
Abstract
Advanced glycation end products (AGEs) enhance microglial activation and intensify the inflammatory response and oxidative stress in the brain. This process may occur due to direct cytotoxicity or interacting with AGEs receptors (RAGE), which are expressed on the surface of microglia. FPS-ZM1 is a high-affinity but nontoxic RAGE-specific inhibitor that has been recently shown to attenuate the Aβ-induced inflammatory response by blocking the ligation of Aβ to RAGE. In this study, we further investigated the effect of FPS-ZM1 on the AGEs/RAGE interaction and downstream elevation of neuroinflammation and oxidative stress in primary microglia cells. The results suggested that FPS-ZM1 significantly suppressed AGEs-induced RAGE overexpression, RAGE-dependent microglial activation, nuclear translocation of nuclear factor kappaB p65 (NF-κB p65), and the expression of downstream inflammatory mediators such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), cyclooxygenase 2 (COX-2)/prostaglandin E2 (PGE2) and inducible nitric oxide synthase (iNOS)/nitric oxide (NO). Furthermore, FPS-ZM1 attenuated AGEs-stimulated NADPH oxidase (NOX) activation and reactive oxygen species (ROS) expression. Finally, FPS-ZM1 elevated the levels of transcription factors nuclear-factor (erythroid-derived 2)-like 2 (Nrf2) and heme oxygenase-1 (HO-1), as well as decreased antioxidant capacity and increased production of oxidative species. Our results suggest that FPS-ZM1 may be neuroprotective through attenuating microglial activation, oxidative stress and inflammation by blocking RAGE.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by the formation of senile plaques and neurofibrillary tangles (NFTs), activation of microglia, elevation of oxidative stress and neuroinflammation [1,2,3]. Microglial activation, which leads to an inflammatory cascade and subsequently causes oxidative stress and cytokine-induced neuron damage, has been demonstrated to play an important role in the pathologic process of AD [4, 5]. Activated microglia have been found to surround amyloid deposits at an early stage and release a broad spectrum of highly cytotoxic substances, including oxygen free radicals, nitric oxide, proteinase and inflammatory mediators [6,7,8,9]. Advanced glycation end products (AGEs) are formed via non-enzymatic reactions between glucose or other carbohydrates and stable protein deposits [10]. The formation of AGEs is widely regarded as an important contributor to the progression of AD by activating microglia and astrocyte to induce oxidative stress and inflammation, as evidence has shown that AGEs are not only elevated in AD brains but also accelerate Aβ deposition and hyperphosphorylation of tau and increase cytotoxicity of glycated proteins [11,12,13,14].
AGEs not only promote protein or lipid cross-linking through direct toxicity, but also through activation of many pathological processes mediated by AGEs receptor (RAGE) [15]. RAGE is a member of the immunoglobulin superfamily, and can bind multiple ligands, including AGEs and β-amyloid peptide (Aβ) [16]. The ligand-receptor interaction activates several signal transduction pathways and regulates cell functions through oxidative stress [13, 17]. AGEs/RAGE interaction induces proinflammatory cytokines such as TNF-α, IL-6, IL-1 and iNOS in N-11 microglia cells [18, 19]. Our previous study found that AGEs induce cell death via oxidative stress and endoplasmic reticulum stress in SH-SY5Y cells and rat cortical neurons [20]. Also, intrahippocampal injection of AGEs induced upregulation of Aβ and amyloid precursor protein (APP) production, inflammation, oxidative stress and cognitive dysfunction in rats [21]. Furthermore, AGEs and RAGE interactions may increase neuroinflammation and oxidative stress via the activation of nuclear factor kappaB (NF-κB) and the production of reactive oxygen species (ROS) [22]. Targeted pharmacological interventions using inhibitors of AGEs, anti-RAGE-antibodies or antagonists of RAGE may be promising therapeutic strategies to slow the progression of cognitive decline in AD.
Previous studies have shown that anti-RAGE treatment using soluble RAGE or anti-RAGE antibodies cannot modify disease progression effectively because they do not cross the blood–brain barrier (BBB) and only block peripheral RAGEs [23, 24]. In a more recent study, FPS-ZM1, a high-affinity but nontoxic RAGE-specific inhibitor, was shown to cross the BBB readily. Further, it blocked RAGE-mediated influx of peripheral Aβ1−40 and Aβ1−42, β-secretase activity, Aβ generation, microglial activation and neuroinflammation [25]. Since the production of AGEs is irreversible and contributes to the pathogenesis of AD [26], it may be more effective to block AGEs simultaneously with Aβ.
In the present study, we explored whether FPS-ZM1 affects the interaction of AGEs-RAGE in primary microglia and investigated the molecular mechanism of AGEs-induced microglial activation, neuroinflammation and oxidative stress in vitro.
