
Abstract
This review summarises the recent findings on metabolic treatments for epilepsy and Amyotrophic Lateral Sclerosis (ALS) in honour of Professor Ursula Sonnewald. The metabolic impairments in rodent models of these disorders as well as affected patients are being discussed. In both epilepsy and ALS, there are defects in glucose uptake and reduced tricarboxylic acid (TCA) cycling, at least in part due to reduced amounts of C4 TCA cycle intermediates. In addition there are impairments in glycolysis in ALS. A reduction in glucose uptake can be addressed by providing the brain with alternative fuels, such as ketones or medium-chain triglycerides. As anaplerotic fuels, such as the triglyceride of heptanoate, triheptanoin, refill the TCA cycle C4/C5 intermediate pool that is deficient, they are ideal to boost TCA cycling and thus the oxidative metabolism of all fuels.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Professor Ursula Sonnewald is among the first important researchers who characterised the dysfunction of metabolic processes such as glucose metabolism, particularly within the tricarboxylic acid (TCA) cycle in models of neurological disorders. She has been very interested in astrocytic vs. global brain metabolism, neuronal-glial interactions, as well as cytosolic vs. mitochondrial metabolic processes. In 2006, my laboratory started to work on alternative fuels and metabolic impairments in epilepsy. With Ursula’s help since 2011 we have been able learn more about this important topic.
This article is an update to our recent review [1] and will first summarise the impairments in glucose metabolism and the TCA cycle in people with epilepsy and animal models of epilepsy. Taken together, the findings discussed strongly suggest that there are impairments in energy metabolism in epileptic brains, which would result in a lack of energy (ATP) production and thus a reduction in energy dependent processes in the brain, which is likely to contribute to seizures. Thus, alternative fuels which can boost ATP generation are well suited to prevent seizure generation. Here, we discuss the anticonvulsant effects of current and potential new metabolic treatments as well as the most recent findings on their metabolic effects on brain energy metabolism and their likely anticonvulsant mechanisms. We will emphasize the work by Ursula and her many colleagues, who together with many others have shown that metabolic treatment approaches are targeted at metabolic impairments in the epileptic brain and are therefore well justified for the treatment of epilepsy without central side effects. We will also mention the effects of metabolic treatment in Amyotrophic Lateral Sclerosis (ALS) models, although their potential for this disease and other neurological disorders with a similar metabolic pathphysiology largely remains to be explored.
Epilepsy
Epilepsy is characterised by the sudden and often unexpected occurrence of seizures based on spontaneous firing of groups of neurons in the brain [2]. The location of these outbursts of extreme neuronal firing in the brain determines the outcome of seizures, such as motor involvement, odd sensations, absences, loss of consciousness and posture. One to two percent of the population suffer from some type of epilepsy and although about 20 different anti-seizure drugs are available, 30% of patients, about 20–40 million people worldwide suffer from medically refractory seizures [3]. Surgery and neurostimulation implants are not suitable for most patients due to high cost or the lack of seizure foci that are amenable to surgical removal.
Metabolic Impairments in Epilepsy
The pathophysiology underlying epilepsy is complicated and complex. Various physiological processes can be out of balance and promote seizures, such as the levels of excitation vs. inhibition, but also inflammation and energy metabolism. This review focusses on the metabolism in between seizures. Growing evidence shows that dysfunction of metabolic processes in the brain including glucose uptake, the TCA cycle and the electron transport chain, can contribute to a lowering of the seizure threshold resulting in epileptic seizures. This is supported by the fact that epileptic seizures are part of the phenotype of patients with mutations resulting in glucose transporter type 1 deficiency [4, 5] or mutations in mitochondrial genes or nuclear genes leading to mitochondrial deficiencies [6]. Using positron emission tomography to detect 18F-fluorodeoxyglucose uptake, it has been repeatedly shown that local cerebral glucose uptake is decreased in epileptic foci in people with epilepsy between seizure activity [7, 8] as well as in rodent epilepsy models [9].
In addition, high brain glucose levels were found in several rodent models of chronic epilepsy, which is an indicator for glucose hypometabolism, defined as decreased glucose consumption [9–13]. In these models, brain lactate levels are highly variable [9, 12–14], suggesting that there is an imbalance in the rate of glycolysis vs. that of the TCA cycle.
Various types of epileptic seizures can occur in people with mutations in TCA cycle enzymes, including pyruvate dehydrogenase [15, 16], α-ketoglutarate dehydrogenase [17], succinyl-CoA synthetase [18], succinate dehydrogenase [19–22] and fumarase [23, 24]. In rodents, Ursula’s group was the first to show that the levels of brain glutamate and aspartate are reduced during chronic epileptic states [11, 14, 25]. This indirectly shows the brain levels of TCA cycle intermediates are reduced, because these amino acids are derived from oxaloacetate and α-ketoglutarate. This was confirmed directly in the mouse pilocarpine model, where the levels of malate [14, 26], succinate and citrate [14] were reduced.
The reduction in the levels of TCA cycle intermediates may be explained by the reduction in glucose uptake discussed above or by additional alterations in TCA cycle activity. Lower TCA cycle intermediate levels will lead to inefficient cycling of the TCA cycle resulting in less reduction of NAD+ and FAD. This will lead to reduced activity of the electron transport chain, and together with reduced glucose metabolism can lead to a decrease in ATP production. As ATP is central to the majority of cellular activities, including the maintenance of membrane potentials, neurotransmitter release and reuptake, defence mechanisms and biomolecular turnover it is conceivable that excitability of neurons will be altered. The anticonvulsant effects of several metabolic treatments, which increase ATP synthesis and are currently used or are being developed, further corroborate this notion.
Intriguingly, the defects in TCA cycling appear to be specific to neurons. Ursula has been very interested in glial metabolism and neuronal-glial interactions. Together with Astrid Nehlig’s laboratory she showed that the metabolism of [1,2-13C]-acetate, which is taken up by astrocytes and oligodendrocytes, was unaltered in a rodent seizure model [11]. This finding however does not rule out other deficiencies in glial metabolism that can contribute to seizures. For example, increased activity of phosphate-activated glutaminase found in MTLE may contribute to impaired glutamate homeostasis [27].
