
Abstract
Memory decline is characteristic of aging and age-related neurodegenerative disorders. This study was designed to investigate the protective effect of hyperbaric oxygen (HBO) against cognitive impairment induced by d-galactose (d-gal) in mice. d-gal was intraperitoneally injected into mice daily for 8 weeks to establish the aging model. HBO was simultaneously administered once daily. The results indicate that HBO significantly reversed D-gal-induced learning and memory impairments. Studies on the potential mechanisms of this action showed that HBO significantly reduced oxidative stress by increasing superoxide dismutase, glutathione peroxidase, and catalase levels, as well as the total anti-oxidation capability, while decreasing the content of malondialdehyde, nitric oxide, and nitric oxide synthase in the hippocampal CA1 region. HBO also inhibited advanced glycation end-product formation and decreased levels of tumor necrosis factor-α and interleukin-6. Moreover, HBO significantly attenuated d-gal-induced pathological injury in the hippocampus, as well as β-amyloid protein1−42 expression and retained BDNF expression. Furthermore, HBO decreased p16, p21 and p53 gene and protein expression in the hippocampus of d-gal-treated mice. In conclusion, the protective effect of HBO against d-gal-induced cognitive impairment was mainly due to its ability to reduce oxidative damage, suppress inflammatory responses, and regulate aging-related gene expression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brain senescence is characterized by gradual loss of cognitive function exhibited by decline in learning and spatial ability [1, 2]. However, the underlying mechanisms of senescence remain unclear. A widely acknowledged theory is the oxidative stress theory [3]. It suggests that the generation of reactive oxygen species (ROS) can trigger mitochondrial dysfunction and cellular impairment, thus resulting in cellular senescence and aging [4, 5]. The concentration of ROS is determined by the balance between the rate of production and clearance by various antioxidant enzymes. Moreover, growth factor deficiency, such as brain-derived neurotrophic factor (BDNF), inhibits neuron proliferation and subsequently leads to senescence [6]. BDNF is a powerful neurotrophic factor of neuronal excitability and synaptic transmission [7] and has been found to promote the survival of all types of neurons related to neurodegenerative disorders [8].
Furthermore, advanced glycation end-products (AGEs) formation is a trigger for the onset of age-related disease. Numerous studies have shown that the interaction of AGEs with its cell surface receptors activates nuclear factor-κB (NF-κB) signaling pathways [9], which increases the production of ROS and certain inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1) and interleukin-6 (IL-6) and enhances microglial cells and astrocyte activation, ultimately resulting in degenerative disorders [10]. In addition, p53-p21-p19 and p16-retinoblastoma signal pathway activation triggered by internal and external factors may result in senescence [11, 12]. The two pathways could promote one another mutually or only one was activated to induce cellular senescence. Therefore, the related drug or therapy for the abovementioned targets may attenuate cognitive impairment and delay aging.
Numerous studies have shown that chronic exposure to d-galactose (d-gal) induces behavioral changes, including deterioration of cognitive and motor symptoms [13, 14]. Moreover, over-dose d-gal induces oxidative stress, glycative stress and apoptotic in vivo [15]. Previous study indicated that d-gal might accelerate the aging process, accounted for 8 weeks d-gal injection induced 24 months aging [16]. Thus, the d-gal-induced senescent mouse mimics many characteristics of the natural process of aging and is regarded as an ideal model to study the aging brain. To prevent oxidative stress-related neurological alterations, vitamin E (Vit E) was chosen as a control, which was consisted with our previous study [17].
Hyperbaric oxygen (HBO) therapy is an adjunctive therapy that has been proposed to improve outcomes following hypoxia and ischemia-related injuries caused by many factors, including carbon monoxide poisoning, myocardial infarction, and cerebral ischemia [18, 19]. Clinically, HBO therapy also has been used to improve neurodegenerative disorders [20, 21]. Our previous work has demonstrated that a combination therapy including HBO and Madopar® could protect against 6-hydroxydopamine (6-OHDA)-induced Parkinson’s disease in rats [22]. Experimental and clinical data suggest that this effect is mediated by reducing oxidative stress, decreasing tissue edema, altering nitric oxide syntheses, and inhibiting neuroinflammation factor expression and apoptotic pathways [23]. Recently, it was found that HBO treatment may alleviate delayed memory impairment after carbon monoxide poisoning by increasing the level of hippocampal BDNF [24]. However, it remains lacking of evidence if HBO could provide protection on cognitive impairment until now.
In our current study, for the purpose of supplying the preclinical data for the clinical application, d-gal-induced senescent mice were used as a model to examine whether HBO provided neuroprotective effects. The Morris water maze was utilized to test behavior. In order to elucidate its possible mechanism, the levels of antioxidants, AGEs, and inflammatory cytokines TNF-α and IL-6 were measured using commercial kits. In addition, immunoreactive cells for BDNF were counted in an immunohistochemical analysis of brain tissue. The expression of the senescence-related proteins (β-amyloid protein (Aβ)1−42, p21 and p53) and mRNA (p16 and p21) was determined using real-time PCR.
