
Abstract
Cancer cells develop and succeed by shifting to different metabolic programs compared with their normal cell counterparts. One of the classical hallmarks of cancer cells is their higher glycolysis rate and lactate production even in the presence of abundant O2 (Warburg effect). Another common metabolic feature of cancer cells is a high rate of glutamine (Gln) consumption normally exceeding their biosynthetic and energetic needs. The term Gln addiction is now widely used to reflect the strong dependence shown by most cancer cells for this essential nitrogen substrate after metabolic reprogramming. A Gln/glutamate (Glu) cycle occurs between host tissues and the tumor in order to maximize its growth and proliferation rates. The mechanistic basis for this deregulated tumor metabolism and how these changes are connected to oncogenic and tumor suppressor pathways are becoming increasingly understood. Based on these advances, new avenues of research have been initiated to find novel therapeutic targets and to explore strategies that interfere with glutamine metabolism as anticancer therapies. In this review, we provided an updated overview of glutamine addiction in glioma, the most prevalent type of brain tumor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Malignant transformation proceeds with changes and modulations (rewiring) of key metabolic routes allowing high rates of growth and proliferation to cancer cells, while also permits adaptation in response to targeted molecular treatments. Metabolic reprograming is now recognized as a major hallmark of cancer and includes dysregulation of normal metabolic pathways and novel activities confined to the tumor [1]. In many tumors, high aerobic glycolysis (Warburg effect) and high rate of glutamine (Gln) consumption for energetic and anaplerotic purposes (glutaminolysis) appear as essential characteristics of their altered metabolism. This anomalous Gln uptake, normally exceeding their biosynthetic and energetic needs, was recognized in pioneer studies of tumor metabolism in animal models [2, 3]. The term Gln addiction was coined to reflect the strong dependence shown by most cancer cells for this essential nitrogen substrate after metabolic reprogramming [4].
Glutamate (Glu) homeostasis is essential for normal cerebral function. The Glu/Gln cycle between neurons and astrocytes at the tripartite synapse is a key mechanism for homeostatic control of Glu, Gln, and GABA concentrations. It plays an essential role by keeping Glu concentrations below excitotoxic levels while supplying an adequate neurotransmitter pool [5, 6]. Thus, the Glu released by neurons is taken up by nearby astrocytes at the synaptic cleft through efficient Glu transport systems and then converted to Gln by glutamine synthetase (GS; EC 6.3.1.2), an enzyme exclusively located in astrocytes [7]. The Glu-derived Gln is finally exported back to neurons where phosphate-activated glutaminase (GA; EC 3.5.1.2) generates neurotransmitter Glu. The importance of GA in glutamatergic synaptic function has been largely recognized. Physiological, biochemical, immunological and NMR spectroscopic data indicate that neurotransmitter Glu is mainly generated through GA reaction [8]. Nevertheless, transamination of α-ketoglutarate involving tricarboxylic acid cycle (TCA) reactions also contributes to generation of neurotransmitter Glu [9].
Brain Glu homeostasis is disrupted in numerous neurological diseases, as well as in most malignant types of cerebral tumors as gliomas [10]. Dysfunction and/or expression changes of Glu transporters, receptors and enzymes involved in Gln/Glu metabolism give rise to altered Glu levels, which have been implicated in the pathology of these diseases. Excitotocixity is frequently seen as a common trait in neurological pathologies [11], including cancer [12]. Also, Gln metabolism exhibits distinct features in brain cancer with great relevance on the metabolic rewiring of proliferating cells. In this article, we review Glu homeostasis in gliomas by focusing on key Gln metabolism-related proteins: glutamine synthetase (GS) and glutaminases (GAs), while Glu receptors will remain out of the scope of this mini-review.
Genomic Landscape of Gliomas
Gliomas are characterized by high invasiveness, rapid proliferation, resistance to apoptosis and short survival times. Gliomas are the most common type of brain tumor in humans, accounting for 80% of malignant CNS tumors [13]. The most deadly and aggressive subtype of glioma is glioblastoma multiforme (GBM) [world health organization (WHO) grade IV astrocytoma] [14]. Gliomas may arise from adult stem and progenitor cells, as well as from differentiated brain cell types like oligodendrocyte precursor cells, astrocytes and neurons [15 and references therein]; nevertheless, adult high-grade gliomas most commonly arise from astrocytic cells in the CNS [16].
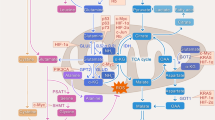
Massive genomics studies led by the Cancer Genome Atlas (TCGA) research network have now generated a comprehensive list of genomic alterations in GBM tumors [17–19]. Glioma driving events have been associated with mutations, deletions, amplifications and translocations of signature genes, including oncogenes such as EGFR, PI3K, PIK3CA, BRAF, FGFR1/2/3, KRAS, NRAS, and tumor-suppressor genes like TP53, PTEN, NF1, PIK3R1 and RB1 [18–20] (Fig. 1). Also, recurrent mutations in the active site of isocitrate dehydrogenase 1 (IDH1) and 2 (IDH2) isoforms are mostly found in a distinct GBM subtype (proneural) with increased overall survival [20, 21]. Newly predicted drivers of gliomagenesis are genes associated with chromatin organization/remodeling (SETD2, ARID2, DNMT3A) and transcriptional regulation (CIC, FUBP1) [20]. Amplifications of PDGFRA and MYC genes are also markers of the proneural subtype of gliomas, which present a survival advantage conferred by its hypermethylated CpG island phenotype [18, 19]. Despite the broad molecular heterogeneity revealed by these genomic analyses, most genetic drivers of glioma can be assigned into main cell signaling pathways, namely Ras-Raf-MEK-ERK, p53/apoptosis, PI3K/AKT/mTOR, chromatin modification, and the anti-apoptotic retinoblastoma pathway [18–20] (Fig. 1). In addition, the maintenance of telomeres seems to be a critical step in GBM pathogenesis; glioma cells may achieve this goal by two alternative mechanisms: either through reactivation of telomerase by telomerase reverse transcriptase (TERT) promoter mutations (increasing TERT expression), or by alternative lengthening of telomeres (ALT) via mutations in the ATRX gene, which encodes an ATP-dependent helicase critically involved in the ALT process [19, 20, and references therein].

Adapted from Brennan et al., 2013 [19]. (Color figure online)
Alterations of critical genes in the PI3K/PTEN, TP53 and RB1 pathways by point mutations, amplifications and homozygous deletions, corresponding to the analysis by whole-exome sequencing of 251 GBM. Mutated genes functionally linked to chromatin modifications are also shown. Numbers under genes identifiers indicate: in red, fraction of tumors with point mutations and/or amplifications in those genes; in green, fraction of tumors with point mutations and/or deletions.
