
Abstract
Parkinson’s disease (PD) is one of the most common degenerative disorders of the central nervous system that produces motor and non-motor symptoms. The majority of cases are idiopathic and characterized by the presence of Lewy bodies containing fibrillar α-synuclein. Small ubiquitin-related modifier (SUMO) immunoreactivity was observed among others in cases with PD. Key disease-associated proteins are SUMO-modified, linking this posttranslational modification to neurodegeneration. SUMOylation and SUMO-mediated mechanisms have been intensively studied in recent years, revealing nuclear and extranuclear functions for SUMO in a variety of cellular processes, including the regulation of transcriptional activity, modulation of signal transduction pathways, and response to cellular stress. This points to a role for SUMO more than just an antagonist to ubiquitin and proteasomal degradation. The identification of risk and age-at-onset gene loci was a breakthrough in PD and promoted the understanding of molecular mechanisms in the pathology. PD has been increasingly linked with mitochondrial dysfunction and impaired mitochondrial quality control. Interestingly, SUMO is involved in many of these processes and up-regulated in response to cellular stress, further emphasizing the importance of SUMOylation in physiology and disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the second most common degenerative disorder of the central nervous system worldwide, with an estimated 7–10 million people affected. After its first description in 1817 by James Parkinson as shaking palsy, PD was considered a degenerative disease of the extrapyramidal system and basal ganglia. However, PD as such does not denote a defined disease but rather comprises a variety of syndromes that are categorized into (i) idiopathic/sporadic syndromes, (ii) familiar/genetic syndromes, (iii) symptomatic/secondary syndromes (e.g., induced by neurotoxins), or (iv) atypical syndromes (within other neurodegenerative diseases such as dementia with Lewy bodies (DLB), multiple-system atrophy, progressive supranuclear palsy, and corticobasal degeneration). Although the incidence of PD increases with age, an estimated four percent of people with PD are diagnosed before the age of 50.
The four cardinal symptoms coincidentally classified as primary motor symptoms are muscular rigidity (stiffness), bradykinesia/akinesia (slowness of movement/inability to initiate movement), resting tremor (involuntary muscle contraction at rest), and postural instability (position instability). Supplementary secondary and non-motor symptoms are dystonia, speech problems, hyposmia (diminished sense of smell), rapid eye movement sleep disturbances, fatigue, depression, fear, anxiety, and cognitive deficits.
On the functional level, the most obvious characteristics are a specific loss of dopaminergic cells, in particular at the substantia nigra pars compacta that projects into the corpus striatum (putamen and caudate nucleus), and a hypoproduction of dopamine. The lack of dopamine is accompanied by an excess of the neurotransmitter glutamate in the basal ganglia that eventually causes motor dysfunction and primary motor symptoms.
Lewy bodies (LBs) were considered as a defining pathological characteristic of PD and DLB. However, not all cases of PD are accompanied by the presence of LBs. LBs were also found in other brain disorders such as Alzheimer’s disease (Goedert et al. 2013), questioning the specific importance of LBs for PD (Schulz-Schaeffer 2012). The accompaniment of neurofilaments, ubiquitin, and β-amyloid in LBs was demonstrated before the main component α-synuclein was identified in 1997 (Spillantini et al. 1997). Later, immunoreactivity of LBs for many other proteins, including parkin and synphilin-1, was shown (Shimura et al. 1999; Wakabayashi et al. 2000).
SUMOylation simultaneously discovered by two groups (Matunis et al. 1996; Mahajan et al. 1997) emerged in recent years as a likely candidate mechanism to regulate a plethora of different processes within the cell. It is well known that ubiquitin and the small ubiquitin-related modifier (SUMO) share similarities in respect to the protein’s tertiary structure and undergo a similar cycle of conjugation and deconjugation. However, there are important differences between both, such as the expression of different SUMO isoforms in mammalians, the existence of a SUMO consensus motif, ubiquitin conjugating enzyme 9 (Ubc9) as exclusive SUMO-E2 conjugation enzyme identified so far, and the dispensability of E3 ligases by direct Ubc9-mediated transfer of SUMO to the target protein (Wilkinson and Henley 2010).