Materials and Methods
Drugs and Reagents
Vit E was provided by Guilin Pharma Co., Ltd (Guilin, China). d-gal was provided by Sigma–Aldrich Co. Ltd (Germany). TNF-α, IL-6 and AGEs ELISA kits were purchased from Wuhan Boster Bio-engineering Co., Ltd. (Wuhan, China). Superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), malondialdehyde (MDA), catalase (CAT), total anti-oxidation capability (T-AOC), nitric oxide (NO), and nitric oxide synthase (NOS) kits were supplied by Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Animals Protocols
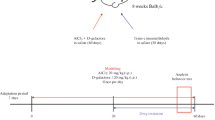
Male Kunming mice, 3 month old, were obtained from the Experimental Animal Center of Guangxi Medical University (Certificate No. SYXK 2009-0002). The research was conducted in accordance with the Animal Ethics Committee of the Guangxi Medical University (Approval No.: 20110501202) and the National Institutes of Health guide for the care and use of Laboratory animals (National Institutes of Health Publications No. 8023, revised 1978). The animals were housed under controlled conditions at 25 ± 2 °C with a relative humidity of 60 ± 10 %, and a 12-h light/dark cycle (lights on from 8:00 AM to 8:00 PM). The mice were given standard rodent chow and free access to water.
A total of 120 mice were randomly assigned to four groups of 30 mice per group: the d-gal model group, consisting of mice that were intraperitoneally injected with 200 mg/kg d-gal (in physiological saline, 150 mg/mL) once daily for 8 weeks; the d-gal + Vit E group, consisting of animals that were intraperitoneally injected with 200 mg/kg d-gal and intragastrically administered Vit E (in 0.1 % Tween 80, 0.2 g/kg), which was initiated in the first day of 6th week of d-gal injection; the d-gal + HBO group, consisting of animals that were intraperitoneally injected with 200 mg/kg d-gal and received HBO treatment, when was initiated the same as Vit E administration; and the normal control group, consisting of mice that were administered equivalent vehicle using the same method.
Hyperbaric Therapies
The animals were placed in hyperbaric chambers (Yantai Hongyuan Co., Ltd.) as previously described [22]. Then, the pressure was increased to 0.25 MPa at a rate of 100 kPa/min, and each treatment lasted for 60 min with pure oxygen. Decompression was performed at a uniform rate over 10 min. HBO treatment was performed once daily for 14 consecutive days.
Morris Water Maze Test
At the end of the treatment period, the behavioral test was performed on next day the same time of day (9:00 am to 3:00 pm).
The apparatus of the Morris water maze was a black stainless steel circular tank with a diameter of 100 cm and a height of 50 cm (Chinese Academy of Medical Sciences). The tank was divided into four equal quadrants. A movable escape platform (10 cm in diameter, 28 cm in height) was located at the center of the 3rd quadrant and hidden 1.5 cm below the surface of the water (25 ± 2 °C). A digital video camera was positioned directly above the pool and connected with a system (SuperMaze software, China) that enabled full collection of the swimming pattern and distance.
The Morris water maze task included two phases (navigation training and spatial probe test) and was administered according to the procedures described for a previous study [14, 25] but with slight modification. The mice performed two trial sessions each day for 5 consecutive days, with each trial having a ceiling time of 60 s and a trial interval of 60 s. Once the mouse located the platform, it was permitted to remain on it for 10 s. The time to reach the platform was measured as the escape latency. If the mouse failed to reach the escape platform within 60 s, it was placed on the platform for 10 s and the escape latency was recorded as 60 s. On day 6, the platform was removed and each mouse was allowed to swim freely for 60 s as the probe test. The latency to reach the target quadrant (where the platform was once hidden), the numbers of crossing the target quadrant and the time spent in target quadrant were recorded for each trial.
Serum and Brain Regions Tissue Sampling
After Morris water maze test, all mice were immediately anesthetized using sodium pentobarbital (30 mg/kg, i.p.), and blood samples were collected from the abdominal aorta. The serum samples were obtained by centrifugation (3500 rpm, 4 °C) for 10 min and were kept frozen at −80 °C until assayed as described below.
After the mice were euthanized, the brain was promptly removed. According to rat brain stereotaxic coordinates [26], the right hippocampal CA1 region was isolated. 8 samples were homogenized with ice-cold physiological saline (10 % w/v) using a homogenizer (Ningbo, China) to measure the antioxidation parameters. Some samples were frozen in liquid nitrogen and preserved at −80 °C for PCR (n = 8) and western blot (n = 8) analysis. The remaining 6 samples were fixed in 4 % paraformaldehyde and prepared for histopathological and immunohistochemical examination.
Biochemical Assays in Hippocampal CA1 Region
Levels of SOD, GSH-Px, MDA, CAT, T-AOC, NO and NOS were measured using commercially available kits (Nanjing Jiancheng Bioengineering Institute, China) according to the manufacturer’s instructions.
Detection of TNF-α, IL-6 and AGEs in the Serum
The levels of TNF-α, IL-6 and AGEs in the serum from each group were measured by ELISA following the manufacturer’s instructions.
Histological and Immunohistochemical Examination
Samples from the hippocampal CA1 region (embedded in paraffin) were cut into five consecutive sections at each stereotaxic level (approximately 4-µm thick), followed by routine hematoxylin and eosin (HE) staining.
Immunohistochemistry was performed as previously described [27]. Using the microwave antigen retrieval method (in sodium citrate antigen retrieval fluid, pH 6.0) to expose the antigenic sites, the sections were then incubated in 0.3 % H2O2 for 30 min at room temperature to eliminate endogenous peroxidase. After blocking in normal goat serum for 30 min at room temperature, the waxed specimens were incubated with rabbit BDNF (Origene, USA; 1:400) monoclonal antibody overnight at 4 °C. The sections were washed in 0.1 mol/L phosphate buffered saline (PBS) three times (5 min each) and incubated with biotinylated goat anti-rabbit immunoglobulin G (Zhongshan Goldenbridge Biological Technology, Beijing, China; 1:400) at 37 °C for 30 min. After washing with 0.1 mol/L PBS three times (5 min each), the specimens were incubated with an avidin–biotin complex at 37 °C for 30 min. Then, they were rinsed with 0.1 mol/L PBS three times (5 min each), incubated with diaminobenzidine for 3 min at room temperature, counterstained with hematoxylin, cleared, mounted and examined.