Materials and Methods
Materials
Bovine serum albumin (BSA), and AGEs-bovine serum albumin (AGEs-BSA) were purchased from BioVision (CA, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT), Triton X-100, poly-l-lysine and 2′,7′-dichlorodihydrofluorescin diacetate (H2DCFH-DA) we\] e purchased from Sigma-Aldrich (Mo, USA). Fetal bovine serum (FBS), Ham F-12 medium, Dulbecco’s modified Eagle’s medium (DMEM), glutamine, penicillin, streptomycin, amphotericin B were obtained from Invitrogen (Carlsbad, Calif). FPS-ZM1 was purchased from Millipore (MA, USA). Rabbit polyclonal antibody against nuclear-factor (erythroid-derived 2)-like 2 (Nrf2) and mouse antibody against inducible NO synthase (iNOS) were purchased from Abcam (CA, USA). Mouse antibody against CD11b, mouse antibody against RAGE, rabbit antibody against heme oxygenase 1 (HO-1), NADPH oxidase 2 (NOX2), p47phox and p67phox were purchased from Santa Cruz Biotechnology (CA, USA). Rabbit polyclonal antibody against NF-κB p65 was purchased from Millipore (MA, USA). Antibody against β-actin, histone H3 and secondary antibodies were purchased from Zhongshan Jinqiao Biotechnology (Beijing, China). The BCA-Protein Assay Kit was obtained from Biocolor BioScience and Technology Company (Shanghai, China). Polyvinylidene difluoride (PVDF) membranes and the enhanced chemiluminescence kit were from Millipore (MA, USA). TNF-α, IL-1β, PGE2 and NO ELISA kits were obtained from R&D Systems (Minneapolis, MN, USA). The COX-2 ELISA kit was from Fitzgerald Industries International (MA, USA).
Primary Microglia Cultures and Treatment
Pregnant Wistar rats were obtained from the Research Center of Experimental Animals of Shandong University. The rats were cared according to the Provisions and General Recommendation of Chinese Experimental Animals Administration Legislation. Primary microglia cultures were prepared from one-day-old newborn Wistar rats according to the procedure described previously [27]. The rat cortical cortices were dissected and digested with 0.25% trypsin for 10 min at 37 °C. After digestion, the cells were plated onto 20 μg/mL poly-l-lysine-coated incubation bottles and cultured in DMEM and Ham F12 mixed medium (1:1), containing 10% FBS, 4 mM glutamine, 100 μg/mL streptomycin, 100 U/mL penicillin and 0.25 μg/mL amphotericin B, in a 37 °C humidified incubator containing 5% CO2 and 20% O2. Media was replaced every 3 days. Fourteen days later, microglia were removed by shaking at 200 revolutions per minute for 3 h. After isolation, microglia were plated onto 6-well plates in culture medium (4 × 105 cells/mL, 1 mL per well). Cells were divided into five groups: (1) Control group (300 μg/mL BSA for 24 h), (2) AGEs group (300 μg/mL AGEs-BSA for 24 h), (3) AGEs + F1 group (25 nM FPS-ZM1 for 1 h, then 300 μg/mL AGEs-BSA for 24 h), (4) AGEs + F2 group (50 nM FPS-ZM1 for 1 h, then 300 μg/mL AGEs-BSA for 24 h), (5) AGEs + F3 group (100 nM FPS-ZM1 for 1 h, then 300 μg/mL AGEs-BSA for 24 h). Cells were treated with or without AGEs-BSA (300 μg/mL) in the absence and presence of FPS-ZM1 (25, 50 or 100 nM) for 24 h. After treatment, cells were collected for immunocytochemistry and ELISA.
Cell Viability Assay
Cell viability was assessed by the MTT assay. Primary microglia cells were plated in 96-well culture plates and exposed to 0, 50, 100, 200, 300, 400 μg/mL BSA or AGEs-BSA for 24 h. The culture medium was replaced by 0.5 mg/mL MTT for another 4 h at 37 °C. The formazan crystals in the cells were solubilized with 150 μL dimethyl sulfoxide (DMSO). The absorbance was detected at 570 nm on a Synergy HT Multi-Mode Microplate Reader (Bio-Teck, USA). The absorbance of normal control group cells was set to 100%, and other groups are expressed as the percentage of MTT reduction.
Immunocytochemistry
Antibody against CD11b was used for microglia staining. Cells were fixed in 0.1 M phosphate-buffered saline (PBS pH 7.4) with 4% paraformaldehyde for 30 min, washed with PBS three times, and then incubated in 0.2% Triton X-100 for 20 min. After three washes, cells were incubated with 3% hydrogen peroxide and blocked with 5% BSA for 30 min. The cells were then incubated overnight at 4 °C with the anti-CD11b antibody (1:100). Rabbit anti-mouse IgG was used as secondary antibody, followed by staining with DAB and hematoxylin. Images were collected under a light microscope at magnifications of ×400.