SUMOylation and ubiquitination are regarded as protein modifications that act differentially on a protein’s function and fate. Surprisingly, research in recent years uncovered the existence of mixing units such as the SUMO-targeted ubiquitin ligases (STUbLs) or E3 ligases with dual functions for SUMO and ubiquitin (Prudden et al. 2007). The mammalian SUMO paralogs SUMO-1 and SUMO-2/SUMO-3, although partially redundant, may fulfill different functions as suggested by various studies on substrate specificity, mono-/polySUMOylation, expression, and oxidative stress, arguing against a generalizing “SUMO function” (Meulmeester et al. 2008; Saitoh and Hinchey 2000; Tatham et al. 2011; Bossis and Melchior 2006).
In several diseases, SUMO immunoreactivity was detected in brain tissues of patients, emphasizing the importance of SUMO in neurodegeneration. SUMO-1 co-localized with neuronal intranuclear inclusions in the brain of patients with polyglutamine disease dentatorubral–pallidoluysian atrophy (DRPLA) (Terashima et al. 2002) and in several cases of sporadic and familial neuronal intranuclear inclusion disease (NIID) (Pountney et al. 2003; Wiltshire et al. 2010). Intranuclear inclusions were also positive for ubiquitin (Mori et al. 2012), and the SUMO substrates promyelocytic leukemia protein (PML), histone deacetylase (HDAC) 4, and Ran GTPase activating protein 1 (RanGAP1) (Takahashi-Fujigasaki and Fujigasaki 2006). Immunocapturing with anti-SUMO-1 antibody and mass spectrometry from intranuclear inclusion bodies from brain tissue of a case with familial NIID identified chaperone heat shock protein (Hsp) 90 and proteins involved in membrane trafficking (Pountney et al. 2008). Furthermore, SUMO-1 immunoreactivity was observed in cytoplasmic inclusions in tissues of multiple-system atrophy cases as well as in LBs in smears of DLB tissue homogenates (Pountney et al. 2005), and SUMO-1 is present in the halo of LBs co-localizing with α-synuclein in the brains of PD and DLB patients (Kim et al. 2011).
This review highlights some of the SUMO-mediated mechanisms that are described in recent years. Interconnections of proteins to the SUMO pathway may occur on distinct levels, including (i) direct SUMO modification of proteins via covalent binding or interaction, (ii) regulation of members of the SUMOylation machinery and deSUMOylating enzymes, (iii) regulation of SUMOylated proteins, or (iv) functional interactions including transcriptional regulation, subcellular localization, interference with other posttranslational modifications, proteasomal function, and protein solubility.
Pathogenesis of Parkinson’s Disease
The majority of PD cases are idiopathic without specific known cause. Around 20 % of the PD cases account for familiar forms of the disease with mutations in several genes. To date, more than a dozen different gene loci have been associated with familial PD (Table 1), but not all of them have been validated so far. Mutations in SNCA and lrrk2 are inherited in an autosomal dominant manner, whereas pink1, parkin, and dj-1 cause autosomal recessive early-onset Parkinsonism. SNCA and lrrk2 alleles were associated with increased risk of sporadic PD, suggesting that idiopathic and familial disease entities share pathophysiological mechanisms. A number of environmental triggers were associated with an increased risk of PD, including neurotoxins and herbicides.
α-Synuclein, DJ-1, and parkin are subject to direct SUMO conjugation and SUMO interaction and will be presented in more detail in the following sections.
α-Synuclein
α-Synuclein is a natively unfolded protein of 140 amino acids (Weinreb et al. 1996) that belongs to the family of synucleins, which also includes β- and γ-synucleins. Alternative splicing of exon 3 and/or 5 of the α-synuclein encoding SNCA gene results in truncated forms of 112 or 126 amino acids (Beyer 2006). α-Synuclein is abundantly expressed in human brain. The protein was first isolated in 1988 using antiserum against cholinergic vesicles. The protein was found to be localized to nuclear envelope and presynaptic terminals of neurons, thus named synuclein (Maroteaux et al. 1988). Later, a non-Aβ component of Alzheimer’s disease amyloid (NAC) was found in amyloid plaques in Alzheimer’s disease patients and identified as homolog of rat synuclein, thus named SNCA (Ueda et al. 1993; Jakes et al. 1994). α-Synuclein is involved in synaptic processes and synaptic vesicle trafficking although the exact physiological function remains to be determined (Chandra et al. 2004, 2005). Structurally, α-synuclein can be divided into 3 domains, an amphipathic N-terminal domain, a central hydrophobic NAC domain with propensity to form β-sheet, and a proline-rich C-terminal tail.