Pathological changes and immunohistochemical observation of the hippocampus CA1 region were analyzed using a light microscope with a magnification of ×400 (Olympus, Germany). Cytoplasm stained brown was the standard of BDNF positive cell. Cell counts were quantified in five independent sections per mouse from all of the experimental groups [28]. The region of interest was captured using a camera and analyzed using ImagePro-Plus software.
Western Blot Analysis
hippocampus CA1 tissue was homogenized in a cold radio immunoprecipitation assay lysis buffer (Solarbio, China) containing 50 mM Tris (pH 7.4), 150 mM sodium chloride, 1 % nonidet P-40, 1 mM ethylene diamine tetraacetic acid, 0.1 % sodium dodecylsulphate, 0.5 % sodium deoxycholate and 1 µg/mL each of aprotinin, leupeptin and pepstatin. The homogenate was centrifuged at 12,000×g for 10 min at 4 °C and the supernatant was transferred to a new tube. After measuring the protein content, the fraction was electrophoresed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were incubated overnight at 4 °C with the primary antibodies: rabbit polyclonal anti-β amyloid 1–42 (Abcam, USA), rabbit polyclonal anti-p21 (Abcam, USA) and rabbit polyclonal anti-p53 (Abcam, USA) then incubated with the secondary antibody diluted 1:10,000 in tris buffered saline with tween (TBST) for 1 h. The immune-reactive bands were detected with HRP staining. β-Actin was the protein loading control. The relative expressions of p21 and p53 proteins were quantified by densitometric scanning using the Odyssey V 3.0 software.
Analysis of p16 and p21 mRNA by Real Time PCR
Total RNA was extracted from hippocampus CA1 tissue using TRIzol reagent (Invitrogen). cDNA was synthesized from total RNA using reverse transcriptase (Roche). As a template, the cDNA was amplified using the 7300 Real Time PCR System (Applied Biosystems) with primers. Then, the levels of p16 and p21 mRNA expression were determined. The parallel amplification of glyceraldehyde-phosphate dehydrogenase (GAPDH) was performed for reference. The sequences of the primers used in this study are as follows: P16 (178 bp, forward primer: 5′-CGGGGACATCAAGACATCGT-3′, reverse primer: 5′-GCCGGATTTAGCTCTGCTCT-3′); P21 (212 bp, forward primer: 5′-CTGTCTTGCACTCTGGTGTCT-3′, reverse primer: 5′-CTAAGGCCGAAGATGGGGAA-3′); and GAPDH (189 bp, forward primer: 5′-ACTTGAAGGGTGGAGCCAAA-3′, reverse primer: 5′-GCCCTTCCACAATGCCAAAG-3′). The final results are described with the relative values (2−∆∆Ct).
Data Analysis
The data are expressed as the mean ± SE. Statistical analyses were performed using Statistics Package for Social Science 13.0 software (SPSS Inc., USA). The data were analyzed using one-way analysis of variance (ANOVA). Post hoc analysis was carried out with the least significant difference (LSD) t test. The level of significance was set at P < 0.05.
Results
HBO Restored Cognitive Impairment Induced by d-gal
Normal control mice rapidly learned the location of the platform, but d-gal-treated mice showed significantly longer escape latencies to the platform on every test day during navigation training [Day 1: 58.58 ± 1.97 vs. 35.04 ± 7.44, F (3, 36) = 53.46, P < 0.05; Day 2: 52.96 ± 5.73 vs. 30.52 ± 6.69, F (3, 36) = 34.199, P < 0.05; Day 3: 50.30 ± 4.04 vs. 23.87 ± 3.13, F (3, 36) = 92.301, P < 0.05; Day 4: 45.86 ± 3.50 vs. 18.32 ± 2.80, F (3, 36) = 135.379, P < 0.05; Day 5: 39.02 ± 3.32 vs. 14.18 ± 1.88, F (3, 36) = 137.912, P < 0.05] (Fig. 1A). Both Vit E and HBO treatment in d-gal-administered mice significantly shortened the escape latency from 3rd to 5th test day [D1: d-gal + Vit E: 57.60 ± 4.65, P > 0.05; d-gal + HBO: 54.38 ± 3.39, P > 0.05; D2: d-gal + Vit E: 47.21 ± 3.28, P > 0.05; d-gal + HBO: 45.63 ± 4.35, P > 0.05; D3: d-gal + Vit E: 39.11 ± 4.09, P < 0.05; d-gal + HBO: 37.00 ± 2.87, P < 0.05; D4: d-gal + Vit E: 31.73 ± 3.09, P < 0.05; d-gal + HBO: 26.56 ± 3.15, P < 0.05; D5: d-gal + Vit E: 24.92 ± 3.86, P < 0.05; d-gal + HBO: 21.31 ± 1.48, P < 0.05].