ELISA
The level of inflammatory mediators tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), cyclooxygenase 2 (COX-2), prostaglandin E2 (PGE2) and nitric oxide (NO) were quantified using specific ELISA kits for rats according to the manufacturer’s instructions.
Protein Measurement and Western Blotting
After treatment, cells were washed with ice-cold 0.1 M PBS (pH 7.4) and lysed in the lysing buffer (500 mM Tris–HCl, pH 7.4, 150 nM NaCl, 1 mM benzamiden, 5 mM EDTA, 1 μg/mL trypsin inhibitor) containing a protease inhibitor cocktail (5 μL/mL) for 40 min at 4 °C. The lysate was centrifuged at 12,000g for 30 min at 4 °C, and the supernatant was collected. Membrane proteins were extracted using a membrane protein extraction kit (BioVision, Mountain View, CA, USA) for detecting the expression of NOX2, p47phox, and p67phox. Protein extraction for Nrf2 and NF-κB p65 was performed using a nuclear extraction kit per the manufacturer’s instructions. Protein concentrations were detected using a BCA kit. Equal amounts of protein (60 μg per lane) were separated on 10% SDS-PAGE gels and then transferred to PVDF membranes. After incubation with primary antibodies overnight at 4 °C, secondary antibodies (1:5000) were incubated for 1 h at room temperature. The enhanced chemiluminescence reagents and an image analyzer (Alpha Innotech, San Leandro, CA, USA) were used for detecting protein bands.
Measurement of Intracellular ROS
The content of intracellular ROS was assessed by fluorescence with H2DCF-DA staining. Cells were stained with 10 μM H2DCF-DA at 37 °C for 30 min. Cells were isolated and washed twice with PBS, and then analyzed by a FACS Vantage flow cytometer. The blue laser with excitation wave length of 488 nm and emission wavelength of 530 nm was used to determine dichlorofluorescein fluorescence. Fluorescent signal intensity was detected by CellQuest software, and the level of ROS was determined using the mean fluorescence intensity (MFI).
Measurement of Intracellular MDA, SOD, and GSH
The cells were washed by ice-cold 0.1 M PBS (pH 7.4) and then lysed in PBS containing 5 μM EDTA, and 0.1% SDS. After homogenization, the homogenate was centrifuged at 1000g for 30 min. The supernatant was used for detection. Malondialdehyde (MDA) content was measured by thiobarbituric acid reactive method. The inhibition of nitro blue tetrazolium reduction represents superoxide dismutase (SOD) activity. GSH content was determined with a 5,5′-dithiobis(2-nitrobenzoic acid) assay.
Statistical Analysis
All data are shown as mean ± standard error of the mean (SEM). Statistical analysis of the results was performed using one-way ANOVA followed by a Tukey–Kramer test for multiple comparisons. The acceptable statistical significance was considered P < 0.05. All statistical analyses were performed using SPSS software, version 21.0 (SPSS, Chicago, IL, USA).
Results
FPS-ZM1 Ameliorated AGEs-BSA-Induced Injury of Primary Microglia Cells
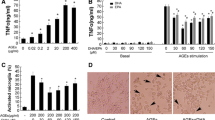
Primary microglia cells were exposed to 0, 50, 100, 200, 300, 400 μg/mL BSA or AGEs-BSA for 24 h. BSA showed almost no effect on cell viability, while 200–400 μg/mL AGEs-BSA decreased cell viability significantly. At a concentration of 300 μg/mL, cell viability showed a reduction of about 50%, so treatment for 300 μg/mL AGEs-BSA was used for cell damage model (Fig. 1a). Pretreatment with FPS-ZM1 for 1 h showed that AGEs-BSA-induced cell damage was alleviated by FPS-ZM1 in a dose-dependent manner (P < 0.01) (Fig. 1b).

