
Abstract
Free radicals induced neural damage is implicated in CNS diseases and rutin isolated form Lonicera japonica are reported to have neuroprotective activity. Previously, we confirmed that rutin exerted neuroprotective effect against sodium nitroprusside (SNP)-induced cell death in PC12 cells. However, the neuroprotective mechanism of rutin is still not fully uncovered. Here, we found that rutin significantly decreased SNP-induced reactive oxygen species in PC12 cells. Rutin reversed the declined GSH/GSSG ratio and mitochondrial membrane potential induced by SNP. Moreover, rutin activated both the protein Akt/mTOR and the extracellular signal-regulated kinase (ERK1/2) signaling pathways and the neuroprotective effects of rutin were blocked by either the specific PI3K inhibitor LY294002 or the MAPK pathway inhibitor PD98059. In summary, these results demonstrated that the neuroprotective effects of rutin might be through activating both the PI3K/Akt/mTOR and ERK1/2 signaling pathways. Our findings support that rutin may have therapeutic potential for the treatment of CNS diseases related to NO neurotoxicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Oxidative stress and mitochondrial impairments are critical patho-logical factors in multiple major neurological diseases, including stroke, Parkinson’s disease (PD), and Alzheimer’s disease (AD) [1–3]. Oxidative damage occurs when the antioxidant defense systems are exceeded by free radical production [4]. It is common sense that excess reactive oxygen/nitrogen species (ROS/RNS) cause damage of cell organelle, oxidation of proteins/lipids, and the activation of the downstream signaling pathways leading to the apoptosis of target cells [5, 6]. Thus, an antioxidant compound would be of greater value if it could also decrease the levels of nitrosative stress [7].
Sodium Nitroprusside (SNP) is a potent nitric oxide (NO) donor. NO is known as a Janus-faced molecule that is physiologically produced through the l-arginine/NO synthase (NOS) pathway. It plays various physiological roles in the central nervous system, including neuromodulation, neurotransmission and synaptic plasticity [8–10]. However, the overproduction of NO is also implicated in the pathogenesis of neurodegenerative disorders and cerebral ischemia–reperfusion injury [11–16]. In central nervous systems, excessive NO reacts with superoxide anions (O2·−) resulting in the formation of peroxynitrite (ONOO−), which induces lipid peroxidation (LPO) linked to the disruption of cell membranes leading to the release of cell organelles [17–20]. ONOO− formation also induces cell damage by its oxidative and apoptotic stimuli and has been implicated in several pathological conditions such as AD and PD [21, 22].
Rutin (quercetin-3-rhamnosyl glucoside) is a common dietary flavonoid glycoside found in buckwheat which is abundantly present in vegetables, passionflower, oranges, and grapes [23]. Cytoprotective effects of rutin after ischemic injury of organs, including the heart, brain, and kidney have been reported [24]. Moreover, rutin inhibits myocardial ischemia/reperfusion-induced apoptosis in vivo and protects H9c2 cells against hydrogen peroxide-mediated injury via ERK1/2 and PI3K/Akt signals in vitro [25]. In particular, rutin inhibited 6-hydroxydopamine (6-OHDA) and A beta-amyloid aggregation -induced neurotoxicity in neuronal cells by improving antioxidant enzyme levels and inhibiting lipid peroxidation [26, 27]. However, there is no report on the effects of rutin against SNP-induced cell death. Previously, we have observed the protective effect of rutin on neuronal cells against SNP-induced neurotoxicity [28]. Given the PI3K/Akt/mTOR and mitogen activated protein kinases (MAPKs) pathway play important roles in cell survival [29–31]. Therefore, the present study was designed to verify the potential neuroprotective effects of rutin against SNP-induced apoptosis and investigated the roles of PI3K/Akt/mTOR and MAPKs pathways in rutin-mediated neuroprotection in PC12 cells.