Anticonvulsant Effects of Current and Potential Metabolic Treatments
Current Ketogenic Therapies
Current metabolic dietary approaches to treat epilepsy include the ketogenic diet [28], the modified Atkins diet [29] and various versions thereof. The classic ketogenic diet consists of mainly long-chain triglycerides and is given in a 4:1 (wt:wt) ratio with 4 g of fat eaten with only 1 g of carbohydrate and protein. This ratio can be lowered to 3:1 or 2:1 or even 1:1 such as in the modified Atkins diet, depending on seizure control and tolerability. Also, slightly more carbohydrates can be consumed while doing the medium-chain triglyceride (MCT) ketogenic diet. In this dietary regime, 50–60% of calories are from MCTs, mostly those of octanoate (8 carbons) and decanoate (10 carbons), which are more ketogenic than long-chain triglycerides [30]. Another alternative is the low glycemic index diet, in which higher amounts of carbohydrates with low glycemic index are allowed [31–33]. Several controlled studies show that these high fat/low carbohydrate diets are effective in many children with epilepsy and some of the children remain seizure-free after stopping dietary treatment [34–36]. Some adults also experience reduction in seizures [37], but unfortunately, the diet regimens are relatively strict and difficult, resulting in low compliance, especially in adults. In addition, these dietary approaches require both dietary and medical supervisions which are not accessible in many countries. Another option, namely ketone esters, which can elevate and sustain blood ketone levels [38] and prevent seizures in various rodent models [39–41] are expected to be available soon. Their anti-seizure efficacy in people with epilepsy still needs to be determined. In summary, these metabolic dietary treatments are far from ideal for long-term approaches to treat adults and children with epilepsy or other disorders and alternative treatment approaches for epilepsy are urgently required.
Mechanisms of Metabolic Anticonvulsant Actions of Ketogenic Therapies
When glucose is in short supply, the liver turns fatty acids into the “C4 ketones”, acetoacetate and β-hydroxybutyrate, which are then excreted into the blood. During ketosis when blood C4 ketone levels are high, human and rodent brains can use substantial amounts of C4 ketones to fuel metabolism instead of glucose (Fig. 1) [42, 43]. This demonstrates that alternative fuels can be effective when glucose metabolism is impaired such as in the epileptic brain. There is also some evidence that a reduction of glycolysis may protect the brain from seizures, which is supported by the anticonvulsant effects of the glycolytic inhibitor 2-deoxyglucose [44–46]. This can also alter transcription regulating neuronal plasticity through NRSF-CtBP [44] and the mTOR pathway [47].

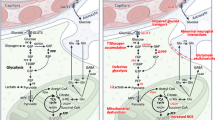
Metabolism of alternative fuels. Glucose is the main energy substrate under physiological conditions and is metabolized by a series of enzymatic reactions through glycolysis into two molecules of pyruvate (black dashed arrow). Pyruvate can be converted into acetyl CoA by pyruvate dehydrogenase (PDH). Acetyl-CoA enters the TCA cycle by condensing with oxaloacetate (OAA) to form citrate and is then further metabolized to generate NADH and subsequently ATP. In astrocytes, pyruvate can be converted into OAA via the anaplerotic enzyme pyruvate carboxylase (PC) to replenish lost TCA cycle intermediates used in the synthesis of neurotransmitters including glutamate (Glu) and γ-aminobutyric acid (GABA). In neurological disorders like epilepsy and ALS where glucose metabolism is believed to be impaired and energy demand is high, alternative fuels such as the ketogenic diet (provides C4 ketones) and medium-chain fatty acid derived from trioctanoin, tridecanoin and triheptanoin can be taken up from the blood and used by the CNS or are converted to ketones by the liver. C4 ketones and medium chain fats produce acetyl-CoA, which enter the TCA cycle (purple and blue dashed arrows). Enhanced levels of ketones in the brain are thought to increase the production of glutamate from α-ketoglutarate (α-KG), which lead to increased conversion of glutamate into glutamine or GABA (blue arrows). Similar to the C4 ketones, the even-chain MCTs trioctanoin (C8) and tridecanoin (C10) provide acetyl CoA when metabolized (purple dashed arrow). Triheptanoin, a C7 medium-chain fuel, is a triglyceride of heptanoate, which is broken down into two acetyl-CoA and one propionyl-CoA molecule(s) by β-oxidation (green dashed arrows). Propionyl-CoA is an anaplerotic molecule that can be further converted into succinyl-CoA and restore lost TCA C4 intermediates and thus promote TCA cycling (Color figure online)
The numerous metabolic and other effects of β-hydroxybutyrate are summarised by [48]. Here, we want to point out that before C4 ketones are metabolised to acetyl-CoA, they are turned into acetoacetyl-CoA by succinyl-CoA transferase using succinyl-CoA, (Fig. 1) [49]. Lowering the amounts of succinyl-CoA during this reaction will relieve its inhibition of citrate synthase, which will then promote the entry of acetyl-CoA into the TCA cycle after condensation with oxaloacetate [50, 51]. This will promote ATP production from any fuels that produce acetyl-CoA, including ketones, medium-chain fats, glucose and lactate. In addition, less oxaloacetate can then be transaminated to aspartate via aspartate transaminase, which uses glutamate as the amine donor. Similarly, enhanced transamination by aspartate transaminase of α-ketoglutarate will produce increased amounts of glutamate and oxaloacetate. Together, this results in higher levels of glutamate, which is then available to be metabolised to glutamine or the inhibitory neurotransmitter GABA. Moreover, Ursula’s team showed that while on the ketogenic diet, the brain can also increase the synthesis oxaloacetate via an increased activity of pyruvate carboxylase, which can refill the deficient C4 intermediates of the TCA cycle (anaplerosis), and thus promote entry and subsequent oxidation of acetyl-CoA [52]. In addition to these metabolic anticonvulsant mechanisms of the ketogenic diet many other physiological pathways, which are likely to contribute to its effects, are altered by this diet (reviewed by [53, 54]).
Even Medium-Chain Fatty Acids as Anticonvulsants?
Valproate is an isomer of octanoic acid and often is used in relatively high doses as the first line treatment for various types of epilepsy. Its similarity to unbranched fatty acids raised the question if medium-chain fatty acids can also be used as anticonvulsants. In patients on the MCT ketogenic diet the blood levels of octanoic acid and decanoic acid reach near millimolar amounts [55–57]. Moreover, medium-chain fatty acids can diffuse directly into the brain and can be metabolised (Fig. 1) [58, 59]. Thus, ketogenesis by the liver should not necessarily be needed to alter brain metabolism while taking medium-chain triglycerides. Interestingly, several studies have demonstrated that specific medium-chain fatty acids can be anticonvulsant in various models in vitro and in vivo [60–63]. In addition, our laboratory has employed the triglycerides of these fatty acids as a suitable formulation to avoid acid overload in patients and animals. Our study in 2014 found anticonvulsant effects after feeding 35% of caloric intake in the form of trioctanoin (triglyceride of octanoate) in the 6-Hz model [64]. While this could not be repeated, we found reproducible effects of tridecanoin (the triglyceride of decanoate) in the acute mouse 6 Hz and fluorothyl models [65].