Notwithstanding the increasing number of genes added to the list of glioma drivers in recent years, only a few molecular factors have shown prognostic utility or predictive value for therapy response in glioma patients. The methylation status of the MGMT promoter is one of them: gene silencing of MGMT, due to methylation of its promoter region, predicts a better response to chemotherapy with alkylating agents like temozolomide in malignant gliomas [17].
Glutamate Homeostasis in Gliomas
Supply of (and dependence on) Glutamine
A century ago Müller reported a negative nitrogen balance in tumor-bearing patients [22]. In 1951, Mider classified tumors as “nitrogen traps” indicating their ability to compete with advantage for host nitrogen compounds [23]. This process produces in the host a negative nitrogen balance and a characteristic weight loss, along with a reciprocal nitrogen increase in the tumor. For example, Shrisvastava and Quastet [24] incubated brain tissue with Ehrlich ascites tumor cells and observed a net Gln flux from brain cortex cells to tumor cells. These experiments were considered as an in vitro model for the role of tumors as nitrogen sinks. Interestingly, many types of tumors behave as “Gln traps” and the source of Gln are mainly the host tissues because tumors usually show repression of their GS activity; however, there are also Gln-independent tumors which satisfy their Gln requirements through GS reaction and show resistance to shortage or depletion of Gln supply [25].
A Gln/Glu cycle between host tissues and the tumor has been proposed from dynamic studies dealing with inter-organ glutamine metabolism in model systems [26, 27]. Support for the existence of this cycle in vivo has come also from studies on enzymatic activities of GS and GA in host tissues during tumor development. Thus, in rats and mice bearing fibrosarcomas, hepatomas, Lewis lung carcinomas and Ehrlich ascitic tumors, the muscle, liver and kidney became net Gln exporters while Gln utilization by the gut was reduced, allowing a net increase in circulating Gln [reviewed in 28–30]. These adaptive changes in gene expression for GS and GA enzymes would explain the high Gln uptake showed by tumor cells. The results suggest a long-term regulation of host enzymes in order to increase the circulating Gln levels needed for tumor growth. Thus, the tumor elicits a specific response in the host nitrogen metabolism so that the whole organism is mobilized to augment circulating Gln levels.
Unlike other peripheral tumors, gliomas seem to have secured an extra supply of Gln in addition to the blood circulating Gln levels. Thus, a recent metabolomics study done with seven GBM patients injected with 13C6-glucose has shown that the fraction of Glc-derived Gln found in the tumor was higher than in the serum, suggesting that tumor Gln can be synthesized in situ and/or provided by neighboring normal astrocytes [31] (Fig. 1). Thus, the microenvironment of gliomas does not seem to pose a serious threat on Gln supply, because of the presence of GS-positive normal astrocytes. Therefore, Gln-limited conditions seem highly improbable in the physiological microenvironment of GBM and, hence, GS expression by gliomas would not be critical for tumor growth [31].
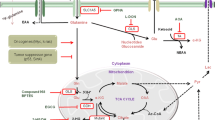
Gliomas are Gln-addicted cells because their increased biosynthetic and energetic needs require consumption of this “conditionally essential” amino acid as an additional fuel, apart from glucose, to sustain their proliferative program. In fact, increased Gln uptake and metabolism has been exploited to monitor gliomas by positron emission tomography (PET) and magnetic resonance imaging [29]. Many cancers show altered glucose metabolism, which constitutes the basis for in vivo PET imaging with (18)F-fluorodeoxyglucose (18F-FDG). However, 18F-FDG is ineffective in evaluating gliomas because of high background uptake in the brain. In contrast, in vivo PET imaging with the Gln analog 4-(18)F-(2S, 4R)-fluoroglutamine (18F-FGln) shows high uptake in gliomas but low background brain uptake, facilitating clear tumor delineation [32]. Of note, Gln transport agencies at the glioma plasma membrane are induced with regard to normal astrocytes to facilitate Gln entry and further catabolism (Fig. 2). Thus, an increased expression of Gln transporter SNAT3 in GBM tissues, compared to lower grade glioma and normal brain, has been reported as a marker of malignant gliomas [33]. Moreover, human GBM cell lines and rat C6 glioma cells also showed upregulation of the ASCT2 (SLC1A5) Gln transporter [34, 35] (Fig. 2). Interestingly, the ASCT2 transporter is responsible for the accumulation of Gln in rapidly growing cells in cultures, especially epithelial cells and multiple types of cancer, and its expression is upregulated by Gln availability [36] and controlled by c-Myc. In addition, the entry of Gln through ASCT2 transporter promotes mammalian target of rapamycin complex 1 (mTORC1) activity: essential amino acids (EAA) as Leu are incorporated into the cell in exchange for Gln and activate mTORC1 to support cell growth and proliferation [37].

Main changes in Gln/Glu-related metabolic pathways and membrane carriers after malignant transformation in gliomas. In the transformation from normal astrocytes to malignant glioma, upregulated proteins are shown in green while downregulated or non-functional proteins are labeled in red. Enhanced Gln uptake takes place through SNAT3 and ASCT2 carriers. The metabolic reprogramming of glioma cells includes GLS overexpression and GLS2 repression. However, expression of GS enzyme is highly variable in gliomas (this fact has been indicated by a dashed arrow on the GS reaction). Several sources of intracellular Glu are indicated. A great part of this Glu exits the cell through the overexpressed Xc− antiporter (its catalytic xCT subunit is shown), but cannot be imported back into the cell because of the lack of functional EAATs transporters. GDH and the aminotransferase BCAT1 are upregulated and can be essential regulators of Glu and α-KG levels. Other important enzymes of nitrogen metabolism shown are transaminases (GOT and GPT) and asparagine synthetase (ASNS). The synthesis of the oncometabolite D-2-hydroxiglutarate (2-HG) (characteristic for a subtype of gliomas with mutant IDH1/2 enzymes) is also indicated, as well as its epigenetic effects in the cell nucleus. Protein abbreviations are indicated in the text. (Color figure online)
The Gln dependence of cancer cells is a key phenotypic trait which needs to be addressed for correct metabolic classification and adequate design of therapeutic strategies. The importance of Gln for some cultured tumor cells was stressed by early reports showing that the oxidative metabolism of Gln becomes the main source of energy with preference to glucose [38, 39]. In tumor cells Gln can serve as an alternative substrate for the TCA cycle and ATP production during aerobic glycolysis. Accordingly, Yuneva and coworkers demonstrated that deficiency in Gln, but not glucose, induced Myc-dependent apoptosis in cultured human fibroblast cells due to an unexpected depletion of most Krebs cycle intermediates [40]. In fact, Myc is another oncogene associated with a poor prognosis in glial tumors [41] and seems to sensitize cancer cells to Gln addiction. In support of this notion, metabolomic studies revealed the use of Gln as the major anaplerotic precursor in human glioma cells [42]. Sound experimental evidences were later found supporting the role of Myc in metabolic reprogramming of SF188 glioma cells (originally isolated from a patient whose tumor displayed Myc amplification) leading to Gln addiction [4]. Even more, Myc-overexpression in mouse embryonic fibroblasts induced key genes of glutaminolysis as Gln transporters (such as ASCT2 and SN2), GA, and lactate dehydrogenase A (LDH-A) which converts Gln-derived pyruvate into lactate [4]. The enhanced cellular dependence on Gln and glutaminolysis induced by Myc was justified by anaplerotic requirement in sustaining the TCA cycle and cell viability [4, 40], and by stimulation of NADPH needed to support biosynthetic purposes for oncogenic growth [42].