Materials and Methods
Materials
Rutin was purchased from China food and Drug Research Institute; 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), Poly-d-lysine, 2′,7′-dichlorodihydrofluorescin diacetate (DCFH-DA) and DMSO were from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and horse serum were purchased from Gibco-BRL (NY, USA). BCA protein assay kit was from Beyotime Institute of Biotechnology (Haimen, China); JC-1 dye (Molecular Probes) was from BestBio (Shanghai, China). Total glutathione (GSH)/oxidized glutathione (GSSG) assay kit was from Jiancheng Biochemical Company (Nanjing, China). Rutin stock solution (100 mg/ml) was dissolved in DMSO and stored at −20 °C. Anti-phospho-Akt (Ser473) antibody, anti-Akt Antibody, anti- phospho-mTOR (Ser2448), anti-mTOR antibody, anti-phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) antibody, anti-ERK1/2 antibody from Cell Signaling Technology (Woburn, USA); LY294002, rapamycin, PD98059, PD169316 and JNK inhibitor were obtained from Calbiochem (La Jolla, CA, USA).
Cell Culture and Treatment
Differentiated PC12 cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) containing 5 % fetal bovine serum (FBS) and 10 % horse serum, 100 µg of streptomycin/ml, and 100 U of penicillin/ml and incubated at 37 °C with 5 % CO2 humidified atmosphere. Cultured media was replaced twice a week with fresh medium as described above. Stock culture was routinely sub-cultured at 1:5 ratio at a weekly interval. For the experiments, cells were pre-incubated with various concentrations of rutin for 2 h without other description, and followed by treatment of SNP for 24 h. Control group was treated with 0.1 % (v/v) DMSO as vehicle control.
MTT Assay
Cell viability was estimated using a 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay as previously described. Briefly, after 24 h treatments, the culture medium was removed and replaced with 90 µl of fresh DMEM. 10 μl of 5 mg/ml MTT in phosphate-buffered saline (PBS) was added to each well and the plates were incubated at 37 °C for another 3 h, then supernatants were discarded. 100 µl of DMSO solutions were added to each wells and the solutions were mixed thoroughly. Then the plates were incubated at 37 °C for another 10 min. Each sample was mixed again and the resultant formazan was measured by its absorbance at 570 nm using a BIO-RAD680 plate reader (Thermo, USA). The experiments were repeated at least 3 times and compared with the control experiment.
Morphologic Changes
PC12 cells grown on 48-well plates were treated with rutin and/or SNP as described above. After that, cells were fixed with 4 % paraformaldehydeand stained with Hoechst 33258 (5 μg/ml) for 10 min at 37 °C in the dark. Then Hoechst 33258 was removed by washing with PBS, and morphologic changes were observed by phase-contrast microscopy and cells images were taken using a fluorescence microscope (IX71, Olympus, Tokyo, Japan).
Measurement of ROS
Intracellular ROS formation was measured by fluorescence using DCFH-DA. Briefly, after treatment, cells were washed and then stained with 10 µM DCFH-DA in serum-free medium for 30 min at 37 °C in the dark. the fluorescence from the DCF was analyzed using a fluorescence plate reader (Flex Station3, Molecular Devices, USA) at excitation and emission wavelengths of 488 and 525 nm, respectively, or taken images using a fluorescence microscope (IX71, Olympus, Tokyo, Japan).
Measurement of GSH and GSSG
Total-GSH was assayed using the 5,5-dithio-bis (2-nitrobenzoic) acid (DTNB)-GSSG reductase recycling. GSSG was measured by measuring 5-thio-2-nitrobenzoic acid (TNB) which was produced from the reaction of reduced GSH with DTNB according to the kit’s manufacturer’s protocols. The concentration of reduced GSH in the sample was obtained by subtracting GSSG from T-GSH. The GSH and GSSG in the cells was assayed by adding the samples to the detection solution in a 96-well plate, respectively. After incubation at room temperature for 25 min, GSH units were determined using a Multiskan Ascent Microplate Reader (Thermo, USA) at 412 nm.