The anticonvulsant mechanisms of medium-chain fats are likely based on the provision of alternative fuels and potentially reduced glucose metabolism. Indeed, there is evidence that trioctanoin reduces glycolysis, After feeding trioctanoin to mice for 3 weeks we found that the steady state levels of several glycolytic metabolites were reduced [64] as well as the maximal activity of phosphofructokinase, the rate limiting enzyme for glycolysis [65]. Strikingly, octanoate appears to be mostly metabolised by astrocytes, based on evidence in cultures showing that only astrocytes produce carbon dioxide from octanoate [66] and in vivo [67, 68]. After infusion of [1-14C]- or [1-13C] octanoate at 15–25% of caloric requirements, the 13C-labelled carbon is mostly found in glutamine relative to glutamate and glutamine is predominantly produced in astrocytes. It remains to be determined if decanoate is also an astrocytic fuel and if it reduces glycolysis, although we recently showed that after feeding tridecanoin there are no alterations in the maximal activity of several glycolytic enzymes investigated [65]. However, surprisingly, tridecanoin is antioxidant and it improves mitochondrial function, which can further support ATP synthesis [65].
MCTs are oils, but are now available in a variety of (medical) food products, in liquid or powdered non-oily formulations or even tasty foods. Several questions remain to be answered. Are MCTs added to a normal diet anticonvulsant in adults and children with epilepsy? Which C8:C10 ratio is most efficacious? Is it possible to achieve seizure control with relatively small amounts of MCTs added to a regular diet, such as 10–20% caloric intake, which was found to be sufficient to increase seizure thresholds in rats [62]?
Uneven Medium-Chain Triglycerides: Triheptanoin as an Anticonvulsant
Triheptanoin is a tasteless uneven medium-chain triglyceride, namely the triglyceride of the seven carbon fatty acid heptanoate. It not only provides acetyl-CoA directly to the mitochondrial TCA cycle, but is also highly effective in producing propionyl-CoA, an alternative substrate for refilling of the TCA cycle within the CNS (and skeletal muscle, Fig. 1). Triheptanoin is already in clinical trials to treat rare metabolic disorders, such as long-chain fatty acid metabolism disorders [69, 70], glycogen storage disorders, Huntington’s disease [71, 72] as well as epilepsy and paroxysmal movements associated with glucose transporter1 deficiency [73, 74]. Recently, in Huntington’s disease patients triheptanoin was shown to normalise metabolic parameters in the brain and to prevent abnormal acidity after exercise in muscle [71, 72].
Triheptanoin is first broken down into glycerol and three heptanoate molecules upon ingestion. Heptanoate can then either enter the blood and brain directly or is metabolised in the liver into glucose or C5 ketone bodies, β-hydroxypentanoate and β-ketopentanoate, which are most likely taken up into cells by monocarboxylate transporters [75]. Heptanoate and C5 ketones are metabolised into acetyl-CoA and the anaplerotic propionyl-CoA [76]. The latter is carboxylated to methylmalonyl-CoA and then metabolised to succinyl-CoA (thereby refilling the C4 intermediates of the TCA cycle, Fig. 1). After going through the TCA cycle while providing reducing equivalents to complexes I and II of the electron transport chain for subsequent ATP production, succinyl-CoA eventually gives rise to oxaloacetate. Increased levels of oxaloacetate will facilitate acetyl-CoA entry into the TCA cycle [77] and will therefore increase ATP production from triheptanoin itself and from other fuels. Hence, triheptanoin provides additional fuel sources and is expected to increase aerobic metabolism in metabolically compromised tissues to increase ATP availability [77].
Noting that ATP is crucial for the maintenance of neuronal membrane potentials, which is thought to be important to prevent the generation of epileptic seizures, we discovered that triheptanoin protects mice against seizures in different epilepsy models. This includes efficacy in three chronic seizure models, namely the corneal kindling model, the second hit pentylenetetrazole test in mice in the chronic “epileptic” phase of the pilocarpine model as well as a genetic absence seizure model [26, 78]. Furthermore, the consistent anticonvulsant effects of triheptanoin in the acute maximal electroshock model [79] revealed a unique anticonvulsant profile. Based on the efficacy in these mouse seizure models, effects against generalised motor and absence seizures, as well as partial and potentially drug-resistant seizures in humans are possible. The unique anticonvulsant profile of triheptanoin in mice motivated three clinical trials in adults and children with epilepsy in Australia (supervised by KB) and all trials are showing promising interim results so far (data unpublished).
Anticonvulsant Mechanism of Triheptanoin
In the healthy mouse brain, [5,6,7-13C]-heptanoate was incorporated into TCA cycle intermediates and metabolites, mostly glutamine (4.3% M+3 enrichment), but also glutamate (1.3% M+3 enrichment). The higher 13C enrichment in glutamine relative to glutamate indicates that heptanoate is mostly metabolised by astrocytes [75]. This apparently low amount of anaplerosis can still be important in regards to the high turnover rate of the TCA cycle [77] and it is expected to be even more crucial when anaplerotic need is high, such as in the “epileptic brain”. Interestingly, the levels of methylmalonyl-CoA were increased in the brains of mice in the chronic “epileptic” stage, but not in healthy mice after the administration of a 35% triheptanoin diet. This suggests that heptanoate or C5 ketones are metabolised in brains with high anaplerotic needs [26]. Also, triheptanoin feeding restored the loss of acetyl-CoA and propionyl-CoA concentrations and increased malate levels in the same brains [26]. Another study showed that triheptanoin did not alter the amounts of glycolytic metabolites in the healthy brain, while trioctanoin led to significant reductions [64]. Feeding triheptanoin for 3 weeks to mice in the chronic stage of the pilocarpine epilepsy model did not alter the steady state levels of TCA cycle intermediates or metabolites, namely succinyl-CoA, aspartate, glutamate and GABA [26], which may be due to fast metabolism of these metabolites. However, together with Ursula we followed the metabolism of [1,2 13C]-glucose and could show that triheptanoin improves metabolism in brains of chronically “epileptic” mice [10]. Specifically, triheptanoin attenuated the drop in the 13C enrichment in malate, citrate and GABA and increased this enrichment in fumarate in “epileptic mice”. Taken together, this demonstrates that triheptanoin is well suited to counteract the loss in the total pool of TCA cycle intermediates and metabolites and that triheptanoin is anaplerotic in “epileptic” brains.