The existence of de novo Gln synthesis through GS reaction shows considerably heterogeneity in gliomas. Thus, exogenous glutamine was limiting for the proliferation of glioma-derived lines D-54 MG, U-118 MG and U-251 MG, but not for glioma-derived lines U-373 MG, D-245 MG, and D-259 MG grown in the absence of supplemental Gln [43]. In vitro cultures of rat glioma C6 cells were highly dependent on Gln and showed a significant upregulation of GS expression after Gln deprivation; hence, authors concluded that coupling glutaminolysis and de novo Gln synthesis was essential for growth and proliferation of these cells [44]. In contrast, newborn rat astrocytes lost their capacity of GS induction after spontaneous neoplastic transformation in culture [45]. In a recent study, with carefully controlled media composition and physiological Gln concentrations, the Gln requirements and GS expression of six established human glioma cell lines were determined [31]. A great variation was found under Gln deprivation conditions; thus, cell growth inhibition ranged from 20% for U251 and SF188 cells to 80% for LN18 cells. In agreement with growth inhibition data, U251 and SF188 glioma cell lines showed the highest GS expression. Furthermore, clear differences in Gln dependency were also found between patient-derived primary human GBM stem-like cells (GSC) and differentiated cells: the expression of GS was considerably higher in GSCs which were also able to grow independently of Gln supplementation [31]. Finally, the same study examined GS expression in human GBM tumors using tissue microarray (TMA) analysis (n = 209 patients). The authors found that GS expression varies greatly between tumors, ranging from negative, comparable to neurons (25% of patients), to high-expression tumors comparable to astrocytes (15%), although GS expression did not predict patient median survival [31]. Finally, human orthotopic gliomas growing in mice synthesized Gln de novo and did not show enhanced glutaminolysis; instead of it, these GBM cells utilized mitochondrial glucose oxidation during aggressive tumor growth in vivo and pyruvate carboxylation for anaplerosis [25].
Generation and Release of Glutamate
Under normal physiological conditions, astrocytes can handle most of the Glu released at the synaptic space through high-capacity glial excitatory amino acid transporters, namely EAAT2 (GLT1) and EAAT1 (GLAST). Then, the cytosolic GS activity of astrocytes converts most of this Glu to Gln for neuronal reuptake. However, marked alterations of glutamate homeostasis occur in gliomas yielding a glutamatergic dysregulation with great repercussions in the progression and metastasis of glioma [10, 45, 46] (Fig. 2). The generation of Glu in gliomas could be mainly ascribed to GA, GDH and aminotransferase enzymes, because gliomas have lost their capacity to take up Glu, one metabolic hallmark shown by their non-malignant counterparts (Fig. 2). Microarray analysis of human glioma biopsies demonstrated the absence of EAAT2, the main Glu transporter in the normal mature brain [47]. Further studies discovered that most commonly established glioma cell lines have no functional activity of any EAAT agency [reviewed in 10].
The absence of synaptic Glu re-uptake and the seemingly well assured supply of Gln coming from the microenvironment suggest an ideal scenario for GA upregulation in gliomas. Four different GA isoenzymes have been described so far in mammalian tissues which are encoded by separate genes in different chromosomes [48–50]. In humans, the GLS gene is located in chromosome 2 and encodes isozymes termed KGA and GAC, whilst the GLS2 gene on chromosome 12 codes for isozymes called GAB and LGA [51]. The human GLS and GLS2 isozymes exhibit distinct tissue distributions and are regulated quite differently [52]. Primary cultures of astrocytes displayed efficient Gln uptake, strong GA activity [53, 54] and expression of GA mRNA transcripts [55]. However, these in vitro results were questioned arguing that GA may somehow be induced by the Gln present in the growth media and by the length of culturing [reviewed in 56]. Nevertheless, normal astrocytes from human and rat brain have been recently reported to express functional GLS (KGA) and GLS2 GA isoforms, although available experimental data predicts an in vivo GA activity considerably lower in astrocytes as compared with that shown by neurons [57].
With regard to gliomas, established cell lines and patient tissues displayed a consistent pattern of GA expression: high levels of GLS isoforms but only traces or lack of GLS2 transcripts [58] (Fig. 2). Although Gln addiction can differ considerably between cancer cell lines, cell proliferation rate is consistently dependent on Gln availability [59]. The major degradative pathway for Gln and the first step in glutaminolysis is carried out in mitochondria and initiated by GA [28]. As mentioned previously, GA also plays a key role in tumorigenesis. Thus, it is well documented that many tumors show an increased GA activity which is positively correlated with their malignancy [30]. The upregulation of GLS isoforms seems a general feature exhibited by many types of cancer and experimental tumors. In gliomas, it has been proposed that c-Myc determines Gln addiction by upregulation of genes coding for proteins required by Gln uptake and metabolism like ASCT2 and GLS [4, 42]. The molecular mechanism connecting GLS upregulation, glutaminolysis activation and oncogene c-Myc was discovered in human Burkitt lymphoma and prostate cancer cells [60] (Fig. 3). Mitochondrial GLS protein was induced tenfold in response to c-Myc, although its mRNA levels did not vary significantly, which suggested a regulation at the posttranscriptional level. Hence, the authors demonstrated an indirect mechanism of regulation through effects on the miRNAs miR23a and miR23b. Normally, these miRNAs bind to the 3′-untranslated region (3′-UTR) of the GLS gene and prevent translation of the message. However, c-Myc suppresses miR-23a/b expression and thus derepresses GLS translation, facilitating Gln oxidation in the mitochondria [60] (Fig. 3). It is currently unknown whether a similar miRNA mechanism would contribute to GLS activation in gliomas.