A mutation in the SNCA gene on chromosome 4q21, causing an A53T amino acid substitution, was described in 1997 and identified as first PD gene (PARK1) (Polymeropoulos et al. 1997). Two other missense mutations (A30P (Kruger et al. 1998) and E46 K (Zarranz et al. 2004)) as well as gene deletions were observed in familial PD. The PD gene PARK4 was described in a large family of causative PD and later identified as gene triplication of SNCA (Farrer et al. 1999; Singleton et al. 2003). Identification of SNCA gene duplication (Chartier-Harlin et al. 2004) followed, suggesting the existence of a gene dosage effect. α-Synuclein is a major constituent of LBs, a hallmark of PD and DLB (Spillantini et al. 1997), giving rise to the term “synucleinopathies”. α-Synuclein undergoes various types of posttranslational modifications such as phosphorylation, acetylation, nitration, O-glycosylation, ubiquitination, and SUMOylation (Fujiwara et al. 2002; Kang et al. 2012; Giasson et al. 2000; Kubo et al. 2001; Tofaris et al. 2003; Dorval and Fraser 2006). The functional consequences of these modifications and their contribution to α-synuclein-mediated toxicity are contrarily discussed.
The SUMOylation of α-synuclein was shown in cultured cells and mammalian brain in vivo (Dorval and Fraser 2006; Krumova et al. 2011). SUMO can be targeted to several lysine residues within the molecule although lysine residues 96 and 102 were identified as major monoSUMOylation sites of α-synuclein. In vitro experiments showed that SUMOylation of α-synuclein significantly delayed or even blocked the aggregation and fibrillation of α-synuclein by an unknown mechanism. Conversely, SUMO deficiency leads to more inclusions within cells. Moreover, preventing α-synuclein from SUMO modification enhanced apoptosis in cells and, even more importantly, resulted in significant less dopaminergic, vesicular monoamine transporter (VMAT)-positive cells in the substantia nigra pars compacta of rats within 12 weeks (Krumova et al. 2011). These results suggest that SUMOylation enhances the solubility of α-synuclein and that aggregation of α-synuclein contributes to its toxicity.
In another study, impaired proteasomal function resulted in the enhanced formation of high molecular weight α-synuclein aggregates containing both ubiquitinated and SUMOylated species (Kim et al. 2011). A follow-up study suggested that human polycomb protein triggered the SUMOylation and subsequent aggregation of α-synuclein, thereby reducing the vulnerability of cells in response to staurosporine (Oh et al. 2011). Hence, the causal relationship between SUMOylation and aggregation and the relevance to α-synuclein toxicity need to be further investigated.
DJ-1
DJ-1 was initially discovered in 1997 as an oncogene that acts cooperatively with H-Ras and whose expression is increased in several types of cancer (Nagakubo et al. 1997) and later as a regulatory subunit of an RNA-binding protein (Hod et al. 1999). The DJ-1 gene is located on chromosome 1p36 and encodes a protein of 189 amino acids length. The solution of the DJ-1 crystal structure revealed that the protein exists as a homodimer (Tao and Tong 2003; Wilson et al. 2003). DJ-1 belongs to the peptidase C56 family of proteins but possesses no protease activity due to a distorted catalytical triad and the occlusion of the putative active site by an additional C-terminal helix (Shendelman et al. 2004). DJ-1 cDNA is ubiquitously expressed in various human tissues such as pancreas, kidney, skeletal muscle, liver, brain and heart (Nagakubo et al. 1997; Zhang et al. 2005). DJ-1 protein is expressed in all brain regions with immunoreactivity in neurons and glial cells/astrocytes. In situ hybridization revealed that DJ-1 mRNA is not expressed specifically in dopaminergic neurons but instead generally in neurons (Galter et al. 2007). DJ-1 is predominantly localized in the cytoplasm, but also in the nucleus and associated with mitochondria (Nagakubo et al. 1997; Hod et al. 1999; Canet-Aviles et al. 2004). Recent evidence was provided for extracellular localization and function of DJ-1 (Kim et al. 2012).