ALS
ALS, also called Motor Neuron Disease (MND), is a progressive neurodegenerative disorder characterized by the degeneration of motor neurons in the spinal cord and brain. This results in muscle weakness culminating in paralysis and finally death usually due to loss of respiratory functions [80]. Most cases are sporadic and only 10% of ALS is hereditary (familiar ALS). A variety of mutations are thought to cause familiar and sporadic ALS, including alterations in superoxide dismutase 1 (SOD1) [81], TAR DNA-Binding Protein (TDP 43) [82], fused in sarcoma (FUS) [83], Ubiquilin2 (UBQLN2) [84], and C9ORF72 [85, 86] and probably many others. Many mechanisms are thought to contribute to the degeneration of motor neurons in ALS, such as glutamate excitotoxicity [87], abnormal protein aggregation [81, 88], impaired axonal transport [89], inflammation [90], oxidative stress [91], altered properties of astrocytes [92] and also abnormalities in energy metabolism [93, 94].
There is a growing body of evidence for metabolic dysfunction in ALS, which contributes to the progression of the disease. Many ALS patients lose substantial amounts of body weight and a low body mass index correlates with shorter survival [95]. Increased energy expenditure has been found in ALS patients as well as in ALS rodent models, which indicates disturbances in energy metabolism [94, 96, 97]. Specifically, altered glucose uptake has been shown in various brain regions and spinal cord [98–102]. Many functional and morphological abnormalities have been found in mitochondria, in addition to reduced amounts of aspartate, glutamine and GABA in the spinal cord of hSOD1G93A mice during early disease stages suggesting that C4-intermediate levels of the TCA cycle are decreased [103]. This is consistent with other findings of reduced oxidative phosphorylation and ATP production in ALS models [101, 104–107].
Moreover, multiple studies have shown compromised neuronal-astrocyte interactions, such as impaired transport of glutamate [108] and loss of the glutamate transporter GLT-1 [109]. Intriguingly, it has been shown that introduction of ALS gene alterations selectively in astrocytes can lead to motor neuron degeneration [92], while in return, non-neuronal cells that do not express mutant SOD1 extended survival of mutant SOD1 expressing motor neurons [110].
Metabolic Therapies for ALS
Based on the findings mentioned above, approaches that target the metabolic impairments found in ALS are warranted. Alternative fuels different to glucose are expected to improve energy supply and meet the high energy demand. Indeed, several approaches that improve TCA cycle function were found to delay disease progression in rodent ALS models (reviewed by [111]).
Dichloroacetate
Pyruvate dehydrogenase kinase inhibits pyruvate dehydrogenase and thus the production of acetyl-CoA from pyruvate and its entry into the TCA cycle [112]. Dichloroacetate (DCA), an inhibitor of PDH kinase, can therefore improve TCA cycle entry and increase aerobic glucose oxidation, which yields high amounts of ATP. In cultured astrocytes from the hSOD1G93A rat model, DCA improved mitochondrial metabolism and prevented toxicity to motor neurons, thereby increasing their survival [113]. Also, in the hSOD1G93A mouse model, DCA improved motor symptoms such as grip strength performance, protected against motor neuron loss and increased survival [113].
Ketogenic Diet and Caprylic Triglycerides (Trioctanoin)
The ketogenic diets used in the treatment of epilepsy all promote body weight loss in adults. In light of the negative correlation between survival and body weight loss, ketogenic diets are therefore contraindicated. On the other hand C4 ketones are excellent alternative fuels and may overcome the problems associated with the decreased ability to use glucose as fuel. Moreover, a variety of other protective mechanisms are ascribed to C4 ketones, such as anti-oxidant and anti-inflammatory properties among others [54], which would be beneficial in ALS. This theory is corroborated by two studies using the hSOD1G93A ALS mouse model. A “mild” ketogenic diet, consisting of 60% fat, 20% carbohydrate and 20% protein, preserved motor performance and protected against motor neurons loss in the spinal cord [114]. Furthermore, addition of the ketogenic medium-chain triglyceride trioctanoin, the triglyceride of octanoate (caprylic acid), also had similar protective effects, although both metabolic treatments did not extend survival in this ALS model [115]. In conclusion, these findings indicate that new approaches that can provide C4 ketones as alternative fuels for patients with ALS are promising.
The Deanna Protocol
The Deanna Protocol uses a variety of alternative fuels, vitamins and antioxidants [116]. There is anecdotal evidence that it has been effective for some patients with ALS. A study in hSOD1G93A mice fed only four main components of the protocol, namely arginine α-ketoglutarate, medium-chain triglycerides, coenzyme Q and GABA [117]. This dietary regimen delayed disease progression and extended survival in hSOD1G93A mice, raising the hope that it may be effective in patients.
Triheptanoin
Triheptanoin is another alternative fuel, which seems to be well suited to overcome the metabolic problems in ALS. Its ability to provide not only acetyl-CoA, but also propionyl-CoA makes it an ideal metabolic treatment for neuronal cells and muscles, which are deficient in TCA cycle intermediates and energy production. Ursula’s former Masters student, Tesfaye W. Tefera, recently found together with others in our laboratory that triheptanoin protects lumbar motor neurons and delays the onset of motor symptoms and body weight loss in the hSOD1G93A ALS mouse model [118]. Clinical studies are now needed to investigate if quality of life of ALS patients can be improved by adding triheptanoin to their diet. Tesfaye has also brought Ursula’s techniques to distinguish between astrocyte vs. total brain metabolism to our laboratory and we are currently investigating to which extent metabolism is affected in the hSOD1G93A model and how it is altered by triheptanoin.
Conclusions
We hope that this article being in honour of Ursula showed her important contribution to the research area of brain metabolism and alternative fuels. Her work indicated the need of anaplerosis in epileptic tissue. She has really driven the field forward by showing that both the ketogenic diet and triheptanoin can address this increased need for anaplerosis in epilepsy. In addition, one of her students brought Ursula’s techniques to the ALS research field, showing that triheptanoin can be beneficial in a model of ALS. It remains to be seen if these findings in models of epilepsy and ALS will improve the treatment of these enigmatic diseases. In response to the critical and urgent need for better and more efficacious treatment of these neurological disorders with fewer side effects compared to traditional medication, controlled clinical studies are needed to address the efficacy of the metabolic treatments discussed. We are hopeful for positive outcomes.