FPS-ZM1 inhibits AGEs-induced cytotoxicity and microglial activation. Cells were treated with different concentrations of AGEs for 24 h. BSA was used as the control. AGEs-induced cytotoxicity was measured by MTT assay (a). Cells were treated with or without AGEs-BSA (300 μg/mL) for 24 h in the absence or presence of pretreatment with FPS-ZM1 (25, 50 or 100 nM, indicated as F1, F2, F3 respectively) for 1 h. The control group received BSA treatment. Cell viability (b), immuohistochemical staining of CD11b (×400 magnification) (c), and RAGE expression (d, e) in primary cultured microglia. The protein expression was analyzed by western blotting and normalized to β-actin. ***P < 0.001 versus control group. ### P < 0.001 versus AGEs group. △△ P < 0.01, △△△ P < 0.001 versus AGEs + F2 group
FPS-ZM1 Suppresses AGEs-Induced Glial Activation and RAGE Expression in Primary Microglia Cultures
In order to further examine the effects of FPS-ZM1 on AGEs-RAGE ligation, we investigated the effects of FPS-ZM1 on AGEs-induced microglial activation and RAGE expression. Primary microglia were treated with AGEs-BSA (300 μg/mL) for 24 h with or without the pretreatment of FPS-ZM1 for 1 h. Increased CD11b expression is a specific marker for microglial activation. As shown in Fig. 1c, in comparison with the control group, AGEs significantly enhanced the expression of CD11b in microglia and FPS-ZM1 significantly attenuated this elevation. AGEs intervention resulted in significant increase in RAGE expression (P < 0.001), which was attenuated by FPS-ZM1 in a dose-dependent manner (Fig. 1d, e). The above results indicated that microglial activation and RAGE expression stimulated by AGEs were significantly reduced by FPS-ZM1.
FPS-ZM1 Alleviates AGEs-Induced Intracellular ROS Production and NOX Activation In Vitro
AGEs-BSA has been demonstrated to induce ROS generation via NOX in SH-SY5Y cells and rat cortical neurons [20]. NOX activation, especially NOX2 induced by microglia, is an important pathological mechanism to induce oxidative stress-related and neuroinflammation-related neurodegeneration. We tested the activation of NOX2 and its two cytosolic subunits, p47phox and p67phox, in primary microglia treated with AGEs. We found that AGEs not only significantly promoted ROS production in primary microglia compared with the control group (P < 0.001) (Fig. 2a), but also significantly increased NOX2, p67phox and p47phox expression via Western blotting (P < 0.001) (Fig. 2b, c). The presence of FPS-ZM1 at concentrations of 25, 50 and 100 nM attenuated the effect of AGEs on ROS production (P < 0.01) and expression of NOX2, p47phox and p67phox (P < 0.05) (Fig. 2).

FPS-ZM1 alleviates AGEs-induced oxidative stress. Primary cultured microglia were treated with or without AGEs-BSA (300 μg/mL) for 24 h in the absence or presence of pretreatment with FPS-ZM1 at three concentrations (25, 50 and 100 nM) for 1 h. ROS production (a), expression of NOX2, p67phox and p47phox (b, c), MDA content (d), GSH content (e), and SOD activity (f). The protein expression was analyzed by western blotting and normalized to β-actin. All data are presented as mean ± SEM from three independent experiments. ***P < 0.001 versus control group. ## P < 0.01, ### P < 0.001 versus AGEs group. △ P < 0.05, △△ P < 0.01, △△△ P < 0.001 versus AGEs + F2 group
FPS-ZM1 Alleviates AGEs-Induced MDA, GSH Content and SOD Activity In Vitro
The effects of FPS-ZM1 on AGEs-induced oxidative damage was determined by MDA, GSH content and SOD activity. More specifically, GSH content and SOD activity represent antioxidant capacity. As shown in Fig. 2, AGEs significantly increased MDA production (P < 0.001), which was attenuated by FPS-ZM1 (P < 0.001) (Fig. 2d). Moreover, FPS-ZM1 reversed AGEs-induced attenuation of GSH content and SOD activity (P < 0.01) (Fig. 2e, f).
FPS-ZM1 Elevates Antioxidative Nrf2 and HO-1 Production
To further investigate the antioxidant effects of FPS-ZM1, we examined the protein levels of Nrf2 and HO-1 by Western blotting. Nrf2 is a pivotal regulator for preventing oxidative stress by regulating Nrf2-related antioxidants, including HO-1. In this study, AGEs significantly increased the expression of Nrf2 and HO-1 (P < 0.05), which was further increased following FPS-ZM1 treatment in a dose-dependent manner (Fig. 3). These results suggested that AGEs stimulated the antioxidant system, and FPS-ZM1 further increased antioxidant levels through Nrf2 and HO-1, which could be the protective mechanism of FPS-ZM1.