Mitochondrial Membrane Potential Determination
Mitochondrial membrane potential was analyzed by using a fluorescent dye JC-1 (BestBio Shanghai China). JC-1 penetrates cells and healthy mitochondria. At low membrane potentials (apoptotic cells), JC-1 exists as a monomer which emits green fluorescence. JC-1 aggregates and emits red fluorescence at higher membrane potentials (non-apoptotic cells). Assays were initiated by incubating PC12 cells with JC-1 (5 mg/L) for 20 min at 37 °C in the dark and the fluorescence of separated cells were captured by inverted fluorescence microscopy (Olympus, Japan, at wavelengths of 490 nm excitation and 530 nm emission for green, and at 540 nm excitation and 590 nm emission for red). The ratios of red/green fluorescence were calculated.
Western Blotting Analysis
Western blotting analysis was performed as previously described [32]. Briefly, Cells from different experimental conditions were lysed with ice-cold RIPA lysis buffer and protein concentration was determined with a BCA protein assay kit according to the manufacturer’s instructions. Equal amounts of lysate protein (20 μg/lane) were subjected to SDS-PAGE with 10 % polyacrylamide gels and electropho-retically transferred to nitrocellulose membranes. Nitrocellulose blots were first blocked with 3 % bovine serum albumin (BSA) in PBST buffer (PBS with 0.01 % Tween 20, pH 7.4), and incubated overnight at 4 °C with primary antibodies in PBST containing 1 % BSA. Immunoreactivity was detected by sequential incubation with horseradish peroxidase-conjugated secondary antibodies, and detected by the enhanced chemiluminescence technique.
Statistical Analysis
All results are reported as means ± SEM for 3–6 experiments. Differences between groups were analyzed using ANOVA, followed by Dunnett’s multi-comparison test with PASW Software (SPSS Inc., Chicago, IL, USA). P values < 0.05 were considered statistically significant.
Results
Protective Effect of Rutin in the PC12 Cells Against SNP Exposure
PC12 cells were treated with various concentrations of rutin as indicated. After 24-h SNP insult, the viability of cells was determined by MTT assay. As shown in Fig. 1b, rutin dose-dependently decreased SNP cytotoxicity, rutin protected PC12 cells against SNP-induced cell death in a dose dependent manner. To determine whether rutin blocked SNP-induced apoptosis, PC12 cells incubated with JC-1 (5 mg/L) was used to evaluate the mitochondrial membrane potential. The shift of fluorescence from red to green indicated by JC-1 reflected the decline of the membrane potential and early apoptosis. As shown in Fig. 2a, treatment with SNP did induce cell apoptosis in PC12 cells, which was indicated by the presence of green cells. Rutin (25 μg/ml) significantly prevented the declined of mitochondrial membrane potential induced by SNP and decreased the number of cells with green cells (Fig. 2b).