References
Tan KN, McDonald TS, Borges K (2015) Metabolic dysfunctions in epilepsy and novel metabolic treatment approaches. In: Preedy V WR (ed) Bioactive nutraceuticals and dietary supplements in neurological and brain disease: prevention and therapy. Elsevier, Amsterdam, pp 461–470
Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, Engel J Jr (2005) Epileptic seizures and epilepsy: definitions proposed by the International league against epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 46:470–472
French JA, Kanner AM, Bautista J, Abou-Khalil B, Browne T, Harden CL, Theodore WH, Bazil C, Stern J, Schachter SC, Bergen D, Hirtz D, Montouris GD, Nespeca M, Gidal B, Marks WJ, Turk WR, Fischer JH, Bourgeois B, Wilner A, Faught RE, Sachdeo RC, Beydoun A, Glauser TA (2004) Efficacy and tolerability of the new antiepileptic drugs, I: treatment of new-onset epilepsy: report of the TTA and QSS subcommittees of the American Academy of Neurology and the American Epilepsy Society. Epilepsia 45:401–409
Mullen SA, Marini C, Suls A, Mei D, Della Giustina E, Buti D, Arsov T, Damiano J, Lawrence K, De Jonghe P, Berkovic SF, Scheffer IE, Guerrini R (2011) Glucose transporter 1 deficiency as a treatable cause of myoclonic astatic epilepsy. Arch Neurol 68:1152–1155
Scheffer IE (2012) GLUT1 deficiency: a glut of epilepsy phenotypes. Neurology 78:524–525
Zsurka G, Kunz WS (2010) Mitochondrial dysfunction in neurological disorders with epileptic phenotypes. J Bioenerg Biomembr 42:443–448
Chugani HT, Chugani DC (1999) Basic mechanisms of childhood epilepsies: studies with positron emission tomography. Adv Neurol 79:883
Kuhl DE, Engel J, Phelps ME, Selin C (1980) Epileptic patterns of local cerebral metabolism and perfusion in humans determined by emission computed tomography of 18FDG and 13NH3. Ann Neurol 8:348–360
Dube C, Boyet S, Marescaux C, Nehlig A (2001) Relationship between neuronal loss and interictal glucose metabolism during the chronic phase of the lithium-pilocarpine model of epilepsy in the immature and adult rat. Exp Neurol 167:227–241
Hadera MG, Smeland OB, McDonald TS, Tan KN, Sonnewald U, Borges K (2013) Triheptanoin partially restores levels of tricarboxylic acid cycle intermediates in the mouse pilocarpine model of epilepsy. J Neurochem 129(1):107–119
Melo TM, Nehlig A, Sonnewald U (2005) Metabolism is normal in astrocytes in chronically epileptic rats: a (13)C NMR study of neuronal-glial interactions in a model of temporal lobe epilepsy. J Cereb Blood Flow Metab 25:1254–1264
Smeland OB, Meisingset TW, Sonnewald U (2012) Dietary supplementation with acetyl-l-carnitine in seizure treatment of pentylenetetrazole kindled mice. Neurochem Int 61:444–454
Hadera MG, Faure JB, Berggaard N, Tefera TW, Nehlig A, Sonnewald U (2014) The anticonvulsant actions of carisbamate associate with alterations in astrocyte glutamine metabolism in the lithium-pilocarpine epilepsy model. J Neurochem 132:532–545
Smeland OB, Hadera MG, McDonald TS, Sonnewald U, Borges K (2013) Brain mitochondrial metabolic dysfunction and glutamate level reduction in the pilocarpine model of temporal lobe epilepsy in mice. J Cereb Blood Flow Metab 33:1090–1097
Barnerias C, Saudubray JM, Touati G, De Lonlay P, Dulac O, Ponsot G, Marsac C, Brivet M, Desguerre I (2010) Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Dev Med Child Neurol 52:e1–e9
Kang HC, Kwon JW, Lee YM, Kim HD, Lee HJ, Hahn SH (2007) Nonspecific mitochondrial disease with epilepsy in children: diagnostic approaches and epileptic phenotypes. Childs Nerv Syst 23:1301–1307
Bonnefont JP, Chretien D, Rustin P, Robinson B, Vassault A, Aupetit J, Charpentier C, Rabier D, Saudubray JM, Munnich A (1992) Alpha-ketoglutarate dehydrogenase deficiency presenting as congenital lactic acidosis. J Pediatr 121:255–258
Elpeleg O, Miller C, Hershkovitz E, Bitner-Glindzicz M, Bondi-Rubinstein G, Rahman S, Pagnamenta A, Eshhar S, Saada A (2005) Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet 76:1081–1086
Bourgeron T, Rustin P, Chretien D, Birch-Machin M, Bourgeois M, Viegas-Pequignot E, Munnich A, Rotig A (1995) Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet 11:144–149
Burgeois M, Goutieres F, Chretien D, Rustin P, Munnich A, Aicardi J (1992) Deficiency in complex II of the respiratory chain, presenting as a leukodystrophy in two sisters with Leigh syndrome. Brain Dev 14:404–408
Horváth R, Abicht A, Holinski-Feder E, Laner A, Gempel K, Prokisch H, Lochmüller H, Klopstock T, Jaksch M (2006) Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA). J Neurol Neurosurg Psychiatry 77:74–76
Parfait B, Chretien D, Rotig A, Marsac C, Munnich A, Rustin P (2000) Compound heterozygous mutations in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Hum Genet 106:236–243
Ezgu F, Krejci P, Wilcox WR (2013) Mild clinical presentation and prolonged survival of a patient with fumarase deficiency due to the combination of a known and a novel mutation in FH gene. Gene 524:403–406
Kerrigan JF, Aleck KA, Tarby TJ, Bird CR, Heidenreich RA (2000) Fumaric aciduria: clinical and imaging features. Ann Neurol 47:583–588
Alvestad S, Hammer J, Eyjolfsson E, Qu H, Ottersen OP, Sonnewald U (2008) Limbic structures show altered glial-neuronal metabolism in the chronic phase of kainate induced epilepsy. Neurochem Res 33:257–266
Willis S, Stoll J, Sweetman L, Borges K (2010) Anticonvulsant effects of a triheptanoin diet in two mouse chronic seizure models. Neurobiol Dis 40:565–572
Eid T, Hammer J, Runden-Pran E, Roberg B, Thomas MJ, Osen K, Davanger S, Laake P, Torgner IA, Lee TS, Kim JH, Spencer DD, Ottersen OP, de Lanerolle NC (2007) Increased expression of phosphate-activated glutaminase in hippocampal neurons in human mesial temporal lobe epilepsy. Acta Neuropathol (Berl) 113:137–152