FPS-ZM1 increases the expression of Nrf-2 and HO-1 in AGEs-induced primary cultured microglia. Cells were treated with or without AGEs-BSA (300 μg/mL) for 24 h in the absence or presence of pretreatment with FPS-ZM1 (25, 50 and 100 nM) for 1 h. The protein expression was normalized to β-actin or histone H3. All data are presented as mean ± SEM from three independent experiments. *P < 0.05, **P < 0.01 versus control group. # P < 0.05, ## P < 0.01, ### P < 0.001 versus AGEs group. △ P < 0.05, △△ P < 0.01 versus AGEs + F2 group
FPS-ZM1 Attenuates AGEs-Induced Activation of NF-κB p65 and Inflammatory Markers in Primary Microglia
By comparing proteins extracted from cytoplasm and the nucleus, we found that AGEs treatment increased NF-κB p65 nuclear translocation from cytoplasm (P < 0.001) (Fig. 4). However, this effect was markedly decreased by FPS-ZM1 in a dose dependent manner (Fig. 4). NF-κB p65 nuclear translocation usually results in upregulation in transcription of genes coding for downstream proinflammatory cytokines. TNF-α, IL-1β, COX-2/PGE2 and iNOS/NO are pivotal biomarkers in inflammatory pathways, which can lead to neurodegeneration. AGEs treatment significantly elevated expressions of TNF-α, IL-1β, COX-2/PGE2 and iNOS/NO in primary microglia compared with the control group (P < 0.001) (Fig. 5). Similarly, the presence of FPS-ZM1 significantly attenuated the upregulation of these pro-inflammatory biomarkers induced by AGEs (P < 0.05) (Fig. 5).

FPS-ZM1 decreases nuclear translocation of NF-κB p65 induced by AGEs in primary cultured microglia. Cells were treated with or without AGEs-BSA (300 μg/mL) for 24 h in the absence or presence of pretreatment with FPS-ZM1 (25, 50 and 100 nM) for 1 h. The control group received BSA treatment. The protein expression was normalized to β-actin or histone H3. All data are presented as mean ± SEM from three independent experiments. ***P < 0.01 versus control group. # P < 0.05, ## P < 0.01, ### P < 0.001 versus AGEs group. △ P < 0.05 versus △△ P < 0.01, △△△ P < 0.001 versus AGEs + F2 group

FPS-ZM1 alleviates the expression of inflammatory biomarkers induced by AGEs in primary cultured microglia. a Representative immunoblots for iNOS in all the treated groups. b Relative density analysis of iNOS protein bands. Quantitative ELISA analysis of TNF-α (d), IL-1β (d), COX-2 (f), PGE2 (e) and NO (c) levels in primary microglia. Cells were treated with or without AGEs-BSA (300 μg/mL) for 24 h in the absence or presence of pretreatment with FPS-ZM1 (25, 50 and 100 nM) for 1 h. The protein expressions by western blotting were normalized to β-actin. All data are presented as mean ± SEM from three independent experiments. ***P < 0.001 versus control group. # P < 0.05, ## P < 0.01, ### P < 0.001 versus AGEs group. △ P < 0.05, △△ P < 0.01, △△△ P < 0.001 versus AGEs + F2 group
Role of NOX, iNOS, Nrf2 and ROS in AGEs-Induced Primary Microglia Oxidative and Inflammatory Damage
Cells were pretreated with the NOX inhibitor apocynin (API), iNOS inhibitor SMT, the activator of Nrf2 sulforaphane (SFP) or the ROS inhibitor NAC, respectively, 1 h prior to the addition of AGEs-BSA. API significantly inhibited the content of ROS (P < 0.001) (Fig. 6a). Moreover, pretreatment with SMT prior to AGEs attenuated production of NO significantly (P < 0.001) (Fig. 6e). Sulforaphane (SFP) significantly increased the activity of SOD (P < 0.001) (Fig. 6g) and HO-1 expression (P < 0.01) (Fig. 6b, f). NAC reduced AGEs-stimulated NF-κB translocation from cytoplasm to nucleus (P < 0.05) (Fig. 6c, d).

Role of NOX, iNOS, Nrf2 and ROS in AGEs-induced primary microglia oxidative and inflammatory damage. Cells were pretreated with API (a), SFP (b, f, g), NAC (c, d), SMT (e) for 1 h before AGEs for 24 h. The protein expressions by western blotting were normalized to β-actin or histone H3. All data are presented as mean ± SEM from three independent experiments. **P < 0.01, ***P < 0.001 versus control group. # P < 0.05, ## P < 0.01, ### P < 0.001 versus AGEs group
Discussion
In the present study, we investigated the effects of FPS-ZM1 on AGEs-induced activation of primary microglia in vitro by measuring the levels of oxidative stress and proinflammatory markers. Our results demonstrated that primary microglia were significantly activated by incubation with AGEs, with increased expression of RAGE, oxidative stress levels and inflammation. FPS-ZM1 blocked the above effects of AGEs.