Behavioral tests. A Latencies to find a hidden platform in the water maze during the 5 days of place navigation training. On the sixth day, another set of tests was performed when the target platform was removed. B The escape latency of the mice when the platform was removed. C The frequency of the mice crossing the target quadrant. D The percentage of time that the mice stayed in the quadrant where the platform was once placed. a P < 0.05 compared to the normal control group. b P < 0.05 compared to the d-gal model group
To assess spatial memory more directly, the mice were subjected to a probe test in which the target platform was removed the day after the navigation training. As shown in Fig. 1B–D, d-gal-administered mice showed a longer time to reach the location of the removed platform (escape latency), lower frequency to cross the location of the platform (the number of target crossings), and less time in the target quadrant (percentage of time in the target quadrant) compared to the normal group [escape latency: 33.73 ± 2.97 vs. 13.15 ± 2.45, F (3, 36) = 118.721, P < 0.05; the number of target crossings: 2.00 ± 0.67 vs. 4.40 ± 0.84, F (3, 36) = 16.923, P < 0.05; percentage of time in the target quadrant: 18.70 ± 3.92 vs. 43.29 ± 2.80, F (3, 36) = 71.789, P < 0.05]. However, these parameters were reversed following Vit E and HBO treatment [escape latency: d-gal + Vit E: 20.91 ± 2.84, P < 0.05; d-gal + HBO: 17.81 ± 1.81, P < 0.05; the number of target crossings: d-gal + Vit E: 2.90 ± 0.74, P < 0.05; d-gal + HBO: 3.20 ± 0.79, P < 0.05; percentage of time in the target quadrant: d-gal + Vit E: 34.40 ± 4.12, P < 0.05; d-gal + HBO: 39.01 ± 4.90, P < 0.05].
HBO Enhanced Antioxidant Activity and Reduced Oxidative Damage
As shown in Fig. 2-gal-treated mice possessed significantly low activities of SOD, GSH-Px, CAT, and T-AOC compared with the control group [SOD: 101.67 ± 12.17 vs. 184.36 ± 6.16, F (3, 28) = 66.362, P < 0.05; GSH-Px: 253.94 ± 29.30 vs. 547.97 ± 31.87, F (3, 28) = 143.534, P < 0.05; CAT: 3.28 ± 0.88 vs. 6.91 ± 0.62, F (3, 28) = 38.551, P < 0.05; T-AOC: 2.76 ± 0.96 vs. 10.30 ± 0.85, F (3, 28) = 128.612, P < 0.05], whereas Vit E and HBO treatment inhibited the reduction induced by d-gal [d-gal + Vit E: SOD: 147.92 ± 15.26, P < 0.05; GSH-Px: 383.74 ± 27.39, P < 0.05; CAT: 5.35 ± 0.45, P < 0.05; T-AOC: 6.17 ± 0.74, P < 0.05; d-gal + HBO: SOD: 169.07 ± 15.21, P < 0.05; GSH-Px: 446.37 ± 28.30, P < 0.05; CAT: 5.79 ± 0.65, P < 0.05; T-AOC: 8.03 ± 0.99, P < 0.05].

Biochemical assays in the hippocampus CA1 region. The results are presented as the mean ± SE, (n = 8). a P < 0.05 compared to the normal control group. b P < 0.05 compared to the d-gal model group
The hippocampal CA1 content of MDA, NO, and NOS was significantly enhanced in the d-gal model group compared with the normal group [MDA: 6.41 ± 0.37 vs. 2.61 ± 0.44, F (3, 28) = 78.518, P < 0.05; NO: 9.08 ± 0.92 vs. 5.08 ± 0.42, F (3, 28) = 39.882, P < 0.05; NOS: 3.72 ± 0.20 vs. 1.85 ± 0.15, F (3, 28) = 56.367, P < 0.05]. In contrast, these increases were attenuated by Vit E and HBO administration [d-gal + Vit E: MDA: 3.78 ± 0.51, P < 0.05; NO: 7.36 ± 0.91, P < 0.05; NOS: 3.08 ± 0.38, P < 0.05; d-gal + HBO: MDA: 3.44 ± 0.61, P < 0.05; NO: 6.43 ± 1.01, P < 0.05; NOS: 2.63 ± 0.41, P < 0.05].
Effect of HBO on TNF-α, IL-6 and AGEs Content in the Serum of Aging Mice Induced by d-gal
Elisa analysis showed that the TNF-α and IL-6 levels in the serum of d-gal-treated mice were significantly higher than those in normal controls [TNF-α: 8.74 ± 0.29 vs. 3.56 ± 0.62, F (3, 36) = 194.338, P < 0.05; IL-6: 76.21 ± 6.30 vs. 37.88 ± 4.89, F (3, 36) = 101.802, P < 0.05]. In contrast, both Vit E and HBO administration suppressed the increase in TNF-α and IL-6 [d-gal + Vit E: TNF-α: 7.45 ± 0.44, P < 0.05; IL-6: 53.08 ± 3.38, P < 0.05; d-gal + HBO: TNF-α: 5.77 ± 0.23, P < 0.05; IL-6: 41.74 ± 2.57, P < 0.05] (Fig. 3).

Effect of HBO on IL-2, IL-6 and AGEs serum concentrations. The results are presented as the mean ± SE, (n = 10). a P < 0.05 compared to the normal control group. b P < 0.05 compared to the d-gal model group
The AGEs level in the d-gal model group was significantly elevated to more than twice the level of that in the normal control group [366.58 ± 30.39 vs. 179.28 ± 27.78, F (3, 36) = 59.387, P < 0.05]. In contrast, both Vit E and HBO administration suppressed the increase in AGEs [d-gal + Vit E: 300.23 ± 24.41, P < 0.05; d-gal + HBO: 257.93 ± 24.75, P < 0.05] (Fig. 3). Notably, d-gal plus HBO administration decreased AGEs expression by approximately 30 % compared to the d-gal treatment alone.