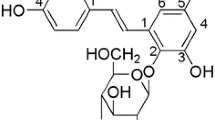
Effects of rutin on SNP-induced cell viability a Structure of rutin; b PC12 cells were treated with rutin (6.25–100 μg/ml) for 2 h and then incubated with 800 μM SNP for a further 24 h. Cells viability were determined by the MTT assay, ## P < 0.01 versus control group, n = 6; **P < 0.01 versus SNP-treated group, (n = 6)

Rutin inhibits SNP-induced morphologic changes and the reduction of mitochondrial membrane potential. PC12 cells were pretreated with or without 25 μg/ml of rutin for 2 h and then treated with or without 800 μM SNP for 24 h. a Rutin significantly attenuated SNP-induced morphologic changes. Representative images were taken by a fluorescence microscope. The images shown are representative of three experiments. b Cells were treated as indicated and the mitochondrial membrane potential was determined as described in Materials and Methods. SNP insult caused the decline of mitochondrial membrane potential of PC12 cells, rutin reversed the effects of SNP insult. ## P < 0.01 versus control group; *P < 0.05, **P < 0.01 versus SNP-treated group, (n = 6)
Effects of Rutin on SNP-Induced ROS Production and Glutathione Level
Previous studies showed that the toxicity of SNP was mediated through the production of ROS [33], rutin was capable of preventing H9c2 cell injury caused by hydrogen peroxide [25]. Therefore, we investigated whether rutin blocked SNP-induced oxidative stress in PC12 cells. Cellular oxidative stress was determined by DCFH-DA staining, a ROS probe [32]. PC12 cells were pretreated with or without 5 μM of rutin for 2 h and then treated with or without 200 μM SNP for 12 h. The microscopy images showed that SNP treatment induced the intracellular production of ROS, which was attenuated by rutin (Fig. 3a, b). To further confirm whether the protection of rutin against oxidative damage is associated with the reversion of GSH depletion induced by SNP, we explored the changes of GSH, GSSG and GSH/GSSG ratio in PC12 cells after 12 h of incubation with rutin in the presence and absence of SNP,Results showed that the GSH levels in PC12 cells were significantly decreased while GSSG levelswere concomitantly increased with the addition of 800 μM SNP. Rutin at 25 μg/ml increased GSH (Fig. 3c) and decreased GSSG levels (Fig. 3d). Consistent with these results, the GSH/GSSG ratios (Fig. 3e) were also decreased by SNP and increased with the addition of rutin.

Effects of rutin on the intracellular ROS, GSH and GSSG levels induced by SNP insult in PC12 cells. a PC12 cells were pretreated with or without 25 μg/ml of rutin for 2 h and then treated with or without 800 μM SNP for 12 h, the fluorescence intensity of DCFH-DA showed that rutin blocked the ROS accumulation induced by SNP. b Histogram showing the ROS level in PC12 cells after expose to SNP in presence or absence of rutin compared to control groups. c PC12 cells were treated with rutin at 25 μg/ml for 2 h, and further exposed to SNP (800 μM) for 12 h. The levels of GSH and GSSG (d) measured using commercial assay kits and the GSH/GSSG ratios (e) were calculated. # P < 0.05, ## P < 0.01 compared with the control group; *P < 0.05,**P < 0.01, compared with the SNP-treated group (n = 6)
Rutin Time- and Dose-Dependently Increased the Phosphorylation Levels of Akt, mTOR and ERK1/2 in PC12 Cells
Given PI3K/Akt/mTOR and MAPKs family members play an important role in oxidative stress-induced neurotoxicity [29, 34, 35], we hypothesized that the activation of the Akt/mTOR and MAPK signaling pathway might be involved in the neuroprotective effects of rutin against SNP-induced cells death. As shown in Fig. 4, rutin increased the phosphorylation of Akt, mTOR and ERK1/2 in PC12 cells in a time- and dose-dependent manner. Rutin increased phosphorylation level of these two kinases were found significant within 20 min and peaked at 40–80 min.

Rutin time- and dose-dependently increased the phosphorylation level of Akt, mTOR and ERK1/2 in PC12 cells. PC12 cells were treated with rutin at a 25 μg/ml for scheduled time (10–80 min); and c scheduled concentrations (6.25–100 μg/ml) for 40 min. The phosphorylation of Akt, mTOR and ERK1/2 in PC12 cells was analyzed by Western blotting. b and d densitometric analysis of the immunoblot was expressed as a percentage of control, *P < 0.05, **P < 0.01 versus control groups, results represent prototypical examples of experiments replicated at least 3 times
Neuroprotective Action of Rutin is Mediated by the Activation of the PI3K/Akt/mTOR and ERK1/2 Signaling pathway in PC12 Cells
Having known that rutin can protect PC12 cells from SNP-induced cell death, we then detect the underlying protective mechanism about rutin, we examined the role of PI3K/Akt/mTOR and MAPK signaling pathways in the protective effect of rutin using LY294002 (PI3K inhibitor), rapamycin (mTOR inhibitor), PD98059 (an ERK1/2 specific inhibitor), PD169316 (a p38 specific inhibitor) and JNK1/2 inhibitor. LY294002, rapamycin, and PD98059 blocked the protective effects of rutin in PC12 cells, while PD169316 and JNK1/2 inhibitor had no effects (Fig. 5a). To further confirm the role of the Akt/mTOR and ERK1/2 pathways in the protective effect of rutin, we found that SNP insult decreased the phosphorylation of both mTOR and ERK1/2 in PC12 cells, while rutin restored the basal levels of phosphorylation of Akt and ERK1/2 which was inhibited by LY294002 and PD98059 respectively (Fig. 5b, c).