Opposing roles of GLS and GLS2 glutaminase isoforms in tumorigenesis. GLS-encoded isoenzymes (mainly the GAC isoform) are upregulated in parallel with the proliferation rate. GLS isoforms are induced in many tumors by the oncogene c-Myc through a miRNA mechanism. In sharp contrast, GLS2-encoded isoenzymes are induced by the tumor suppressor p53 and related proteins p63 and p73, and are related to quiescent, nonproliferating, and differentiated cell states
Interestingly, GLS isoforms are also upregulated by other oncogenic signaling pathways such as the small Rho GTPases, through activation of nuclear factor-kappa B (NF-κB), an important regulator of cell survival, proliferation, and differentiation frequently involved in malignant transformation [61]. Also, in relation with signaling pathways controlling GLS isoforms, a synergistic cross-talk between KGA-mediated glutaminolysis and epidermal growth factor (EGF)-activated Raf-Mek-Erk signaling was reported in human 293 T cells [62]. Upregulation of GLS seems to be a mechanism by which cancer cells gain selective advantages for using alternative sources of carbon favoring their adaptation to changing metabolic environments. Therefore, tumor microenvironment is crucial to understand the metabolic adaptations underpinning the malignant program of cancer cells. For example, the activation of GLS expression, paralleled by GS repression in some types of tumors, is strictly dependent on the genetic lesion and the tissue of origin [62]. Thus, Yuneva and co-workers demonstrated that in MYC-induced liver tumors, the expression of GS was suppressed and expression of the GLS2-encoded LGA isoform was replaced by the catalytically more potent GLS isoform. Moreover, MYC-induced tumors exhibited increased expression of the high-affinity Gln transporter ASCT2 (SLC1A5). In sharp contrast to liver tumors, MYC-induced lung tumors display increased expression of both GS and GLS and accumulate Gln [62]. In line with this, SF188 glioma cells overexpressing c-Myc also showed high levels of GS mRNA and protein [31], supporting the view that variable Gln metabolism in MYC-induced tumors depends on the tissue of origin. In addition to tissue microenvironment, the oncogene also influences the metabolic adaptations regarding Gln/Glu metabolism, as was clearly demonstrated in liver tumors: MET-induced hepatic tumors produced Gln, while Myc-induced catabolized it [62].
In contrast to GLS isoforms, the role of GLS2 isoenzymes in tumor cells is greatly unknown. Co-expression of GLS and GLS2 transcripts has been reported in established cancer cell lines of colon, hepatoma, leukemia and breast, although protein data suggest that GLS isoforms would account for the majority of GA activity in these human tumor cells [63, 64]. In fact, GLS2 expression is downregulated in highly malignant glioblastoma [58], as well as in human liver and colon cancers [65–67]. Indeed, GLS2 was confirmed as a target gene of the tumor suppressor p53 in both non-tumor and tumor cells [65, 66] (Fig. 3). Therefore, its repression in cancer cells might be explained by p53 mutations and lack of function frequently found in gliomas and other highly malignant tumors (Fig. 1). However, the GLS2 silencing mechanism so far identified in glioblastoma, liver and colorectal cancer is promoter methylation, with independency of the p53 status [67, 68]. Although repression of GLS2 is a frequent trait associated with tumorigenesis, GLS2 is also upregulated in some types of cancer. For example, the expression of GLS2 was significantly enhanced in cervical carcinoma; even more, this upregulation was related to therapeutic resistance [69]. Of note, GLS2 was the main GA isoform induced in MYC-N amplified human neuroblastomas, instead of GLS, and correlated with unfavorable patient survival [70].
As mentioned previously, the intracellular Glu pool in glioma cells can be supplied by other enzymatic activities distinct of GAs. Mitochondrial Glu can be converted to α-KG by glutamate dehydrogenase (GDH) or intramitochondrial aminotransferases, including glutamate pyruvate transaminase (GPT, alanine aminotransferase) and glutamate oxaloacetate transaminase (GOT, aspartate aminotransferase) (Fig. 2). Both GDH and transaminases can also operate in the reverse direction, that is, synthesis of Glu from α-KG; again, context-dependent and tumor-specific regulation will determine whether these enzymes contribute to Glu or α-KG generation. Thus, while quiescent cells displayed enhanced levels of GDH and reduced levels of transaminases, in proliferating breast cancer cells GDH mRNA negatively correlates with proliferative gene signatures, whereas transaminases were highly expressed in proliferative tumors which catabolize Glu via transaminases to synthesize non-essential amino acids (NEAAs) [71]. However, in human GBM the pattern was the opposite: GDH expression and activity were upregulated in human glioma cell lines and tissues, being considered as an oncogenic factor whose high expression levels predicts poor outcome. Of note, GDH act as a key regulator for intracellular α-KG in glioma cells and becomes a key salvage pathway to survive impairments of glucose metabolism [72]. The mRNA and protein levels of GDH were particularly increased in IDH1 mutant GBM relative to IDH1 wild-type GBM [17, 73], probably to replenish the α-KG lost by conversion to 2-HG (Fig. 2). Finally, GDH has been recently identified as the molecular connection between glutaminolysis and mTORC1 signaling. Activation of mTORC1, a key regulator of nutrient uptake and cellular proliferation, has been linked to Gln addiction in cancer cells: mTORC1 stimulates Gln metabolism by inducing GDH [74], while glutaminolysis and cellular level of αKG also activates mTORC1 thereby promoting cell growth and inhibiting autophagy [75].
Other two enzymes of nitrogen metabolism which may contribute to the intracellular Glu pool in gliomas are asparagine synthetase (ASNS) and branched-chain amino acid transaminase 1 (BCAT1) (Fig. 2). Thus, in a recent study analyzing gliomas from 156 patients, the gene expression patterns for GLS, BCAT1 and ASNS were determined [76]. ASNS and GLS transcripts were twofold higher in GBMs compared to anaplastic gliomas, while BCAT1 mRNA expression was also higher in GBM. Further, newly diagnosed GBMs showed poorer survival if any of these three genes had increased expression. Branched-chain amino acids have recently emerged as important external source of amino groups for the synthesis of brain Glu (reviewed in [77]). It is noteworthy that expression of high levels of BCAT1 accelerates amino acid catabolism and promotes cell proliferation in gliomas [78]. Interestingly, overexpression of BCAT1 was exclusive to tumors carrying wild-type IDH1 and IDH2 genes (Fig. 2), dependent on the concentration of αKG and suppressed by ectopic overexpression of mutant IDH1, revealing a link between IDH1 function and BCAT1 upregulation [78]. The central role of BCAT1 in glioma pathogenesis was further underscored by reduced tumor growth in vivo and decreased proliferation and invasiveness in vitro after suppression of BCAT1 expression.