Effects of HBO on Histopathological Changes in the Hippocampus
The neuropathological features of hippocampal CA1 sections are presented in Fig. 4. The pyramidal cells in the CA1 region were tightly arranged and exhibited round nuclei in normal animals. Pyramidal neuron shrinkage and chromatin condensation of nuclei were also observed in d-gal model animals. Treatment with Vit E and HBO resulted in an improvement in the structure of these neurons.

Histopathology in the hippocampus CA1 region of mice (HE, magnification ×400). The pyramidal cells in the CA1 region were tightly arranged and exhibited rounded nuclei in the normal control group (A). Extensive damage and loss of pyramidal cells were observed in the d-gal model animals (B). Vit E and HBO were found to restore the structure of neurons (C, D). E The number of surviving neurons in the hippocampus CA1 region. Bar graph of neuronal counts relative to the number of surviving neurons/mm2 CA1 pyramidal layer (scale bar 50 μm). The data are expressed as the mean ± SE (n = 6). a P < 0.05 compared to the normal control group. b P < 0.05 compared to the d-gal model group. c P < 0.05 compared to the d-gal + Vit E group
A decline in the number of surviving neurons in the CA1 region was observed in the d-gal model group in relation to the normal control [48.51 ± 12.52 vs. 203.81 ± 17.35, F (3, 20) = 252.003, P < 0.05] (Fig. 4). Treatment with Vit E produced a trend of increase in the number of hippocampal CA1 neurons [65.20 ± 6.42, P > 0.05] although it had no statistic difference. Interestingly, HBO treatment produced a significant improvement in the number of surviving neurons in the CA1 region compared with the d-gal model animals [146.20 ± 9.70, P < 0.05]. Moreover, number of surviving neurons was 2.24 times higher in HBO treatment group than that in the Vit E group (P < 0.05).
BDNF Positive Cells in the Hippocampus
Immunohistochemical analysis showed that the number of BDNF-positive cells in the mouse hippocampal CA1 was significantly decreased in the d-gal-treated mice when compared with the normal control animals [77.00 ± 19.88 vs. 229.00 ± 19.49, F (3, 20) = 131.592, P < 0.05] (Fig. 5). Vit E and HBO treatment significantly increased the number of BDNF-positive cells compared to the d-gal model group [d-gal + Vit E: 120.00 ± 7.91, P < 0.05; d-gal + HBO: 157.20 ± 10.43, P < 0.05]. Additionally, the number of BDNF-positive cells was 1.3 times higher in the HBO treatment group than that in the Vit E group (P < 0.05).

Effect of HBO on BDNF expression in the hippocampus CA1 region (scale bar 50 μm). A Normal control group; B d-gal model group; C d-gal-Vit group; D d-gal-HBO group. E Bar represents the mean ± SE (n = 6). a P < 0.05 compared to the normal control group. b P < 0.05 compared to the d-gal model group. c P < 0.05 compared to the d-gal + Vit E group
Effect of HBO on the Senescence-Related Protein Expression in the Hippocampus
The western blot analysis is shown in Fig. 6. Compared with the normal control group, the expression of Aβ1−42, p21 and p53 proteins was induced in the hippocampus in the d-gal model group [Aβ1−42: 0.89 ± 0.05 vs. 0.37 ± 0.08, F (3, 36) = 58.440, P < 0.05; p21: 1.38 ± 0.08 vs. 0.65 ± 0.07, F (3, 36) = 71.906, P < 0.05; p53: 0.66 ± 0.08 vs. 0.22 ± 0.01, F (3, 36) = 102.103, P < 0.05]. Compared with the d-gal model group, the expression of Aβ1−42, p21 and p53 proteins was inhibited in the d-gal plus Vit E and d-gal plus HBO group [d-gal + Vit E: Aβ1−42: 0.61 ± 0.08, P < 0.05; p21: 0.94 ± 0.07, P < 0.05; p53: 0.40 ± 0.04, P < 0.05; d-gal + HBO: Aβ1−42: 0.52 ± 0.06, P < 0.05; p21: 0.80 ± 0.13, P < 0.05; p53: 0.31 ± 0.03, P < 0.05]. Moreover, the expression of p53 was decreased by 23.08 % in the HBO treatment group than that in the Vit E group (P < 0.05).

Effect of HBO on senescence-related protein expression in the hippocampus CA1 region. The bands are from a representative blot. Lane 1 normal control group; lane 2 d-gal model group; lane 3 d-gal-Vit group; lane 4 d-gal-HBO group. The data values are expressed as the mean ± SE (n = 8). a P < 0.05 compared to the normal control group. b P < 0.05 compared to the d-gal model group. c P < 0.05 compared to the d-gal + Vit E group
Effect of HBO on the Senescence-Related Gene Expression in the Hippocampus
As shown in Fig. 7, compared with the normal control group, the expression of p16 and p21 mRNA in the hippocampus was up-regulated in the d-gal model group [p16: 1.49 ± 0.15 vs. 0.46 ± 0.10, F (3, 36) = 60.152, P < 0.05; p21: 1.05 ± 1.12 vs. 0.54 ± 0.03, F (3, 36) = 34.954, P < 0.05]. Compared with the d-gal model group, the expression of p16 and p21 mRNA was down-regulated in the d-gal plus Vit E treated and d-gal plus HBO treated groups [d-gal + Vit E: p16: 0.57 ± 0.09, P < 0.05; p21: 0.69 ± 0.02, P < 0.05; d-gal + HBO: p16: 1.05 ± 0.07, P < 0.05; p21: 0.83 ± 0.04, P < 0.05]. Compared with the d-gal plus Vit E group, the expression of p16 and p21 was more down-regulated in the d-gal plus HBO group (P < 0.05).

