Lo EH, Ericsson M, Moskowitz MA (2008) Acute plasmalemma permeability and protracted clearance of injured cells after controlled cortical impact in mice. J Cereb Blood Flow Metab 28(3):490–505
Wang Y, Han R, Liang ZQ, Wu JC, Zhang XD, Gu ZL, Qin ZH (2008) An autophagic mechanism is involved in apoptotic death of rat striatal neurons induced by the non-N-methyl-D-aspartate receptor agonist kainic acid. Autophagy 4(2):214–226
Edinger AL, Thompson CB (2004) Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 16(6):663–669
Werner C, Engelhard K (2007) Pathophysiology of traumatic brain injury. Br J Anaesth 99(1):4–9
Xu X, Chua CC, Zhang M, Geng D, Liu CF, Hamdy RC, Chua BH (2010) The role of PARP activation in glutamate-induced necroptosis in HT-22 cells. Brain Res 1343:206–212
Bell BD, Leverrier S, Weist BM, Newton RH, Arechiga AF, Luhrs KA, Morrissette NS, Walsh CM (2008) FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci USA 105(43):16677–16682
Ch’en IL, Beisner DR, Degterev A, Lynch C, Yuan J, Hoffmann A, Hedrick SM (2008) Antigen-mediated T cell expansion regulated by parallel pathways of death. Proc Natl Acad Sci USA 105(45):17463–17468
Christofferson DE, Yuan J (2010) Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol 22(2):263–268
Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304(5676):1500–1502
Clark RS, Bayir H, Chu CT, Alber SM, Kochanek PM, Watkins SC (2008) Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy 4(1):88–90
Itakura E, Kishi C, Inoue K, Mizushima N (2008) Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 19(12):5360–5372
Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T (2004) LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 117(Pt 13):2805–2812
Tanida I, Ueno T, Kominami E (2008) LC3 and autophagy. Methods Mol Biol 445:77–88
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131(6):1149–1163
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282(33):24131–24145
Pattingre S, Levine B (2006) Bcl-2 inhibition of autophagy: a new route to cancer? Cancer Res 66(6):2885–2888
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122(6):927–939
Erlich S, Shohami E, Pinkas-Kramarski R (2006) Neurodegeneration induces upregulation of Beclin 1. Autophagy 2(1):49–51
Sadasivan S, Dunn WA Jr, Hayes RL, Wang KK (2008) Changes in autophagy proteins in a rat model of controlled cortical impact induced brain injury. Biochem Biophys Res Commun 373(4):478–481
Zhang X, Chen Y, Jenkins LW, Kochanek PM, Clark RS (2005) Bench-to-bedside review: apoptosis/programmed cell death triggered by traumatic brain injury. Crit Care 9(1):66–75
Cheema ZF, Wade SB, Sata M, Walsh K, Sohrabji F, Miranda RC (1999) Fas/Apo [apoptosis]-1 and associated proteins in the differentiating cerebral cortex: induction of caspase-dependent cell death and activation of NF-kappaB. J Neurosci 19(5):1754–1770
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X (1997) Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90(3):405–413
Graham SH, Chen J, Clark RS (2000) Bcl-2 family gene products in cerebral ischemia and traumatic brain injury. J Neurotrauma 17(10):831–841
Han W, Xie J, Li L, Liu Z, Hu X (2009) Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis. Apoptosis 14(5):674–686
Declercq W, Vanden Berghe T, Vandenabeele P (2009) RIP kinases at the crossroads of cell death and survival. Cell 138(2):229–232
Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P (2007) RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ 14(3):400–410
Xu X, Chua CC, Kong J, Kostrzewa RM, Kumaraguru U, Hamdy RC, Chua BH (2007) Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J Neurochem 103(5):2004–2014
Kim S, Dayani L, Rosenberg PA, Li J (2010) RIP1 kinase mediates arachidonic acid-induced oxidative death of oligodendrocyte precursors. Int J Physiol Pathophysiol Pharmacol 2(2):137–147
Zhu S, Zhang Y, Bai G, Li H (2011) Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington’s disease. Cell Death Dis 2:e115
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (30872666 and 81172911) and the Shanghai Forensic Key Lab Foundation (KF1005).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Yao-Qi Wang and Long Wang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Wang, YQ., Wang, L., Zhang, MY. et al. Necrostatin-1 Suppresses Autophagy and Apoptosis in Mice Traumatic Brain Injury Model. Neurochem Res 37, 1849–1858 (2012). https://doi.org/10.1007/s11064-012-0791-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-012-0791-4