Effects of rutin on the PI3K/Akt/mTOR and MAPKs pathway in PC12 cells. a PC12 cells were treated with various inhibitors for 30 min, then treated with rutin for 24 h. MTT assay was used to determine cell viability. # P < 0.05 versus control group; *P < 0.05, **P < 0.01 versus serum-free SNP group, & P < 0.05, && P < 0.01 versus SNP + rutin-treated group. b Protective effects of rutin on SNP-induced phosphorylated levels of Akt, mTOR and ERK1/2 MAPKs in PC12 cells. PC12 cells were pretreated with different inhibitors for 30 min and then exposed to rutin and SNP for 12 h. Levels of phosphorylated Akt, mTOR and ERK1/2 proteins were evaluated by Western blot analysis. c Densitometric analysis of the immunoblot was expressed as a percentage of control. **P < 0.01 versus control groups, n = 3; ## P < 0.01 versus SNP-treated group; && P < 0.01 versus SNP + rutin group n = 3. Results represent prototypical examples of experiments replicated at least 3 times
Discussion
To the best of our knowledge, this is the first report to show that rutin significantly attenuated SNP-induced neurotoxicity by decreasing the production of ROS and reversing GSH depletion in PC12 cells. Meanwhile, rutin significantly promoted survival of PC12 cells via activating both Akt/mTOR and ERK1/2 pathways.
In neurological diseases (e.g., stroke, Alzheimer’s disease, or PD), antioxidant therapy should be an attractive strategy against neuronal loss [36, 37]. Mitochondrial membrane is one of the important target affected by pathological levels of ROS, which is an intracellular process contributing to apoptosis [38]. The mechanisms of protective effects of rutin against ischemic tissue injury have been focused on the enzymes that reduce oxygen free radicals, such as superoxide dismutase (SOD) [39]. It has been reported that rutin protects the testes from I–R injury by scavenging ROS through increasing SOD and catalase activities [40]. However, no previous studies have been conducted regarding the neuroprotective mechanism of rutin on SNP injury. In the current investigation, our results showed that SNP increased the production of ROS in PC12 cells, which is consistent with a previous report showing that the toxicity of SNP was mediated through the production of ROS [41]. Rutin attenuated accumulation of intracellular ROS in PC12 cells supported that rutin has antioxidant properties [42]. In addition, we observed that SNP insult was followed by loss of the mitochondrial membrane potential, which was significantly reversed rutin. Moreover, rutin increased ratio of GSH/GSSG in SNP insulted PC12 cells supported further the conclusion that rutin can protect against PC12 cell apoptosis induced by SNP. No doubt, rutin is capable of protecting against this insult and can be used as an antioxidant.
Substantial evidences have shown that both survival (Akt/mTOR) and death (JNK) pathways are the perpetrators of the loss of mitochondrial potential to an apoptosis-mediated cell death in different in vivo and in vitro models of pathophysiology in several neurologic disorders [43, 44]. Rutin significantly increased ERK1/2, cAMP response element-binding protein (CREB) and brain-derived neurotrophic factor (BDNF) gene expression on beta-amyloid induced neurotoxicity in rats [45]. In the current research, rutin time and concentration-dependently stimulated the phosphrylation of Akt/mTOR and ERK1/2, moreover, rutin significantly reversed the SNP-induced inhibition against phosphrylation of Akt/mTOR and ERK1/2. Interestingly, LY294002, rapmycin and PD98059 abrogated the rutin-induced phosphrylation of Akt, mTOR and ERK1/2 respectively while blocked the neuroprotective effect of rutin on the survival of PC12 cells significantly but not completely. All these data support the proposition that rutin promotes the survival of PC12 cells from SNP-induced injury via the PI3K/Akt/mTOR and ERK1/2. Results showed that rutin modulated of Akt/mammalian target of rapamycin (mTOR) and ERK1/2 pathways.