The induction of BCAT1 occurs only in gliomas expressing wt IDH1/2. Therefore, this subtype of gliomas might have a constant supply of Glu (and αKG) through BCAT1 and GLS activities. However, in mutant IDH1/2 gliomas, BCAT1-induction disappears [73, 78] and Glu production must rely essentially on GLS, because GDH has been reported to operate chiefly in the generation of αKG, particularly in mutant IDH1/2 GBM [73]. Therefore, it is tempting to speculate that this strong dependence on GLS for Glu generation in mutant IDH1/2 gliomas explains why this tumor subtype is particularly susceptible to GLS inhibition [79].
In agreement with the previously cited Gln/Glu cycle occurring between peripheral tumors and host tissues, where the enhanced uptake of Gln by tumors is usually accompanied by an efflux of Glu and ammonia toward the host tissues, the Glu generated by the altered nitrogen metabolism of gliomas can also exit the cell because, unlike their non-malignant counterparts, gliomas release Glu (Fig. 2). Cultured glioma cells secrete excitotoxic levels of Glu [80] and implanted glioma cells continue to secrete neuro-excitotoxic amounts of Glu in vivo, thus promoting their growth in the brain [81]. This exacerbated Glu efflux takes place mainly through the cystine/Glu antiporter or system Xc −, a transporter importing cystine in exchange for Glu which is overexpressed in human gliomas (Fig. 2) [46]. Cystine is a precursor for the cellular synthesis of reduced glutathione (GSH) that protect cells from oxidative stress; for example, this system is upregulated under stress conditions and protects gliomas against the radiation-induced damage (Fig. 2). Clinical studies have shown that Glu concentration in cerebrospinal fluid from glioma patients may rise up to 400 µM, which is 400-fold the physiological value and well above the neurotoxicity level [reviewed in 10]. The Glu-enriched microenvironment of glioma cells confers several adaptive advantages. These tumors have a distinct growth advantage because the excess of Glu acts as an excitotoxic weapon killing surrounding neurons by over-activation of N-methyl-D-aspartate receptors (NMDARs) and vacating space needed for glioma proliferation in the cranial cavity [45]. The increased extracellular Glu concentration is responsible for tumor-associated seizures and epileptic activity observed in glioma patients [12]. Also, increased uptake of cystine facilitates GSH production and enables ROS detoxification [10]. Finally, with regard to invasiveness properties, the secreted Glu has been shown to possess an autocrine/paracrine effect promoting cell invasion, probably mediated through α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) [82].
Targeting GA Isoforms as Anticancer Strategy in Gliomas
We first postulated a completely different role for GLS and GLS2 isoforms in cancer (Fig. 3). The hypothesis was put forward before knowing the relationship of GA isoforms with oncogenes and tumor suppressor genes and based on experimental evidences obtained from tumor and non-tumor cells, which demonstrated that isoforms encoded by GLS are upregulated in parallel with the proliferation rate, whereas isoforms encoded by GLS2 are related to quiescent, non-proliferating, and differentiated cell states [64]. Consequently, we proposed that the process of malignant transformation shifts the pattern of GA expression in such way that GLS becomes up-regulated while GLS2 is frequently repressed (Fig. 3). Then, a molecular basis for this hypothesis came from recent studies in human cancer cell lines linking GLS to oncogene c-Myc [60] and GLS2 to tumor suppressor p53 [65, 66].
In line with the above mentioned working hypothesis, blocking of GLS expression was thought as a plausible gene therapy strategy targeting GA isoforms mainly responsible of the Gln-addicted phenotype of some type of tumors. We first reported that inhibition by antisense technology of Gls expression (KGA isoform), an enzyme linked to neoplastic transformation, allowed reversion of tumor cells to a more differentiated and less malignant phenotype. Thus, EATC transfected with antisense KGA cDNA constructs (0.28AS-2 cell line) were markedly impaired in their growth and proliferation capacity, showed marked changes in their morphology and lost their tumorigenic capacity in vivo [83]. Moreover, knocking down Gls induced apoptosis in 0.28AS-2 cells, caused oxidative stress and sensitized the cells to methotrexate [84]. Silencing GLS in GBM cells using RNAi technology also suppressed, but did not eliminate, their growth in culture and in vivo [85]. In fact, GLS was required for maximal growth of GBM cells in culture and in vivo: colony formation and growth of s.c. xenografts were significantly reduced after GLS knockdown [85].
We also hypothesize that tumor could be inhibited by GLS silencing or, alternatively, by GLS2 overexpression. Human GBM T98G cell line expresses high amounts of GLS transcripts, while GLS2 transcripts are hardly detectable in these cells [58]. Furthermore, in view of the nuclear location of GLS2 in neurons [86] and astrocytes [57] and its presumed role in modulation of gene transcription [86], we hypothesized that its deficit has implications for the physiology of glia-derived tumors, perhaps driving them toward a malignant phenotype. To address this question, human glioblastoma T98G cells (having negligible levels of GLS2 expression) were stably transfected with the full GAB cDNA coding sequence and the effects of transfection on basic physiological parameters were assessed: proliferation, migration and survival. The transfected cells (T98-GAB cells) showed a 40% decrease of cell survival, a 45% reduction of cell migration and a 47% decrease in the proliferation index. Microarray analysis revealed a significantly altered expression of 85 genes in T98-GAB, but not in sham-transfected or control cells. Microarray data, which included over 47,000 transcripts, were confirmed by qPCR analysis for eight genes potentially relevant to glioma malignancy: S100A16, CAPN2, FNDC3B, DYNC1LI1, TIMP4, MGMT, ADM, and TIMP1 [87]. Indeed, GLS2 has been shown to act as a tumor suppressor in human cancer cells from liver, colon and lung [66, 67, 88].
An intriguing question arose whether or not combination of GLS silencing and GLS2 overexpression would increase the inhibition of cell proliferation and survival of glioblastoma cells elicited by individual manipulations. To answer this question, the expression of KGA and GAC isoforms was knocked down with siRNA in a human glioblastoma cell line that was (T98-GAB cells) or was not (T98G cells) previously transfected with GAB cDNA, respectively [87]. Then, cell viability and proliferation were investigated in so treated cells with a graded inhibition of KGA and GAC, in order to analyze the correlation between the phenotypic changes and the Gln content of the cells as a marker of the intensity of its consumption. In both T98G and T98-GAB cell lines, silencing of GLS decreased cell viability and proliferation in a different, sequence-dependent degree, and the observed decreases were in either cell line highly correlated with increase of intracellular Gln, a parameter manifesting decreased Gln degradation [89]. The results show that combination of negative modulation of GA isoforms arising from GLS gene with the introduction of the GLS2 gene product, GAB, may in the future provide a useful means to curb glioblastoma growth in situ. At the same time, the results underscore the critical role of Gln degradation mediated by KGA in the manifestations of aggressive glial tumor phenotype.