Wilder RM (1921) The effects of ketonemia on the course of epilepsy. Mayo Clin Proc 2:307–308
Kossoff EH, Krauss GL, McGrogan JR, Freeman JM (2003) Efficacy of the Atkins diet as therapy for intractable epilepsy. Neurology 61:1789–1791
Huttenlocher PR (1976) Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatr Res 10:536–540
Coppola G, D’Aniello A, Messana T, Di Pasquale F, della Corte R, Pascotto A, Verrotti A (2011) Low glycemic index diet in children and young adults with refractory epilepsy: first Italian experience. Seizure 20:526–528
Muzykewicz DA, Lyczkowski DA, Memon N, Conant KD, Pfeifer HH, Thiele EA (2009) Efficacy, safety, and tolerability of the low glycemic index treatment in pediatric epilepsy. Epilepsia 50:1118–1126
Pfeifer HH, Thiele EA (2005) Low-glycemic-index treatment: a liberalized ketogenic diet for treatment of intractable epilepsy. Neurology 65:1810–1812
Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, Whitney A, Cross JH (2008) The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol 7:500–506
Sharma S, Sankhyan N, Gulati S, Agarwala A (2013) Use of the modified Atkins diet for treatment of refractory childhood epilepsy: a randomized controlled trial. Epilepsia 54:481–486
Martin K, Jackson CF, Levy RG, Cooper PN (2016) Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst Rev 2:CD001903
Klein P, Tyrlikova I, Mathews GC (2014) Dietary treatment in adults with refractory epilepsy: a review. Neurology 83:1978–1985
Kashiwaya Y, Pawlosky R, Markis W, King MT, Bergman C, Srivastava S, Murray A, Clarke K, Veech RL (2010) A ketone ester diet increases brain malonyl-CoA and Uncoupling proteins 4 and 5 while decreasing food intake in the normal Wistar Rat. J Biol Chem 285:25950–25956
D’Agostino DP, Pilla R, Held HE, Landon CS, Puchowicz M, Brunengraber H, Ari C, Arnold P, Dean JB (2013) Therapeutic ketosis with ketone ester delays central nervous system oxygen toxicity seizures in rats. Am J Physiol Reg Integr Comp Physiol 304:R829–R836
Viggiano A, Pilla R, Arnold P, Monda M, D’Agostino D, Coppola G (2015) Anticonvulsant properties of an oral ketone ester in a pentylenetetrazole-model of seizure. Brain Res 1618:50–54
Ciarlone SL, Grieco JC, D’Agostino DP, Weeber EJ (2016) Ketone ester supplementation attenuates seizure activity, and improves behavior and hippocampal synaptic plasticity in an Angelman syndrome mouse model. Neurobiol Dis 96:38–46
Courchesne-Loyer A, Croteau E, Castellano CA, St-Pierre V, Hennebelle M, Cunnane SC (2016) Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: A dual tracer quantitative positron emission tomography study. J Cereb Blood Flow Metab. doi:10.1177/0271678X16669366
Zhang Y, Kuang Y, Xu K, Harris D, Lee Z, LaManna J, Puchowicz MA (2013) Ketosis proportionately spares glucose utilization in brain. J Cereb Blood Flow Metab 33:1307–1311
Garriga-Canut M, Schoenike B, Qazi R, Bergendahl K, Daley TJ, Pfender RM, Morrison JF, Ockuly J, Stafstrom C, Sutula T (2006) 2-Deoxy-d-glucose reduces epilepsy progression by NRSF-CtBP–dependent metabolic regulation of chromatin structure. Nat Neurosci 9:1382–1387
Gasior M, Yankura J, Hartman AL, French A, Rogawski MA (2010) Anticonvulsant and proconvulsant actions of 2-deoxy-d-glucose. Epilepsia 51:1385–1394
Stafstrom CE, Ockuly JC, Murphree L, Valley MT, Roopra A, Sutula TP (2009) Anticonvulsant and antiepileptic actions of 2,deoxy-d-glucose in epilepsy models. Ann Neurol 65:435–447
Potter WB, O’Riordan KJ, Barnett D, Osting SM, Wagoner M, Burger C, Roopra A (2010) Metabolic regulation of neuronal plasticity by the energy sensor AMPK. PLoS One 5:e8996
Achanta LB, Rae CD (2016) β-Hydroxybutyrate in the brain: one molecule, multiple mechanisms. Neurochem Res. doi:10.1007/s11064-016-2099-2
Yudkoff M, Daikhin Y, Nissim I, Horyn O, Lazarow A, Luhovyy B, Wehrli S, Nissim I (2005) Response of brain amino acid metabolism to ketosis. Neurochem Int 47:119–128
Yudkoff M, Daikhin Y, Nissim I, Grunstein R, Nissim I (1997) Effects of ketone bodies on astrocyte amino acid metabolism. J Neurochem 69:682–692
Yudkoff M, Daikhin Y, Nissim I, Lazarow A, Nissim I (2004) Ketogenic diet, brain glutamate metabolism and seizure control. Prostaglandins Leukot Essent Fatty acids 70:277–285
Melo TM, Nehlig A, Sonnewald U (2006) Neuronal-glial interactions in rats fed a ketogenic diet. Neurochem Int 48:498–507
Bough KJ, Rho JM (2007) Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 48:43–58
Masino SA, Rho JM (2012) Mechanisms of ketogenic diet action. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV (eds) Jasper’s basic mechanisms of the epilepsies, 4th edn., Bethesda
Dean HG, Bonser JC, Gent JP (1989) HPLC analysis of brain and plasma for octanoic and decanoic acids. Clin Chem 35:1945–1948
Haidukewych D, Forsythe WI, Sills M (1982) Monitoring octanoic and decanoic acids in plasma from children with intractable epilepsy treated with medium-chain triglyceride diet. Clin Chem 28:642–645
Sills MA, Forsythe WI, Haidukewych D, MacDonald A, Robinson M (1986a) The medium chain triglyceride diet and intractable epilepsy. Arch Dis Child 61:1168–1172
Kuge Y, Yajima K, Kawashima H, Yamazaki H, Hashimoto N, Miyake Y (1995) Brain uptake and metabolism of [1-11C] octanoate in rats: pharmacokinetic basis for its application as a radiopharmaceutical for studying brain fatty acid metabolism. Ann Nucl Med 9:137–142
Oldendorf WH (1973) Carrier-mediated blood–brain barrier transport of short-chain monocarboxylic organic acids. Am J Physiol 224:1450–1453