Oxidative stress is involved in the progression of AD [28]. Activation of microglia induces oxidative stress via NOX2 activation, resulting in apoptosis of surrounding neurons [29]. Microglia produce ROS through NOX, especially NOX2 [30]. NOX2 activation can be regulated by its two cytoplasm subunits: p47phox and p67phox [30]. In the present study, FPS-ZM1 inhibited the AGEs-induced production of NOX2 and ROS, as well as cytosolic translocation of p47phox and p67phox to the membrane. Our findings demonstrated that NOX was an effector protein of RAGE during AGEs-induced ROS production in primary microglia cultures, and FPS-ZM1 effectively attenuated AGEs-induced RAGE dependent oxidative stress. Consistent with our study, AGEs have been found to induce oxidative stress of murine hepatic stellate cells, SH-SY5Y cells, vascular endothelial cells, and rat cortical neurons by modulating the RAGE-NADPH oxidase pathway [20, 31, 32]. In order to determine the role of NOX in AGEs stimulated primary microglia, we pretreated cells with a NOX inhibitor prior to AGEs treatment. Our results showed that API significantly inhibited the content of ROS, suggesting that AGEs interact with RAGE and subsequently induce NOX activation to promote ROS production.
Nrf2 is a pivotal redox regulator that integrates with antioxidant response elements (ARE) in promotor regions of the antioxidant target genes, including HO-1 [33]. Nrf2 and HO-1 can protect cells from oxidative stress [34]. Previous research has found that AGEs lead to Nrf2 mediated HO-1 induction in endothelial cells [35]. In the present study, AGEs increased the production of Nrf2 and HO-1 in primary microglia, indicating that AGEs could activate the compensatory protective effects of Nrf2 and HO-1. However, MDA content, which represents the products of lipid peroxidation, increased. In addition, GSH content and SOD activity, which are antioxidant markers, decreased significantly in AGEs-stimulated microglia. The presence of FPS-ZM1 decreased the oxidative stress state by elevating the production of Nrf2 and HO-1 while decreasing MDA content and elevating GSH content and SOD activity. Our findings suggest that inhibition of RAGE by FPS-ZM1 decrease AGEs-induced microglia-mediated oxidative stress through elevating the expression of Nrf2 and HO-1. Pretreatment with a Nrf2 activator prior to AGEs treatment significantly enhanced SOD activity and protein expression of HO-1, indicating that Nrf2 was a pivotal upstream regulatory factor of SOD and HO-1.
Microglial activation mediates the inflammatory response in AD [36]. Stimulation of N-11 microglia by AGEs activated the transcription factor NF-κB, resulting in transcription of genes coding downstream inflammatory markers such as IL-1, TNF-α and IL-6 [18, 19]. A previous report has shown that AGEs-induced COX-2 and iNOS overexpression induces the expression of PGE2 and NO in human osteoarthritic chondrocytes [37]. In the hippocampus of an AD brain, increased amounts of these inflammatory mediators have been detected in association with plaques and tangles [38, 39]. Further, high levels of iNOS and NO from activated microglia may result in neurodegeneration [40]. Similarly, COX-2 and PGE2 contribute to the initiation and progression of AD [41]. Thus, anti-neuroinflammatory agents have been considered to play a neuroprotective role in AD. A previous study has shown that FPS-ZM1 substantially suppresses microglial activation and NF-κB-related expression of microglia-induced proinflammatory cytokines, including TNF-α and IL-1β, in the hippocampus and cortex of APP transgenic mice [25]. Our results showed that AGEs augmented inflammatory responses in primary microglia by inducing nuclear translocation of NF-κB p65 and subsequently increasing expression of TNF-α, IL-1β, COX-2/PGE2 and iNOS/NO levels. FPS-ZM1 attenuated the expression of all these inflammatory mediators. Our results demonstrated that AGEs-induced RAGE dependent microglial activation and inflammatory response were blocked by FPS-ZM1. Thus, strategies such as blocking AGEs-RAGE ligation might exert neuroprotective effects against microglia-induced neuroinflammation. Other evidence showed that ROS could directly activate NF-κB [42]. Adding ROS inhibitor prior to AGEs treatment significantly inhibited nuclear translocation of NF-κB, indicating that ROS might induce an inflammatory release in AGEs-stimulated primary microglia.
In summary, our results suggest that FPS-ZM1, a new RAGE inhibitor, apart from downregulating RAGE expression, can also protect primary microglia from AGEs-induced RAGE/NF-κB-related oxidative stress and inflammatory cell injury by blocking the AGEs-RAGE interaction. Such a multifunctional agent may be a promising therapeutic approach for the treatment of AD. Further investigation should be conducted to explore the therapeutic effect of FPS-ZM1 on AGEs in vivo in AD mice.