Taken together, our results indicate that rutin exerts strikingly protective effects against SNP-induced cytotoxicity in PC12 cells through activating the PI3K/Akt/mTOR and ERK1/2 pathways, which might be helpful for extending the usage of rutin for NO-associated diseases, such as PD.
References
Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott M, Meigs J, Mozaffarian D, Nichol G, O’Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y, American Heart Association Statistics C, Stroke Statistics S (2009) Heart disease and stroke statistics–2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 119:e21–e181
Yousuf S, Atif F, Ahmad M, Hoda N, Ishrat T, Khan B, Islam F (2009) Resveratrol exerts its neuroprotective effect by modulating mitochondrial dysfunctions and associated cell death during cerebral ischemia. Brain Res 1250:242–253
Ying W (1997) Deleterious network: a testable pathogenetic concept of Alzheimer’s disease. Gerontology 43:242–253
Butterfield DA, Castegna A, Lauderback CM, Drake J (2002) Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging 23:655–664
Poon HF, Calabrese V, Scapagnini G, Butterfield DA (2004) Free radicals and brain aging. Clin Geriatr Med 20:329–359
Culmsee C, Gerling N, Landshamer S, Rickerts B, Duchstein HJ, Umezawa K, Klumpp S, Krieglstein J (2005) Nitric oxide donors induce neurotrophin-like survival signaling and protect neurons against apoptosis. Mol Pharmacol 68:1006–1017
Silva JP, Proenca F, Coutinho OP (2008) Protective role of new nitrogen compounds on ROS/RNS-mediated damage to PC12 cells. Free Radical Res 42:57–69
Guix FX, Uribesalgo I, Coma M, Munoz FJ (2005) The physiology and pathophysiology of nitric oxide in the brain. Prog Neurobiol 76:126–152
Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML (2000) Caspase-2 mediates neuronal cell death induced by beta-amyloid. J Neurosci 20:1386–1392
Contestabile A, Ciani E (2004) Role of nitric oxide in the regulation of neuronal proliferation, survival and differentiation. Neurochem Int 45:903–914
Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM (2007) Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 8:766–775
Chan CS, Gertler TS, Surmeier DJ (2009) Calcium homeostasis, selective vulnerability and Parkinson’s disease. Trends Neurosci 32:249–256
Emerit J, Edeas M, Bricaire F (2004) Neurodegenerative diseases and oxidative stress. Biomed Pharmacother 58:39–46
Dawson VL, Dawson TM (1996) Nitric oxide neurotoxicity. J Chem Neuroanat 10:179–190
Kuppusamy P, Ohnishi ST, Numagami Y, Ohnishi T, Zweier JL (1995) Three-dimensional imaging of nitric oxide production in the rat brain subjected to ischemia-hypoxia. J Cereb Blood Flow Metab 15:899–903
Bondy SC, Naderi S (1994) The formation of reactive oxygen species in a fraction of rat brain by metabolism of nitric oxide. Neurosci Lett 168:34–36
Zhang F, Casey RM, Ross ME, Iadecola C (1996) Aminoguanidine ameliorates and l-arginine worsens brain damage from intraluminal middle cerebral artery occlusion. Stroke 27:317–323
Neufeld AH (1999) Nitric oxide: a potential mediator of retinal ganglion cell damage in glaucoma. Surv Ophthalmol 43(Suppl 1):S129–S135