DJ-1 is a multifunctional protein that, under oxidative stress, exerts profound cytoprotective functions in its capacity as redox-dependent molecular chaperone, transcriptional co-activator, and regulator of androgen receptor signaling.
The involvement of DJ-1 with neurodegeneration became apparent when the gene locus was identified as PARK7 causing early-onset autosomal recessive Parkinsonism (van Duijn et al. 2001; Bonifati et al. 2003). Data regarding the cellular expression of DJ-1 mRNA and protein in the brain of PD patients compared to control cases remain inconsistent (Galter et al. 2007; Olzmann et al. 2007a; Kumaran et al. 2009). DJ-1 protein does not accumulate in LBs or Lewy neurites. Instead, it is strongly expressed in reactive astrocytes of sporadic PD cases (Bandopadhyay et al. 2004; Rizzu et al. 2004). In this context, it is interesting that DJ-1 is also strongly expressed in reactive astrocytes adjacent to brain infarcts, indicating its importance in oxidative stress conditions (Neumann et al. 2004; Mullett et al. 2009).
Several causative mutations were identified in the DJ-1 gene that account for 1–2 % of early-onset cases of PD. The most severe missense mutation L166P is located near the dimer interface and disrupts α-helix G of the structurally important C-terminal helix–kink–helix motif (Bonifati et al. 2003; Gorner et al. 2007), leading to a dramatic destabilization of the homodimer and functional loss of DJ-1L166P. Other sequence alterations such as M26I, E64D, and E163K result in only slight disturbances of the DJ-1 structure, and their effects on DJ-1 function are less clear.
DJ-1 is posttranslationally modified by oxidation, S-nitrosylation, and SUMOylation (Canet-Aviles et al. 2004; Akhtar et al. 2012; Tao and Tong 2003). DJ-1 is considered as an atypical peroxiredoxin-like peroxidase that scavenges peroxides by oxidizing cysteine residue 106 (Andres-Mateos et al. 2007) and enabling mitochondrial translocation. The role of SUMO modification with respect to DJ-1 function involves both the SUMOylation of DJ-1 itself and DJ-1 regulation of other proteins’ SUMOylation, thereby regulating their activities. DJ-1 protein was shown to be covalently modified by SUMO-1 at lysine residue 130 as the main SUMOylation site. Already before PARK7 was identified as DJ-1, the SUMO-1 modification of DJ-1 at K130 was found to be necessary for DJ-1 to manifest full activities for cell transformation and stimulation of cell growth (Takahashi et al. 2001). UV irradiation stimulates the expression and SUMOylation of DJ-1, as well as its oxidation and shuttling into the nucleus, suggesting that DJ-1 SUMOylation is important for its protective effect from UV-induced apoptosis. Mutation of K130 to arginine has a minimal impact on the structure of the protein (Tao and Tong 2003), but leads to multi-/polySUMOylation elsewhere in the DJ-1 protein. Mutated DJ-1 recovers in insoluble fractions and results in sensitization of cells against UV irradiation (Shinbo et al. 2006). This suggests that improper SUMOylation rather than structural alterations is responsible for the dysfunction of DJ-1. Intriguingly, also the dysfunctional disease-associated mutant DJ-1L166P showed improper SUMOylation (Shinbo et al. 2006). These studies illustrate that mutation of a physiological SUMOylation site might lead to improper SUMOylation at other sites within the target protein. Mutations of any amino acid, not necessarily a lysine residue, in a disease-related protein, might result in altered SUMOylation.