In another recently published study, glioma SFxL and LN229 cells, with silenced GLS expression [85], were employed along with glioma T98-GAB cell line to ascertain whether modulations of GA expression may synergize with oxidative stress against proliferation of glioma cells [90]. GLS-silenced glioma cells showed lower survival ratios and a reduced GSH-dependent antioxidant capacity. Silencing GLS or overexpressing GLS2 genes decreased glioma cell survival. This effect was increased by an oxidative insult. Furthermore, ROS generation by treatment with oxidizing agents synergized with either GLS silencing or GLS2 overexpression to suppress malignant properties of glioma cells, including the reduction of cellular mobility. Of note, blocking GLS or overexpressing GLS2 evoked lower c-Myc and Bcl-2 expression, as well as higher pro-apoptotic Bid expression [90]. In conclusion, combination of modulation of GA expression and treatment with oxidizing agents may become a therapeutic strategy for gliomas and other intractable cancers.
On the other hand, pharmacological strategies to inhibit Gln metabolism in cancer cells are rapidly evolving from Gln analogs, like acivicin, azaserine and 6-diazo-5-oxo-l-norleucine (DON) [28], to novel isoform-specific allosteric inhibitors discovered in recent years like BPTES [bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide], compound 968, CB-839 and Ebselen (Fig. 2) [29, 61, 91, 92]. While Gln analogs lack specificity and induce important secondary effects and toxicity, some of the new GLS inhibitors are not drug-like compounds because of their high molecular weight, poor solubility and low bioavailability [92]. Notwithstanding the limitations of these novel drugs for human use, they are becoming useful tools for selective inhibition of glutaminolysis in tumors, in order to ascertain the relevance of Gln and GA in cancer metabolic reprogramming. For example, two recent studies have attracted attention at combined anticancer therapies using GLS inhibitors in T-cell acute lymphoblastic leukemia (T-ALL) [93] and human GBM [94]. Anti-NOTCH1 therapy in T-ALL and mTOR inhibitor treatment in human GBM found resistance in many patients due to compensatory Gln metabolism induced by metabolic reprogramming. In both cases, GLS upregulation and enhanced glutaminolysis were key determinants of the response to each therapy. Remarkably, combined genetic and/or pharmacological inhibition of NOTCH1/GLS and mTOR/GLS in T-ALL and GBM, respectively, resulted in massive synergistic tumor cell death and growth inhibition in tumor-bearing mice [93, 94].
Future Directions
Overwhelming evidences now support the key role of GA isoforms in cancer cell growth, proliferation and metastasis. In gliomas, enhanced glutamine catabolism may be essential for energy generation, anaplerosis, nucleotide/lipid biosynthesis, generation of GSH and invasive properties. The enhanced glutaminolysis seems a hallmark of in vitro glioma cell lines, but differences in Gln dependence and glutaminolysis rates have been found in studies dealing with in vivo GBM model, because the particular tumor microenvironment in brain must be taken into account.
In summary, GA is essential to the metabolic phenotype of growing tumors, including gliomas. The proliferative programme of cancer cells can be stopped either by knocking-down GLS or by up-regulating GLS2 isoforms, in agreement with their seemingly opposing roles in cancer. Although it is presently unknown how GA isozymes may undergo such different roles in tumor biology, the control of GA isozyme expression may prove to be a key tool to alter both metabolic and oxidative stress in cancer therapy. At first glance, these contrasting roles of GA isozymes may appear inconsistent, as well as the fact that glutaminolysis in cancer can be activated by c-Myc for tumorigenesis and also by p53 for tumor suppression. However, there are some hints that may help to explain this apparently puzzling behavior. Recently, novel GA isoforms and extramitochondrial locations for these proteins have been discovered: identifying the function of each isozyme is essential for understanding the role of GA in tumors. In addition, the interactome of GA isoforms is starting to be uncovered adding a new level of regulatory complexity with important functional consequences, including selective and regulated targeting to concrete cellular locations. Clearly, GLS and GLS2 show distinct kinetics, molecular and immunological properties that make the consequences of their enhanced expression quite different and strongly dependent on factors that include signal, environment, and cell/tissue type. Elucidation of the molecular mechanisms associated with Gln catabolism in gliomas will certainly improve the few effective therapeutic options available to date.
There is a strong need for better and more specific GA inhibitors. In this sense, brain GA should be added to the list of novel candidates for the pharmacotherapy of gliomas. However, the lack of potent and specific GA inhibitors with good brain penetrant behavior has precluded so far testing GA inhibition in the pharmacotherapy of gliomas. In spite of this limitation, studies using animal models bearing human tumors suggest that GA inhibition would not be effective as a single-arm therapy; instead of it, GA inhibitors may exert a sound synergistic anticancer effect when combined with inhibitors of known oncogenic drivers of gliomas.
Abbreviations
- α-KG:
-
2-oxoglutarate
- AMPARs:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors
- BPTES:
-
[bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide]
- NEAA:
-
Non-essential amino acids
- DON:
-
6-diazo-5-oxo-l-norleucine
- EATC:
-
Ehrlich ascites tumor cells
- GBM:
-
Glioblastoma multiforme
- GDH:
-
Glutamate dehydrogenase
- GS:
-
Glutamine synthetase
- GSH:
-
Reduced glutathione
- HDAC:
-
Histone deacetylase
- IDH1:
-
Isocitrate dehydrogenase 1
- mTORC1:
-
Mammalian target of rapamycin complex 1
- NF-κB:
-
Nuclear factor-kappa B
- NMDARs:
-
N-methyl-d-aspartate receptors
- OXPHOS:
-
Oxidative phosphorylation
- ROS:
-
Reactive oxygen species
- T-ALL:
-
T-cell acute lymphoblastic leukemia
- TCA:
-
Tricarboxylic acid cycle
References
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Kvamme E, Svenneby G (1961) The effect of glucose on glutamine utilization by Ehrlich ascites tumor cells. Cancer Res 21:92–98
Kovacevic Z, Morris HP (1972) The role of glutamine in the oxidative metabolism of malignant cells. Cancer Res 32:326–333
Wise DR, DeBerardinis RJ, Mancuso A et al (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 105:18782–18787