Ferreira SM, Lerner SF, Brunzini R, Evelson PA, Llesuy SF (2004) Oxidative stress markers in aqueous humor of glaucoma patients. Am J Ophthalmol 137:62–69
Kumar DM, Agarwal N (2007) Oxidative stress in glaucoma: a burden of evidence. J Glaucoma 16:334–343
Aoyama K, Matsubara K, Fujikawa Y, Nagahiro Y, Shimizu K, Umegae N, Hayase N, Shiono H, Kobayashi S (2000) Nitration of manganese superoxide dismutase in cerebrospinal fluids is a marker for peroxynitrite-mediated oxidative stress in neurodegenerative diseases. Ann Neurol 47:524–527
Chung KK, Dawson TM, Dawson VL (2005) Nitric oxide, S-nitrosylation and neurodegeneration. Cell Mol Biol 51:247–254
Manach C, Morand C, Demigne C, Texier O, Regerat F, Remesy C (1997) Bioavailability of rutin and quercetin in rats. FEBS Lett 409:12–16
Araujo JR, Goncalves P, Martel F (2011) Chemopreventive effect of dietary polyphenols in colorectal cancer cell lines. Nutr Res 31:77–87
Jeong JJ, Ha YM, Jin YC, Lee EJ, Kim JS, Kim HJ, Seo HG, Lee JH, Kang SS, Kim YS, Chang KC (2009) Rutin from Lonicera japonica inhibits myocardial ischemia/reperfusion-induced apoptosis in vivo and protects H9c2 cells against hydrogen peroxide-mediated injury via ERK1/2 and PI3 K/Akt signals in vitro. Food Chem Toxicol 47:1569–1576
Magalingam KB, Radhakrishnan A, Haleagrahara N (2013) Rutin, a bioflavonoid antioxidant protects rat pheochromocytoma (PC-12) cells against 6-hydroxydopamine (6-OHDA)-induced neurotoxicity. Int J Mol Med 32:235–240
Wang SW, Wang YJ, Su YJ, Zhou WW, Yang SG, Zhang R, Zhao M, Li YN, Zhang ZP, Zhan DW, Liu RT (2012) Rutin inhibits beta-amyloid aggregation and cytotoxicity, attenuates oxidative stress, and decreases the production of nitric oxide and proinflammatory cytokines. Neurotoxicology 33:482–490
Zhang L, Lai YC, Wang HT, Wang RK, Meng Q, Zheng WH (2014) [Protective effect of rutin against oxidative injury in neuronal cells]. J Chin Med Mater 37:640–644
Kimura R, Okouchi M, Fujioka H, Ichiyanagi A, Ryuge F, Mizuno T, Imaeda K, Okayama N, Kamiya Y, Asai K, Joh T (2009) Glucagon-like peptide-1 (GLP-1) protects against methylglyoxal-induced PC12 cell apoptosis through the PI3 K/Akt/mTOR/GCLc/redox signaling pathway. Neuroscience 162:1212–1219
Zhao ZY, Luan P, Huang SX, Xiao SH, Zhao J, Zhang B, Gu BB, Pi RB, Liu J (2013) Edaravone protects HT22 neurons from H2O2-induced apoptosis by inhibiting the MAPK signaling pathway. CNS Neurosci Ther 19:163–169
Ortuno-Sahagun D, Gonzalez RM, Verdaguer E, Huerta VC, Torres-Mendoza BM, Lemus L, Rivera-Cervantes MC, Camins A, Zarate CB (2014) Glutamate excitotoxicity activates the MAPK/ERK signaling pathway and induces the survival of rat hippocampal neurons in vivo. J Mol Neurosci MN 52:366–377
Wang R, Yang J, Peng L, Zhao J, Mu N, Huang J, Lazarovici P, Chen H, Zheng W (2015) Gardenamide A attenuated cell apoptosis induced by serum deprivation insult via the ERK1/2 and PI3 K/AKT signaling pathways. Neuroscience 286:242–250
Zhang H, Mak S, Cui W, Li W, Han R, Hu S, Ye M, Pi R, Han Y (2011) Tacrine(2)-ferulic acid, a novel multifunctional dimer, attenuates 6-hydroxydopamine-induced apoptosis in PC12 cells by activating Akt pathway. Neurochem Int 59:981–988