Parkin
Parkin is a ubiquitin ligase of 465 amino acids that is encoded in humans by the PARK2 gene on chromosome 6q (Kitada et al. 1998; Shimura et al. 2000). Parkin belongs to the RBR family (RING, in between RING, RING) of atypical E3 ligases. An ubiquitin-like domain (UBL) is located at the amino terminus of parkin. The C-terminal half of the protein comprises two RING finger motifs (RING1 and RING2). Both flank the cysteine-rich in between RING fingers (IBR) motif (Morett and Bork 1999). The atypical E3 ligase character refers to the fact that parkin binds the E2-conjugating enzyme UBE2L3 via the first RING domain, but requires a conserved cysteine residue in the second RING domain in order to transfer ubiquitin in a HECT-type E3-like manner (Wenzel et al. 2011). Alternative splicing results in at least 7 different parkin isoforms (Beyer et al. 2008; Kitada et al. 1998, 2000) diverging for the presence or absence of the above-mentioned domains (Dagata and Cavallaro 2004). Parkin is present in tissues such as heart, testis, and skeletal muscle and is highly expressed in the brain. Intracellularly, parkin is localized in the cytosol (Shimura et al. 1999) and associated with the Golgi apparatus (Kubo et al. 2001). A defined portion of parkin can be recruited to mitochondria and the nucleus (Stichel et al. 2000; Cookson et al. 2003). Parkin is a component of a multiprotein E3 ubiquitin ligase complex that targets misfolded proteins for degradation and protects cells against various forms of cellular stress. The subcellular localization of parkin was suggested to define its specific functions such as gene regulation, mitochondrial quality control, neutralization of oxidative stress, and ER stress (Narendra et al. 2008; Chen et al. 2010a). Parkin can promote monoubiquitination of substrates as well as polyubiquitination linked via K27, K48, or K63 of ubiquitin (Doss-Pepe et al. 2005; Geisler et al. 2010). Polyubiquitin chains are built of at least four K48-linked ubiquitin molecules attached to a target protein, are recognized by the 26S proteasome subunit, and promote degradation. Monoubiquitination and polyubiquitination via K63 or K27 direct the localization of proteins. Intriguingly, parkin undergoes autoubiquitination in an E2-dependent manner, leading to its own degradation (Um and Chung 2006; Lim et al. 2005; Choi et al. 2000).
Mutations in the PARK2 gene are the most common autosomal recessive cause of early-onset PD and typically not associated with α-synuclein and ubiquitin-positive LBs (Kitada et al. 1998; Abbas et al. 1999; Hattori et al. 1998; Leroy et al. 1998). However, parkin immunoreactivity was present in the central core of a majority of LBs from sporadic and non-parkin familial PD cases, suggesting that parkin is a key factor in the biogenesis of LB inclusions (van de Warrenburg et al. 2001; Schlossmacher et al. 2002; Shimura et al. 2000; Bandopadhyay et al. 2005).
About 50 % of the mutations in the PARK2 gene account for genomic rearrangements such as duplications and deletions (Elfferich et al. 2011). The remaining 50 % represent small point mutations, most of which located within the RING domains. Two mutations each were found in the UBL domain (P37L and R42P) and IBR motif (G328E and R334C) (Geisler et al. 2010).
Parkin selectively interacts with SUMO-1, but not with SUMO-2. The interaction between parkin and SUMO-1 was observed in mammalian central nervous system and in neuronal cells. The association of SUMO-1 with parkin results in increased autoubiquitination and ubiquitin–proteasome system (UPS)-mediated degradation. SUMO-1 promotes the shuttling of a small pool of parkin protein from the cytosol into the nucleus. Proteasome inhibition resulted in the enhanced nuclear shuttling of parkin in the presence of SUMO-1 (Um and Chung 2006). Numerous putative substrates and interaction partners of parkin have been identified in recent years, which will be discussed in more detail below.
Molecular Mechanisms for Parkinson’ Disease
The majority of PD cases are idiopathic, which means “a disease of its own”, of unknown causes. Indeed, the cause and pathogenesis of PD remain elusive. However, starting with the discovery of environmental triggers such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or herbicides through to the discovery of risk genes in familiar PD, various mechanisms have been suggested in recent years that might be involved in or are even responsible for the pathology of PD in all entities, independent of their origin. Recent findings implicate mitochondrial dysfunction, oxidative damage, subcellular mislocalization of proteins, and abnormal protein accumulation as key molecular mechanisms compromising dopamine neuronal function and survival (Pilsl and Winklhofer 2012; Hauser and Hastings 2013; Patel and Chu 2011; Kalia et al. 2013; Cuervo et al. 2010; Michel et al. 2013). This review focuses on PD pathology in light of SUMOylation-dependent processes.