Hertz L (1979) Functional interactions between neurons and astrocytes I. Turnover and metabolism of putative amino acid transmitters. Prog Neurobiol 13:277–323
Berl S, Clarke DD (1983) The metabolic compartmentation concept. In: Hertz L, Kvamme E, McGeer EG, Schousboe A (eds) Glutamine, glutamate and GABA in the central nervous system. Liss, New York, pp 205–217
Norenberg MD, Martínez-Hernández A (1979) Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res 161:303–310
Hertz L (2004) Intercellular metabolic compartmentation in the brain: past, present and future. Neurochem Int 45:285–296
Waagepetersen HS, Qu H, Sonnewald U, Shimamoto K, Schousboe A (2005) Role of glutamine and neuronal glutamate uptake in glutamate homeostasis and synthesis during vesicular release in cultured glutamatergic neurons. Neurochem Int 47:92–102
Robert SM, Sontheimer H (2014) Glutamate transporters in the biology of malignant gliomas. Cell Mol Life Sci 71:1839–1854
Eid T, Lee TSW, Wang Y, Peréz E et al (2013) Gene expression of glutamate metabolizing enzymes in the hippocampal formation in human temporal lobe epilepsy. Epilepsia 54:228–238
Buckingham SC, Campbell SL, Haas BR, Montana V et al (2012) Glutamate release by primary brain tumors induces epileptic activity. Nat Med 17:1269–1274
Ostrom QT, Bauchet L, Davis FG et al (2014) The epidemiology of glioma in adults: A “state of the science” review. Neuro-Oncology 16:896–913
Louis DN, Ohgaki H, Wiestler OD et al (2007) The 2007 who classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
Cuddapah VA, Robel S, Watkins S, Sontheimer H (2014) A neurocentric perspective on glioma invasion. Nat Rev Neurosci 15:455–465
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D et al (2016) The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131:803–820
The Cancer Genome Atlas (TCGA) Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068
Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110
Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA et al (2016) Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164:550–563
Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Müller F (1889) Stoffwechseluntersuchungen bei Krebskranken. Ztschr f klin Med 16:496–549
Mider GB (1951) Some aspects of nitrogen and energy metabolism in cancerous subjects: a review. Cancer Res 11:821–829
Shrivastava GC, Quastel JH (1962) Malignancy and tissue metabolism. Nature 196:876–880
Marin-Valencia I, Yang C, Mashimo T et al (2012) Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab 15:827–837
Márquez J, Sánchez-Jiménez F, Medina MA et al (1989) Nitrogen metabolism in tumor bearing mice. Arch Biochem Biophys 268:667–675
Rivera S, Azcón-Bieto J, López-Soriano FJ et al (1988) Amino acid metabolism in tumour-bearing mice. Biochem J 249:443–449
Medina MA, Sánchez-Jiménez F, Márquez J, Quesada AR, Núñez de Castro I (1992) Relevance of glutamine metabolism to tumor cell growth. Mol Cell Biochem 113:1–15
Hensley CT, Wasti AT, DeBerardinis RJ (2013) Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Investig 123:3678–3684
Márquez J, Matés JM, Alonso FJ, Martín-Rufián M, Lobo C, Campos-Sandoval JA (2015) Canceromics studies unravel tumor’s glutamine addiction after metabolic reprogramming. In: Mazurek S, Shoshan M (eds) Tumor cell metabolism: patways, regulation and biology. Springer Verlag, Vienna, pp 257–286
Tardito S, Oudin A, Ahmed SU et al (2015) Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol 17:1556–1568
Venneti S, Dunphy MP, Zhang H et al (2015) Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med 7:274ra17
Sidoryk M, Matyja E, Dybel A et al (2004) Increased expression of a glutamine transporter SNAT3 is a marker of malignant gliomas. Neuroreport 15:575–578
Kobayashi M, Mizutani A, Nishi K et al (2016) Differences in accumulation and the transport mechanism of l-and d-methionine in high-and low-grade human glioma cells. Nucl Med Biol 44:78–82
Dolińska M, Dybel A, Zabłocka B, Albrecht J (2003) Glutamine transport in C6 glioma cells shows ASCT2 system characteristics. Neurochem Int 43:501–507
Bungard CI, McGivan JD (2004) Glutamine availability up-regulates expression of the amino acid transporter protein ASCT2 in HepG2 cells and stimulates the ASCT2 promoter. Biochem J 382:27–32
Nicklin P, Bergman P, Zhang B et al (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136:521–534
Reitzer LJ, Wice BM, Kennell D (1979) Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 254:2669–2676
Goossens V, Grooten J, Fiers W (1996) The oxidative metabolism of glutamine. A modulator of reactive oxygen intermediate-mediated cytotoxicity of tumor necrosis factor in L929 fibrosarcoma cells. J Biol Chem 271:192–196
Yuneva M, Zamboni N, Oefner P et al (2007) Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 178:93–105
Ben-Porath I, Thomson MW, Carey VJ et al (2008) An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 40:499–507
DeBerardinis RJ, Mancuso A, Daikhin E et al (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 104:19345–19350
Dranoff G, Elion GB, Friedman HS, Campbell GL, Bigner DD (1985) Influence of glutamine on the growth of human glioma and medulloblastoma in culture. Cancer Res 45:4077–4081
He Q, Shi X, Zhang L et al (2016) De novo glutamine synthesis: importance for the proliferation of glioma cells and potentials for its detection with 13N-ammonia. Mol Imaging 15:1–9
de Groot J, Sontheimer H (2011) Glutamate and the biology of gliomas. Glia 59:1181–1189
Watkins S, Sontheimer H (2012) Unique biology of gliomas: challenges and opportunities. Trends Neurosci 35:546–556
de Groot JF, Liu TJ, Fuller G, Yung WK (2005) The excitatory amino acid transporter-2 induces apoptosis and decreases glioma growth in vitro and in vivo. Cancer Res 65:1934–1940
Márquez J, de la López Oliva AR, Matés JM et al (2006) Glutaminase: a multifaceted protein not only involved in generating glutamate. Neurochem Int 48:465–471
de la Rosa V, Campos-Sandoval JA, Martín-Rufián M (2009) A novel glutaminase isoform in mammalian tissues. Neurochem Int 55:76–84
Martín-Rufián M, Tosina M, Campos-Sandoval JA et al (2012) Mammalian glutaminase Gls2 gene encodes two functional alternative transcripts by a surrogate promoter usage mechanism. PLoS ONE 7:e38380
Aledo JC, Gómez-Fabre PM, Olalla L et al (2000) Identification of two human glutaminase loci and tissue-specific expression of the two related genes. Mamm Genome 11:1107–1110
Matés JM, Segura JA, Martín-Rufián M et al (2013) Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr Mol Med 13:514–534
Schousboe A, Hertz L, Svenneby G, Kvamme E (1979) Phosphate activated glutaminase activity and glutamine uptake in primary cultures of astrocytes. J Neurochem 32:943–950