Li Y, Hu Z, Chen B, Bu Q, Lu W, Deng Y, Zhu R, Shao X, Hou J, Zhao J, Li H, Zhang B, Huang Y, Lv L, Zhao Y, Cen X (2012) Taurine attenuates methamphetamine-induced autophagy and apoptosis in PC12 cells through mTOR signaling pathway. Toxicol Lett 215:1–7
Xu B, Chen S, Luo Y, Chen Z, Liu L, Zhou H, Chen W, Shen T, Han X, Chen L, Huang S (2011) Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS one 6:e19052
van Muiswinkel FL, Kuiperij HB (2005) The Nrf2-ARE Signalling pathway: promising drug target to combat oxidative stress in neurodegenerative disorders. Curr Drug Targets CNS Neurol Disord 4:267–281
Jomova K, Vondrakova D, Lawson M, Valko M (2010) Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem 345:91–104
Paradies G, Petrosillo G, Paradies V, Ruggiero FM (2011) Mitochondrial dysfunction in brain aging: role of oxidative stress and cardiolipin. Neurochem Int 58:447–457
Bhandary B, Piao CS, Kim DS, Lee GH, Chae SW, Kim HR, Chae HJ (2012) The protective effect of rutin against ischemia/reperfusion-associated hemodynamic alteration through antioxidant activity. Arch Pharm Res 35:1091–1097
Wei SM, Yan ZZ, Zhou J (2011) Protective effect of rutin on testicular ischemia-reperfusion injury. J Pediatr Surg 46:1419–1424
Wang R, Yang J, Liao S, Xiao G, Luo J, Zhang L, Little PJ, Chen H, Zheng W (2014) Stereoselective reduction of 1-o-isopropyloxygenipin enhances its neuroprotective activity in neuronal cells from apoptosis induced by sodium nitroprusside. ChemMedChem 9:1397–1401
Jimenez-Aliaga K, Bermejo-Bescos P, Benedi J, Martin-Aragon S (2011) Quercetin and rutin exhibit antiamyloidogenic and fibril-disaggregating effects in vitro and potent antioxidant activity in APPswe cells. Life Sci 89:939–945
Rodriguez-Blanco J, Martin V, Garcia-Santos G, Herrera F, Casado-Zapico S, Antolin I, Rodriguez C (2012) Cooperative action of JNK and AKT/mTOR in 1-methyl-4-phenylpyridinium-induced autophagy of neuronal PC12 cells. J Neurosci Res 90:1850–1860
Chen L, Xu B, Liu L, Luo Y, Yin J, Zhou H, Chen W, Shen T, Han X, Huang S (2010) Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab Invest 90:762–773
Moghbelinejad S, Nassiri-Asl M, Farivar TN, Abbasi E, Sheikhi M, Taghiloo M, Farsad F, Samimi A, Hajiali F (2014) Rutin activates the MAPK pathway and BDNF gene expression on beta-amyloid induced neurotoxicity in rats. Toxicol Lett 224:108–113
Acknowledgments
This study was supported by college projects (No. 2014BS012), funding from the Jiangxi University of Traditional Chinese Medicine, and Key projects of Natural Science Foundation of Jiangxi Province (No. 20151BDH80081) to R. Wang, the Program for Science and Technology of Wuhan (No. 2013062301010816) to L. Wang.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Rights and permissions
About this article

Cite this article
Wang, R., Sun, Y., Huang, H. et al. Rutin, A Natural Flavonoid Protects PC12 Cells Against Sodium Nitroprusside-Induced Neurotoxicity Through Activating PI3K/Akt/mTOR and ERK1/2 Pathway. Neurochem Res 40, 1945–1953 (2015). https://doi.org/10.1007/s11064-015-1690-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-015-1690-2