Chang P, Terbach N, Plant N, Chen PE, Walker MC, Williams RS (2013) Seizure control by ketogenic diet-associated medium chain fatty acids. Neuropharmacology 69:105–114
Perlman BJ, Goldstein DB (1984) Membrane-disordering potency and anticonvulsant action of valproic acid and other short-chain fatty acids. Mol Pharmacol 26:83–89
Wlaz P, Socala K, Nieoczym D, Luszczki JJ, Zarnowska I, Zarnowski T, Czuczwar SJ, Gasior M (2012) Anticonvulsant profile of caprylic acid, a main constituent of the medium-chain triglyceride (MCT) ketogenic diet, in mice. Neuropharmacology 62:1882–1889
Wlaz P, Socala K, Nieoczym D, Zarnowski T, Zarnowska I, Czuczwar SJ, Gasior M (2015) Acute anticonvulsant effects of capric acid in seizure tests in mice. Prog NeuroPsychopharmacol Biol Psychiatry 57:110–116
McDonald TS, Tan KN, Hodson MP, Borges K (2014) Alterations of hippocampal glucose metabolism by even versus uneven medium chain triglycerides. J Cereb Blood Flow Metab 34:153–160
Tan KN, Carrasco-Pozo C, McDonald TS, Puchowicz M, Borges K (2016) Tridecanoin is anticonvulsant, antioxidant, and improves mitochondrial function. J Cereb Blood Flow Metab. doi:10.1177/0271678X16659498
Edmond J, Robbins R, Bergstrom J, Cole R, De Vellis J (1987) Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J Neurosci Res 18:551–561
Cremer JE, Teal HM, Heath DF, Cavanagh JB (1977) The Influence of portocaval anastomosis on the metabolism of labeled octanoate, butyrate and leucine in rat brain. J Neurochem 28:215–222
Ebert D, Haller RG, Walton ME (2003) Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J Neurosci 23:5928–5935
Roe CR, Mochel F (2006) Anaplerotic diet therapy in inherited metabolic disease: therapeutic potential. J Inherit Metab Dis 29:332–340
Vockley J, Marsden D, McCracken E, DeWard S, Barone A, Hsu K, Kakkis E (2015) Long-term major clinical outcomes in patients with long chain fatty acid oxidation disorders before and after transition to triheptanoin treatment: a retrospective chart review. Mol Genet Metab 116:53–60
Mochel F, Duteil S, Marelli C, Jauffret C, Barles A, Holm J, Sweetman L, Benoist JF, Rabier D, Carlier PG, Durr A (2010) Dietary anaplerotic therapy improves peripheral tissue energy metabolism in patients with Huntington’s disease. Eur J Hum Genet 18:1057–1060
Adanyeguh IM, Rinaldi D, Henry PG, Caillet S, Valabregue R, Durr A, Mochel F (2015) Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 84(5):490–495
Mochel F, Hainque E, Gras D, Adanyeguh IM, Caillet S, Heron B, Roubertie A, Kaphan E, Valabregue R, Rinaldi D, Vuillaumier S, Schiffmann R, Ottolenghi C, Hogrel JY, Servais L, Roze E (2016) Triheptanoin dramatically reduces paroxysmal motor disorder in patients with GLUT1 deficiency. J Neurol Neurosurg Psychiatry 87:550–553
Pascual JM, Liu P, Mao D, Kelly DI, Hernandez A, Sheng M, Good LB, Ma Q, Marin-Valencia I, Zhang X, Park JY, Hynan LS, Stavinoha P, Roe CR, Lu H (2014) Triheptanoin for glucose transporter type I deficiency (G1D): modulation of human ictogenesis, cerebral metabolic rate, and cognitive indices by a food supplement. JAMA Neurol 71:1255–1265
Marin-Valencia I, Good LB, Ma Q, Malloy CR, Pascual JM (2013) Heptanoate as a neural fuel: energetic and neurotransmitter precursors in normal and glucose transporter I-deficient (G1D) brain. J Cereb Blood Flow Metab 33:175–182
Kinman RP, Kasumov T, Jobbins KA, Thomas KR, Adams JE, Brunengraber LN, Kutz G, Brewer WU, Roe CR, Brunengraber H (2006) Parenteral and enteral metabolism of anaplerotic triheptanoin in normal rats. Am J Physiol Endocrinol Metab 291:E860–E866
Brunengraber H, Roe CR (2006) Anaplerotic molecules: current and future. J Inherit Metab Dis 29:327–331
Kim TH, Borges K, Petrou S, Reid CA (2013) Triheptanoin reduces seizure susceptibility in a syndrome-specific mouse model of generalized epilepsy. Epilepsy Res 103:101–105
Thomas NK, Willis S, Sweetman L, Borges K (2012) Triheptanoin in acute mouse seizure models. Epilepsy Res 99:312–317
Baumer D, Talbot K, Turner MR (2014) Advances in motor neurone disease. J R Soc Med 107:14–21
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH Jr (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL, Mugnaini E, Pericak-Vance MA, Siddique T (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477:211–215
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Consortium I, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268
Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT (1990) Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol 28:18–25
Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281:1851–1854
Julien JP, Beaulieu JM (2000) Cytoskeletal abnormalities in amyotrophic lateral sclerosis: beneficial or detrimental effects? J Neurol Sci 180:7–14
McGeer PL, McGeer EG (2002) Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 26:459–470
Barber SC, Mead RJ, Shaw PJ (2006) Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim Biophys Acta 1762:1051–1067
Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S (2007) Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 10:615–622
Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP (2004) Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci USA 101:11159–11164
Dupuis L, Pradat PF, Ludolph AC, Loeffler JP (2011) Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol 10:75–82
Paganoni S, Deng J, Jaffa M, Cudkowicz ME, Wills AM (2011) Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve 44:20–24
Korner S, Hendricks M, Kollewe K, Zapf A, Dengler R, Silani V, Petri S (2013) Weight loss, dysphagia and supplement intake in patients with amyotrophic lateral sclerosis (ALS): impact on quality of life and therapeutic options. BMC Neurol 13:84