References
Duyckaerts C, Delatour B, Potier MC (2009) Classification and basic pathology of Alzheimer disease. Acta Neuropathol (Berl) 118(1):5–36. doi:10.1007/s00401-009-0532-1
Giulian D (1999) Microglia and the immune pathology of Alzheimer disease. Am J Hum Genet 65(1):13–18. doi:10.1086/302477
Zhu X, Su B, Wang X, Smith MA, Perry G (2007) Causes of oxidative stress in Alzheimer disease. Cell Mol Life Sci 64(17):2202–2210. doi:10.1007/s00018-007-7218-4
Wilkinson BL, Landreth GE (2006) The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer’s disease. J Neuroinflamm 3:30. doi:10.1186/1742-2094-3-30
Blasko I, Grubeck-Loebenstein B (2003) Role of the immune system in the pathogenesis, prevention and treatment of Alzheimer’s disease. Drugs Aging 20(2):101–113
Lee YB, Nagai A, Kim SU (2002) Cytokines, chemokines, and cytokine receptors in human microglia. J Neurosci Res 69(1):94–103. doi:10.1002/jnr.10253
Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C (1993) Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia 7(1):75–83. doi:10.1002/glia.440070113
Stollg G, Jander S (1999) The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol 58(3):233–247
Arends Y, Duyckaerts C, Rozemuller J, Eikelenboom P, Hauw J (2000) Microglia, amyloid and dementia in Alzheimer disease: a correlative study. Neurobiol Aging 21(1):39–47
Singh R, Barden A, Mori T, Beilin L (2001) Advanced glycation end-products: a review. Diabetologia 44(2):129–146. doi:10.1007/s001250051591
Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, Manogue K, Cerami A (1994) Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci 91(11):4766–4770
Li XH, Lv BL, Xie JZ, Liu J, Zhou XW, Wang JZ (2012) AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol Aging 33(7):1400–1410. doi:10.1016/j.neurobiolaging.2011.02.003
Munch G, Thome J, Foley P, Schinzel R, Riederer P (1997) Advanced glycation endproducts in ageing and Alzheimer’s disease. Brain Res Rev 23(1–2):134–143
Gasic-Milenkovic J, Loske C, Deuther-Conrad W, Munch G (2001) Protein “AGEing”–cytotoxicity of a glycated protein increases with its degree of AGE-modification. Z Gerontol Geriatr 34(6):457–460
Srikanth V, Maczurek A, Phan T, Steele M, Westcott B, Juskiw D, Munch G (2011) Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol Aging 32(5):763–777. doi:10.1016/j.neurobiolaging.2009.04.016
Cai Z, Liu N, Wang C, Qin B, Zhou Y, Xiao M, Chang L, Yan LJ, Zhao B (2015) Role of RAGE in Alzheimer’s disease. Cell Mol Neurobiol. doi:10.1007/s10571-015-0233-3
Guglielmotto M, Aragno M, Tamagno E, Vercellinatto I, Visentin S, Medana C, Catalano MG, Smith MA, Perry G, Danni O (2012) AGEs/RAGE complex upregulates BACE1 via NF-κB pathway activation. Neurobiol Aging 33(1):196, e113–e196, e127
Dukic-Stefanovic S, Gasic-Milenkovic J, Deuther-Conrad W, Münch G (2003) Signal transduction pathways in mouse microglia N-11 cells activated by advanced glycation endproducts (AGEs). J Neurochem 87(1):44–55. doi:10.1046/j.1471-4159.2003.01988.x
Berbaum K, Shanmugam K, Stuchbury G, Wiede F, Korner H, Munch G (2008) Induction of novel cytokines and chemokines by advanced glycation endproducts determined with a cytometric bead array. Cytokine 41(3):198–203. doi:10.1016/j.cyto.2007.11.012
Yin QQ, Dong CF, Dong SQ, Dong XL, Hong Y, Hou XY, Luo DZ, Pei JJ, Liu XP (2012) AGEs induce cell death via oxidative and endoplasmic reticulum stresses in both human SH-SY5Y neuroblastoma cells and rat cortical neurons. Cell Mol Neurobiol 32(8):1299–1309. doi:10.1007/s10571-012-9856-9
Hong Y, Shen C, Yin Q, Sun M, Ma Y, Liu X (2016) Effects of RAGE-specific inhibitor FPS-ZM1 on amyloid-beta metabolism and AGEs-induced inflammation and oxidative stress in rat hippocampus. Neurochem Res 41(5):1192–1199. doi:10.1007/s11064-015-1814-8
Vicente Miranda H, Outeiro TF (2010) The sour side of neurodegenerative disorders: the effects of protein glycation. J Pathol 221(1):13–25. doi:10.1002/path.2682
Schmidt AM, Sahagan B, Nelson RB, Selmer J, Rothlein R, Bell JM (2009) The role of RAGE in amyloid-beta peptide-mediated pathology in Alzheimer’s disease. Curr Opin Investig Drugs 10(7):672–680
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B (2003) RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 9(7):907–913. doi:10.1038/nm890
Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, Love R, Perry S, Paquette N, Deane RJ (2012) A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest 122(4):1377
Lapolla A, Traldi P, Fedele D (2005) Importance of measuring products of non-enzymatic glycation of proteins. Clin Biochem 38(2):103–115. doi:10.1016/j.clinbiochem.2004.09.007
Tseng Y-T, Hsu Y-Y, Shih Y-T, Lo Y-C (2012) Paeonol attenuates microglia-mediated inflammation and oxidative stress-induced neurotoxicity in rat primary microglia and cortical neurons. Shock 37(3):312–318
Mohmmad Abdul H, Sultana R, Keller JN, St Clair DK, Markesbery WR, Butterfield DA (2006) Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid β-peptide (1–42), H2O2 and kainic acid: implications for Alzheimer’s disease. J Neurochem 96(5):1322–1335
Qin B, Cartier L, Dubois-Dauphin M, Li B, Serrander L, Krause KH (2006) A key role for the microglial NADPH oxidase in APP-dependent killing of neurons. Neurobiol Aging 27(11):1577–1587. doi:10.1016/j.neurobiolaging.2005.09.036
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87(1):245–313. doi:10.1152/physrev.00044.2005
Niiya Y, Abumiya T, Yamagishi S, Takino J, Takeuchi M (2012) Advanced glycation end products increase permeability of brain microvascular endothelial cells through reactive oxygen species-induced vascular endothelial growth factor expression. J Stroke Cerebrovasc Dis 21(4):293–298. doi:10.1016/j.jstrokecerebrovasdis.2010.09.002
Guimaraes EL, Empsen C, Geerts A, van Grunsven LA (2010) Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J Hepatol 52(3):389–397. doi:10.1016/j.jhep.2009.12.007
Cremers NA, Lundvig DM, van Dalen SC, Schelbergen RF, van Lent PL, Szarek WA, Regan RF, Carels CE, Wagener FA (2014) Curcumin-induced heme oxygenase-1 expression prevents H2O2-induced cell death in wild type and heme oxygenase-2 knockout adipose-derived mesenchymal stem cells. Int Mol Sci 15(10):17974–17999. doi:10.3390/ijms151017974
Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J (2016) Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci 73(17):3221–3247. doi:10.1007/s00018-016-2223-0
He M, Siow RC, Sugden D, Gao L, Cheng X, Mann GE (2011) Induction of HO-1 and redox signaling in endothelial cells by advanced glycation end products: a role for Nrf2 in vascular protection in diabetes. Nutr Metabol Cardiovasc Dis 21(4):277–285. doi:10.1016/j.numecd.2009.12.008
Cameron B, Landreth GE (2010) Inflammation, microglia, and alzheimer’s disease. Neurobiol Dis 37(3):503–509. doi:10.1016/j.nbd.2009.10.006
Nah SS, Choi IY, Lee CK, Oh JS, Kim YG, Moon HB, Yoo B (2008) Effects of advanced glycation end products on the expression of COX-2, PGE2 and NO in human osteoarthritic chondrocytes. Rheumatology 47(4):425–431. doi:10.1093/rheumatology/kem376
Ojala J, Alafuzoff I, Herukka S-K, van Groen T, Tanila H, Pirttilä T (2009) Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol Aging 30(2):198–209
Phillips EC, Croft CL, Kurbatskaya K, O’Neill MJ, Hutton ML, Hanger DP, Garwood CJ, Noble W (2014) Astrocytes and neuroinflammation in Alzheimer’s disease. Biochem Soc Trans 42(5):1321–1325
Brown GC (2007) Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem Soc Trans 35(Pt 5):1119–1121. doi:10.1042/BST0351119
Hoozemans J, Rozemuller J, Van Haastert E, Veerhuis R, Eikelenboom P (2008) Cyclooxygenase-1 and-2 in the different stages of Alzheimer’s disease pathology. Curr Pharm Des 14(14):1419–1427
Chandel NS, Trzyna WC, McClintock DS, Schumacker PT (2000) Role of oxidants in NF- B activation and TNF-gene transcription induced by hypoxia and endotoxin. J Immunol 165(2):1013–1021. doi:10.4049/jimmunol.165.2.1013
Acknowledgements
This work was supported by research grants from the National Natural Science Foundation of China (No. 30971036) and Shandong Provincial Natural Science Foundation of China (No. Y2008C13).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shen, C., Ma, Y., Zeng, Z. et al. RAGE-Specific Inhibitor FPS-ZM1 Attenuates AGEs-Induced Neuroinflammation and Oxidative Stress in Rat Primary Microglia. Neurochem Res 42, 2902–2911 (2017). https://doi.org/10.1007/s11064-017-2321-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-017-2321-x