Effect of HBO on senescence-related gene expression in the hippocampus CA1 region. p16 and p21 mRNA expression were assayed using real-time PCR. The data values are expressed as the mean ± SE (n = 8). a P < 0.05 compared to the normal control group. b P < 0.05 compared to the d-gal model group. c P < 0.05 compared to the d-gal + Vit E group
Discussion
Memory decline, an aging characteristic, is thought to cause gradual loss of spatial memory. Chronic systemic d-gal injection induces memory deficits and decreases antioxidant enzyme activity, which results in neurodegeneration and poor immune responses [29]. In this study, the Morris water maze test was used to evaluate spatial memory and learning in d-gal mice, which is mainly dependent on hippocampal functions. Our results showed that administration of d-gal impaired the performance of mice in the Morris water maze test, which was in agreement with previous findings [14]. The administration of HBO or Vit E significantly reduced the escape latency and improved the deficits in platform crossings for the target quadrant. This result indicates that HBO and Vit E have the potential to ameliorate cognitive deficits induced by d-gal.
Oxidative stress and ROS have been proposed as being critical causes of aging [30]. The antioxidant activity has been reported to be beneficial with respect to slowing this aging process [31]. In the present study, d-gal treatment caused notable oxidative damage, including a decrease in SOD, GSH-Px, CAT, and T-AOC, as well as an increase in MDA level in the hippocampus of mice, which was similar to previous reports [17]. SOD converts superoxide anions into peroxides, which are then converted into water by CAT and GSH-Px. GSH-Px reduces toxic peroxide into a nontoxic hydroxyl compound and preserves the structure and function of the cell membrane [32]. CAT decomposes hydrogen peroxide (H2O2) into molecular oxygen and water [33]. T-AOC reflects the capacity of the non-enzymatic intracellular antioxidant defense system [34]. MDA is a major biomarker that appears during the final stages of lipid peroxidation initiated by excessive ROS and indicates indirect impairment at the cellular level [35]. Both of the HBO and Vit E treatment significantly reversed the changes of SOD, GSH-Px, CAT, T-AOC, and MDA levels, suggesting that the neuroprotective effect of HBO scavenged ROS mainly by enhancing antioxidant activity and consequently decreasing lipid peroxidative damage. Moreover, NO plays a role in maintaining ‘‘antioxidant homeostasis’’ and impaired NO bioactivity is a pathogenic factor in aging [36]. We observed that accompanying with the increase of SOD, GSH-Px, CAT, and T-AOC levels in the mice hippocampal CA1 region, the activities of NO and NOS were significantly decreased by HBO and Vit E treatment. These findings indicated that HBO mobilized its protective mechanism to inhibit the overproduction of NO in the mice hippocampus.
Following d-gal intraperitoneally injected for 8 weeks, a variety of proinflammatory cytokines such as IL-1β, IL-6, and TNF-a are released in the aged brain [37]. Besides, it has been reported that microglia activation stimulates the release of inflammatory mediators including IL-6 and TNF-α [38]. In the present study, we confirmed that d-gal administration significantly increased AGEs, TNF-α and IL-6 levels in the hippocampus, which may be involved in impairment of spatial learning and memory ability. However, HBO and Vit E administration suppressed AGEs formation, which in turn diminished production of TNF-α and IL-6. Our study also showed that HBO treatment retained hippocampal BDNF expression, which might be beneficial to neural survival and synaptic integrity for d-gal-treated mice. Even the d-gal + HBO group exhibited the higher expression of BDNF positive neurons than the d-gal + Vit E group.
Aging is associated with degeneration of hippocampal CA1 neurons. Deposition of Aβ in the brain is known to increase oxidative stress, leading to neurodegeneration and disrupted cognitive function [39, 40]. We found that the expression of Aβ1−42 proteins in the hippocampal CA1 region of d-gal model mice was significantly increased compared to normal controls. It is interesting to note that HBO treatment significantly decreased the expression of Aβ1−42 proteins. Moreover, it was reported that pyramidal cells of the hippocampus are sensitive to d-gal-induced neuronal injury [41]. Our histopathological results also showed that extensively damaged neurons and the loss of pyramidal cells in the hippocampal CA1 region were observed in the d-gal model group, and HBO significantly attenuated pathological injury. However, Vit E treatment group did not showed significantly more surviving neurons when compared with the model group. These results support the concept that HBO has a neuroprotective effect on d-gal-induced mimetic aging.
Ectopic overexpression of p21 or p53 was previously observed in tumor cell lines [42]. Recently, it has been demonstrated that the activation of p53 can activate downstream p21 to maintain cell cycle arrest for DNA repair [43, 44]. Moreover, p16 deficiency partially prevented an age-induced decline in cell proliferation and tissue function [45]. In this study, we found that p16, p21 and p53 expression was significantly higher in the aging model than normal controls. Moreover, HBO and Vit E significantly decreased p16, p21 and p53 expression. Importantly, we found the expression of p53 protein as well as p16 and p21 gene were lower in rats treated with HBO than Vit E.
These results indicate that HBO may regulate the expression of genes that delay senescence.
Conclusion
In conclusion, our present study provides evidence that HBO treatment can prevent cognitive impairment and hippocampal senescence in a mouse model of d-gal-induced aging, suggesting that HBO is involved in anti-oxidation, anti-inflammation, and modulation of aging-related gene expression. To some extent, HBO administration exhibited better effect in cognitive improvement and senescence-related protein and gene expression.