Desport JC, Preux PM, Magy L, Boirie Y, Vallat JM, Beaufrere B, Couratier P (2001) Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am J Clin Nutr 74:328–334
Dalakas MC, Hatazawa J, Brooks RA, Di Chiro G (1987) Lowered cerebral glucose utilization in amyotrophic lateral sclerosis. Ann Neurol 22:580–586
Hatazawa J, Brooks RA, Dalakas MC, Mansi L, Di Chiro G (1988) Cortical motor-sensory hypometabolism in amyotrophic lateral sclerosis: a PET study. J Comput Assist Tomogr 12:630–636
Ludolph AC, Langen KJ, Regard M, Herzog H, Kemper B, Kuwert T, Bottger IG, Feinendegen L (1992) Frontal lobe function in amyotrophic lateral sclerosis: a neuropsychologic and positron emission tomography study. Acta Neurol Scand 85:81–89
Browne SE, Yang L, DiMauro JP, Fuller SW, Licata SC, Beal MF (2006) Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiol Dis 22:599–610
Cistaro A, Valentini MC, Chio A, Nobili F, Calvo A, Moglia C, Montuschi A, Morbelli S, Salmaso D, Fania P, Carrara G, Pagani M (2012) Brain hypermetabolism in amyotrophic lateral sclerosis: a FDG PET study in ALS of spinal and bulbar onset. Eur J Nucl Med Mol Imaging 39:251–259
Niessen HG, Debska-Vielhaber G, Sander K, Angenstein F, Ludolph AC, Hilfert L, Willker W, Leibfritz D, Heinze HJ, Kunz WS, Vielhaber S (2007) Metabolic progression markers of neurodegeneration in the transgenic G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Eur J Neurosci 25:1669–1677
Wiedemann FR, Manfredi G, Mawrin C, Beal MF, Schon EA (2002) Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem 80:616–625
Jung C, Higgins CM, Xu Z (2002) Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J Neurochem 83:535–545
Mattiazzi M, D’Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G (2002) Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem 277:29626–29633
Menzies FM, Cookson MR, Taylor RW, Turnbull DM, Chrzanowska-Lightowlers ZM, Dong L, Figlewicz DA, Shaw PJ (2002) Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain 125:1522–1533
Rothstein JD, Martin LJ, Kuncl RW (1992) Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med 326:1464–1468
Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 38:73–84
Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH Jr, Julien JP, Goldstein LS, Cleveland DW (2003) Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302:113–117
Ngo S, Steyn F, McCombe P, Borges K (2015) High caloric diets for amyotrophic lateral sclerosis. In: RWaV Preedy (ed) Bioactive nutraceuticals and dietary supplements in neurological and brain disease: prevention and therapy. Elsevier, Amsterdam, pp 461–470
Holness MJ, Sugden MC (2003) Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans 31:1143–1151
Miquel E, Cassina A, Martinez-Palma L, Bolatto C, Trias E, Gandelman M, Radi R, Barbeito L, Cassina P (2012) Modulation of astrocytic mitochondrial function by dichloroacetate improves survival and motor performance in inherited amyotrophic lateral sclerosis. PLoS One 7:e34776
Zhao Z, Lange DJ, Voustianiouk A, MacGrogan D, Ho L, Suh J, Humala N, Thiyagarajan M, Wang J, Pasinetti GM (2006) A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci 7:29
Zhao W, Varghese M, Vempati P, Dzhun A, Cheng A, Wang J, Lange D, Bilski A, Faravelli I, Pasinetti GM (2012) Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PloS one 7:e49191
Group AL, Fournier C, Bedlack B, Hardiman O, Heiman-Patterson T, Gutmann L, Bromberg M, Ostrow L, Carter G, Kabashi E, Bertorini T, Mozaffar T, Andersen P, Dietz J, Gamez J, Dimachkie M, Wang Y, Wicks P, Heywood J, Novella S, Rowland LP, Pioro E, Kinsley L, Mitchell K, Glass J, Sathornsumetee S, Kwiecinski H, Baker J, Atassi N, Forshew D, Ravits J, Conwit R, Jackson C, Sherman A, Dalton K, Tindall K, Gonzalez G, Robertson J, Phillips L, Benatar M, Sorenson E, Shoesmith C, Nash S, Maragakis N, Moore D, Caress J, Boylan K, Armon C, Grosso M, Gerecke B, Wymer J, Oskarsson B, Bowser R, Drory V, Shefner J, Lechtzin N, Leitner M, Miller R, Mitsumoto H, Levine T, Russell J, Sharma K, Saperstein D, McClusky L, MacGowan D, Licht J, Verma A, Strong M, Lomen-Hoerth C, Tandan R, Rivner M, Kolb S, Polak M, Rudnicki S, Kittrell P, Quereshi M, Sachs G, Pattee G, Weiss M, Kissel J, Goldstein J, Rothstein J, Pastula D, Gleb L, Ogino M, Rosenfeld J, Carmi E, Oster C, Barkhaus P, Valor E (2013) ALS Untangled No. 20: the Deanna protocol. Amyotroph Lateral Scler Frontotemporal Degener 14:319–323
Ari C, Poff AM, Held HE, Landon CS, Goldhagen CR, Mavromates N, D’Agostino DP (2014) Metabolic therapy with Deanna protocol supplementation delays disease progression and extends survival in amyotrophic lateral sclerosis (ALS) mouse model. PLoS One 9:e103526
Tefera TW, Wong Y, Barkl-Luke ME, Ngo ST, Thomas NK, McDonald TS, Borges K (2016) Triheptanoin protect motor neurons and delays symptom onset in a mouse model of ALS model. PLOS One 11(8):e0161816
Acknowledgements
We are grateful for scholarships from UQ international scholarships (KNT, TWT) and APA (TSM) and funding from the Australian National Health and Medical Research Council (Grant 1044007 to KB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
KB has filed for patents for the use of triheptanoin in seizure disorders and ALS.
Rights and permissions
About this article

Cite this article
Tefera, T.W., Tan, K.N., McDonald, T.S. et al. Alternative Fuels in Epilepsy and Amyotrophic Lateral Sclerosis. Neurochem Res 42, 1610–1620 (2017). https://doi.org/10.1007/s11064-016-2106-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-016-2106-7