Kvamme E, Svenneby G, Hertz L, Schousboe A (1982) Properties of phosphate activated glutaminase in astrocytes cultured from mouse brain. Neurochem Res 7:761–770
Szeliga M, Matyja E, Obara M et al (2008) Relative expression of mRNAs coding for glutaminase isoforms in CNS tissues and CNS tumors. Neurochem Res 33:808–813
Márquez J, Matés JM, Campos-Sandoval JA (2016) Glutaminases. In: Sonnewald U, Schousboe A (eds) Advances in neurobiology. The glutamate/GABA/glutamine cycle: amino acid neurotransmitter homeostasis. Springer Verlag, Vienna, pp 133–171
Cardona C, Sánchez-Mejías E, Dávila JC et al (2015) Expression of Gls and Gls2 glutaminase isoforms in astrocytes. Glia 63:365–382
Szeliga M, Sidoryk M, Matyja E et al (2005) Lack of expression of the liver-type glutaminase (LGA) mRNA in human malignant gliomas. Neurosci Lett 374:171–173
Collins CL, Wasa M, Souba WW et al (1998) Determinants of glutamine dependence and utilization by normal and tumor-derived breast cell lines. J Cell Physiol 176:166–178
Gao P, Tchernyshyov I, Chang TC et al (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458:762–765
Wang J-B, Erickson JW, Fuji R et al (2010) Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18:207–219
Yuneva M, Fan TWM, Allen TD (2012) The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 15:157–170
Turner A, McGivan JD (2003) Glutaminase isoform expression in cell lines derived from human colorectal adenomas and carcinomas. Biochem J 370:403–408
Pérez-Gómez C, Campos-Sandoval JA, Alonso FJ et al (2005) Co-expression of glutaminase K and L isoenzymes in human tumour cells. Biochem J 386:535–542
Hu W, Zhang C, Wu R et al (2010) Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA 107:7455–7460
Suzuki S, Tanaka T, Poyurovsky MV et al (2010) Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA 107:7461–7466
Zhang J, Wang C, Chen M et al (2013) Epigenetic silencing of glutaminase 2 in human liver and colon cancers. BMC Cancer 13:601
Szeliga M, Bogacinska-Karas M, Kuzmicz K et al (2016) Downregulation of GLS2 in glioblastoma cells is related to DNA hypermethylation but not to the p53 status. Mol Carcinog 55:1309–1316
Xiang L, Xie G, Liu C, Zhou J, Chen J, Yu S, Li J, Pang X, Shi H, Liang H (2013) Knock-down of glutaminase 2 expression decreases glutathione, NADH, and sensitizes cervical cancer to ionizing radiation. Biochim Biophys Acta - Mol Cell Res 1833(12):2996–3005
Xiao D, Ren P, Su H et al (2015) Myc promotes glutaminolysis in human neuroblastoma through direct activation of glutaminase 2. Oncotarget 6:40655–40666
Coloff JL, Murphy JP, Braun CR et al (2016) Differential glutamate metabolism in proliferating and quiescent mammary epithelial cells. Cell Metab 23:867–880
Yang C, Sudderth J, Dang T et al (2009) Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res 69:7986–7993
Nagashima H, Tanaka K, Sasayama T et al (2016) Diagnostic value of glutamate with 2-hydroxyglutarate in magnetic resonance spectroscopy for IDH1 mutant glioma. Neuro-Oncology 1–10. doi:10.1093/neuonc/now090
Csibi A, Fendt SM, Li C et al (2013) The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153:840–854
Duran RV, Oppliger W, Robitaille AM et al (2012) Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell 47:349–358
Panosyan EH, Lasky JL, Lin HJ et al (2016) Clinical aggressiveness of malignant gliomas is linked to augmented metabolism of amino acids. J Neurooncol 128:57–66
Yudkoff M (2016) Interactions in the metabolism of glutamate and the branched-chain amino acids and ketoacids in the CNS. Neurochem Res. doi:10.1007/s11064-016-2057-z
Tönjes M, Barbus S, Park YJ et al (2013) BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med 19:901–908
Seltzer MJ, Bennett BD, Joshi AD et al (2010) Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res 70:8981–8987
Ye ZC, Sontheimer H (1999) Glioma cells release excitotoxic concentrations of glutamate. Cancer Res 59:4383–4391
Takano T, Lin JH, Arcuino G et al (2001) Glutamate release promotes growth of malignant gliomas. Nat Med 7:1010–1015
Lyons SA, Chung WJ, Weaver AK et al (2007) Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res 67:9463–9471
Lobo C, Ruiz-Bellido MA, Aledo JC et al (2000) Inhibition of glutaminase expression by antisense mRNA decreases growth and tumourigenicity of tumour cells. Biochem J 348:257–261
Lora J, Alonso FJ, Segura JA et al (2004) Antisense glutaminase inhibition decreases glutathione antioxidant capacity and increases apoptosis in Ehrlich ascitic tumour cells. Eur J Biochem 271:4298–4306
Cheng T, Sudderth J, Yang C et al (2011) Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci USA 108:8674–8679
Olalla L, Gutiérrez A, Campos JA et al (2002) Nuclear localization of l-glutaminase in mammalian brain. J Biol Chem 277:38939–38944
Szeliga M, Obara-Michlewska M, Matyja E et al (2009) Transfection with liver-type glutaminase (LGA) cDNA alteres gene expression and reduces viability, migration and proliferation of T98G glioma cells. Glia 57:1014–1023
Liu J, Zhang C, Lin M et al (2014) Glutaminase 2 negatively regulates the PI3K/AKT signaling and shows tumor suppression activity in human hepatocellular carcinoma. Oncotarget 5:2635–2647
Szeliga M, Bogacińska-Karaś M, Różycka A et al (2013) Silencing of GLS and overexpression of GLS2 genes cooperate in decreasing the proliferation and viability of glioblastoma cells. Tumour Biol 35:1855–1862
Martín-Rufián M, Nascimento-Gomes R, Higuero A et al (2014) Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells. J Mol Med 92:277–290
Gross MI, Demo SD, Dennison JB et al (2014) Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther 13:890–901
Thomas AG, Rojas C, Tanega C et al (2013) Kinetic characterization of ebselen, chelerythrine and apomorphine as glutaminase inhibitors. Biochem Biophys Res Commun 438:243–248
Herranz D, Ambesi-Impiombato A, Sudderth J et al (2015) Metabolic reprograming induces resistance to anti-NOTCH1 therapies in acute lymphoblastic leukemia. Nat Med 21:1182–1189
Tanaka K, Sasayama T, Irino Y et al (2016) Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J Clin Invest 125:1591–1602
Acknowledgements
The work performed by the Canceromics group and cited in this article was financially supported by Grant SAF2015-64501-R from the Spanish Ministry of Economy and Competitivity (to JM and JMM) and Grant RD12/0028/0013 (JM) of the RTA RETICS network from the Spanish Health Institute Carlos III.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Márquez, J., Alonso, F.J., Matés, J.M. et al. Glutamine Addiction In Gliomas. Neurochem Res 42, 1735–1746 (2017). https://doi.org/10.1007/s11064-017-2212-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-017-2212-1