References
Wild K, Howieson D, Webbe F, Seelye A, Kaye J (2008) Status of computerized cognitive testing in aging: a systematic review. Alzheimers Dement 4:428–437
Keller JN (2006) Age-related neuropathology, cognitive decline, and Alzheimer’s disease. Ageing Res Rev 5:1–13
Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H (2007) Trends in oxidative aging theories. Free Radic Biol Med 43:477–503
Pak JW, Herbst A, Bua E, Gokey N, McKenzie D, Aiken JM (2003) Mitochondrial DNA mutations as a fundamental mechanism in physiological declines associated with aging. Aging Cell 2:1–7
Ralay Ranaivo H, Hodge JN, Choi N, Wainwright MS (2012) Albumin induces upregulation of matrix metalloproteinase-9 in astrocytes via MAPK and reactive oxygen species-dependent pathways. J Neuroinflammation 9:1742–2094
Joseph J, Cole G, Head E, Ingram D (2009) Nutrition, brain aging, and neurodegeneration. J Neurosci 29:12795–12801
Takeda M, Takahashi M, Matsumoto S (2014) Inflammation enhanced brain-derived neurotrophic factor-induced suppression of the voltage-gated potassium currents in small-diameter trigeminal ganglion neurons projecting to the trigeminal nucleus interpolaris/caudalis transition zone. Neuroscience 261:223–231
Sun M-K, Nelson TJ, Alkon DL (2015) Towards universal therapeutics for memory disorders. Trends Pharmacol Sci 36:384–394
Lu J, Wu DM, Zheng YL, Hu B, Zhang ZF, Ye Q, Liu CM, Shan Q, Wang YJ (2010) Ursolic acid attenuates D-galactose-induced inflammatory response in mouse prefrontal cortex through inhibiting AGEs/RAGE/NF-kappaB pathway activation. Cereb Cortex 20:2540–2548
Srikanth V, Maczurek A, Phan T, Steele M, Westcott B, Juskiw D, Munch G (2011) Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol Aging 32:763–777
Campo-Trapero J, Cano-Sanchez J, Palacios-Sanchez B, Llamas-Martinez S, Lo Muzio L, Bascones-Martinez A (2008) Cellular senescence in oral cancer and precancer and treatment implications: a review. Acta Oncol 47:1464–1474
Sharpless NE (2004) Ink4a/Arf links senescence and aging. Exp Gerontol 39:1751–1759
Budni J, Pacheco R, da Silva S, Garcez ML, Mina F, Bellettini-Santos T, de Medeiros J, Voss BC, Steckert AV, Valvassori SS, Quevedo J (2015) Oral administration of d-galactose induces cognitive impairments and oxidative damage in rats. Behav Brain Res 31:30350–30358
Liu L, Lu Y, Kong H, Li L, Marshall C, Xiao M, Ding J, Gao J, Hu G (2012) Aquaporin-4 deficiency exacerbates brain oxidative damage and memory deficits induced by long-term ovarian hormone deprivation and D-galactose injection. Int J Neuropsychopharmacol 15:55–68
Wei H, Li L, Song Q, Ai H, Chu J, Li W (2005) Behavioural study of the D-galactose induced aging model in C57BL/6 J mice. Behav Brain Res 157:245–251
Song X, Bao M, Li D, Li YM (1999) Advanced glycation in D-galactose induced mouse aging model. Mech Ageing Dev 108:239–251
Doan VM, Chen C, Lin X, Nguyen VP, Nong Z, Li W, Chen Q, Ming J, Xie Q, Huang R (2015) Yulangsan polysaccharide improves redox homeostasis and immune impairment in D-galactose-induced mimetic aging. Food Funct 6:1712–1718
Dave KR, Prado R, Busto R, Raval AP, Bradley WG, Torbati D, Perez-Pinzon MA (2003) Hyperbaric oxygen therapy protects against mitochondrial dysfunction and delays onset of motor neuron disease in Wobbler mice. Neuroscience 120:113–120
Chen C, Chen W, Nong Z, Ma Y, Qiu S, Wu G (2016) Cardioprotective effects of combined therapy with hyperbaric oxygen and diltiazem pretreatment on myocardial ischemia–reperfusion injury in rats. Cell Physiol Biochem 38:2015–2029
Iwamoto K, Ikeda K, Mizumura S, Tachiki K, Yanagihashi M, Iwasaki Y (2014) Combined treatment of methylprednisolone pulse and memantine hydrochloride prompts recovery from neurological dysfunction and cerebral hypoperfusion in carbon monoxide poisoning: a case report. J Stroke Cerebrovasc Dis 23:592–595
Stetler RA, Leak RK, Gan Y, Li P, Zhang F, Hu X, Jing Z, Chen J, Zigmond MJ, Gao Y (2014) Preconditioning provides neuroprotection in models of CNS disease: paradigms and clinical significance. Prog Neurobiol 114:58–83
Pan X, Chen C, Huang J, Wei H, Fan Q (2015) Neuroprotective effect of combined therapy with hyperbaric oxygen and madopar on 6-hydroxydopamine-induced Parkinson’s disease in rats. Neurosci Lett 600:220–225
Godman CA, Joshi R, Giardina C, Perdrizet G, Hightower LE (2010) Hyperbaric oxygen treatment induces antioxidant gene expression. Ann N Y Acad Sci 1197:178–183
Zhai X, Lin H, Chen Y, Chen X, Shi J, Chen O, Li J, Sun X (2016) Hyperbaric oxygen preconditioning ameliorates hypoxia–ischemia brain damage by activating Nrf2 expression in vivo and in vitro. Free Radic Res 4:1–34
Zhu J, Mu X, Zeng J, Xu C, Liu J, Zhang M, Li C, Chen J, Li T, Wang Y (2014) Ginsenoside Rg1 prevents cognitive impairment and hippocampus senescence in a rat model of D-galactose-induced aging. PLoS One 9:e101291
George P, Charles W (1998) The rat brain in stereotaxic coordinates. Qingchuan Zhuge translate. People’s Medical Publishing House, Beijing, 2007 32
Wu W, Wang X, Xiang Q, Meng X, Peng Y, Du N, Liu Z, Sun Q, Wang C, Liu X (2014) Astaxanthin alleviates brain aging in rats by attenuating oxidative stress and increasing BDNF levels. Food Funct 5:158–166
Zhou Y, Dong Y, Xu Q, He Y, Tian S, Zhu S, Zhu Y, Dong X (2013) Mussel oligopeptides ameliorate cognition deficit and attenuate brain senescence in D-galactose-induced aging mice. Food Chem Toxicol 59:412–420
Lu J, Zheng YL, Wu DM, Luo L, Sun DX, Shan Q (2007) Ursolic acid ameliorates cognition deficits and attenuates oxidative damage in the brain of senescent mice induced by D-galactose. Biochem Pharmacol 74:1078–1090
Silva JP, Coutinho OP (2010) Free radicals in the regulation of damage and cell death—basic mechanisms and prevention. Drug Discov Ther 4:144–167
Hsia CH, Wang CH, Kuo YW, Ho YJ, Chen HL (2012) Fructo-oligosaccharide systemically diminished D-galactose-induced oxidative molecule damages in BALB/cJ mice. Br J Nutr 107:1787–1792
Prisila Dulcy C, Singh HK, Preethi J, Rajan KE (2012) Standardized extract of Bacopa monniera (BESEB CDRI-08) attenuates contextual associative learning deficits in the aging rat’s brain induced by D-galactose. J Neurosci Res 90:2053–2064
Wang BS, Juang LJ, Yang JJ, Chen LY, Tai HM, Huang MH (2012) Antioxidant and antityrosinase activity of flemingia macrophylla and glycine tomentella roots. Evid Based Complement Alternat Med 431081:10
Parameshwaran K, Irwin MH, Steliou K, Pinkert CA (2010) D-galactose effectiveness in modeling aging and therapeutic antioxidant treatment in mice. Rejuvenation Res 13:729–735
Zhong SZ, Ge QH, Qu R, Li Q, Ma SP (2009) Paeonol attenuates neurotoxicity and ameliorates cognitive impairment induced by d-galactose in ICR mice. J Neurol Sci 277:58–64
Shi Y, Camici GG, Luscher TF (2010) Cardiovascular determinants of life span. Pflugers Arch 459:315–324
Tsai SJ, Yin MC (2012) Anti-glycative and anti-inflammatory effects of protocatechuic acid in brain of mice treated by D-galactose. Food Chem Toxicol 50:3198–3205
Tatar CL, Appikatla S, Bessert DA, Paintlia AS, Singh I, Skoff RP (2010) Increased Plp1 gene expression leads to massive microglial cell activation and inflammation throughout the brain. ASN Neuro 2:00043
Hardy J, Selkoe DJ (2002) Medicine—The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356
Arendash GW, Schleif W, Rezai-Zadeh K, Jackson EK, Zacharia LC, Cracchiolo JR, Shippy D, Tan J (2006) Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience 142:941–952
Cui X, Zuo P, Zhang Q, Li X, Hu Y, Long J, Packer L, Liu J (2006) Chronic systemic D-galactose exposure induces memory loss, neuro degeneration, and oxidative damage in mice: protective effects of R-alpha-lipoic acid. J Neurosci Res 84:647–654
Towers CG, Guarnieri AL, Micalizzi DS, Harrell JC, Gillen AE, Kim J, Wang CA, Oliphant MU, Drasin DJ, Guney MA, Kabos P, Sartorius CA, Tan AC, Perou CM, Espinosa JM, Ford HL (2015) The Six1 oncoprotein downregulates p53 via concomitant regulation of RPL26 and microRNA-27a-3p. Nat Commun 6:10077
Bieging-Rolett KT, Johnson TM, Brady CA, Beaudry VG, Pathak N, Han S, Attardi LD (2016) p19 is required for the cellular response to chronic DNA damage. Oncogene 4:490
Gire V, Dulic V (2015) Senescence from G2 arrest, revisited. Cell Cycle 14:297–304
Ding Y, Chen J, Okon IS, Zou MH, Song P (2015) Absence of AMPKalpha2 accelerates cellular senescence via p16 induction in mouse embryonic fibroblasts. Int J Biochem Cell Biol 71:72–80
Acknowledgments
This work was supported by the Guangxi Sanitation and Family Planning Committee Project (No. Z2016582). The authors wish to thank Dr. Jun Chen for his technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that there are no conflicts of interest.
Additional information
Xiaoyu Chen and Yaoxuan Li have contribute equally to this work.
Rights and permissions
About this article

Cite this article
Chen, X., Li, Y., Chen, W. et al. Protective Effect of Hyperbaric Oxygen on Cognitive Impairment Induced by d-Galactose in Mice. Neurochem Res 41, 3032–3041 (2016). https://doi.org/10.1007/s11064-016-2022-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-016-2022-x