Mitochondrial Homeostasis and Function
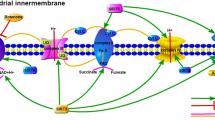
Mitochondria supply most of the energy for the brain and are of peculiar interest in neurodegeneration. Contrary to the obsolete view of mitochondria as static bodies, these are highly dynamic organelles that undergo constant cycles of fission and fusion to maintain their morphology and function (Chen and Chan 2009; Youle and van der Bliek 2012; Zungu et al. 2011). The process of mitochondrial fusion allows DNA replication, complementation of protein/lipid contents, and distribution of metabolic intermediates. Fission is necessary for the redistribution of mitochondrial DNA during cell division. It increases the number and capacity of mitochondria and enables to segregate damaged mitochondrial parts for autophagy by the lysosomal system, known as “mitophagy”. There is growing evidence that SUMO plays a role in regulating mitochondrial fission and fusion (Fig. 1).

SUMO is involved in mitochondrial processes. Cellular stress leads to the formation of ROS and up-regulation of global SUMOylation, which in turn influences various mitochondrial processes. Three pathways are exemplarily shown and indicated by white numbers on black circles. (1) Drp1 translocation from the cytosol to the mitochondrial membrane is necessary for Drp1 to exert its function as mediator of mitochondrial fission. SUMOylation promotes and deSUMOylation blocks the translocation of Drp1. Moreover, SUMO-1 overexpression results in a more stable active pool of Drp1. (2) SUMOylation of the cytoprotective protein DJ-1 facilitates its subsequent oxidization at C106 and translocation to the mitochondrial membrane where it inhibits Fis1, another key player in mitochondrial fission. (3) PINK1 accumulates at the outer membrane of depolarized damaged mitochondria and recruits the E3 ligase parkin. Parkin-mediated ubiquitination of proteins such as the pore-forming VDACs promotes mitophagy. Ubiquitination of the parkin substrate Drp1 promotes Drp1 degradation and counteracts mitochondrial fission. SUMOylation of parkin reduces substrate ubiquitination in favor of parkin autoubiquitination and drives parkin translocation into the nucleus. ROS reactive oxygen species, SUMO small ubiquitin-related modifier, SENP sentrin-/SUMO-specific protease; PINK1 PTEN-induced putative kinase 1, Drp1 dynamin-related protein 1, Fis1 fission protein 1, VDAC voltage-dependent anion channel, Ub ubiquitin
Dynamin-related protein 1 (Drp1) is a key regulator of mitochondrial fission and the first identified mitochondrial SUMO target. Drp1 is recruited from the cytosol to the mitochondrial outer membrane to coordinate membrane scission (Shaw and Nunnari 2002). Drp1 interacts with the SUMO-conjugating enzyme Ubc9 via multiple regions and is a direct target of SUMO modification by all three SUMO isoforms at non-canonical conjugation sites. The activity of Drp1, rather than the mitochondrial recruitment, seems to be linked to SUMOylation (Figueroa-Romero et al. 2009). SUMO-1 overexpression dramatically increases mitochondrial fragmentation, while Drp1 is protected from degradation under the same conditions, resulting in a more stable active pool of Drp1 (Harder et al. 2004). A cytosolic pool of sentrin-/SUMO-specific protease (SENP) 5 catalyzes the cleavage of SUMO-1 from a number of mitochondrial substrates, including Drp1. Overexpression of SENP5 rescues SUMO-1-induced mitochondrial fragmentation. By contrast, the silencing of SENP5 results in fragmented and altered mitochondrial morphology (Zunino et al. 2007). These studies suggest that SUMOylation of Drp1 leads to increased Drp1-mediated fission (Fig. 1). The mitochondrial fission enables the cell to isolate defective segments and to prevent further damage as response to minor cell stress.
PTEN-induced putative kinase 1 (PINK1) is an atypical member of the family of serine/threonine kinases and encoded by the early-onset PD gene PARK6 (Valente et al. 2004). The 581-amino-acid-long protein is characterized by an N-terminal mitochondrial targeting sequence, a transmembrane helix, a central kinase domain, and a C-terminal domain with possibly autoregulatory function (Beilina et al. 2005). PINK1 mRNA is expressed in the brain with highest levels in the hippocampus, substantia nigra, and cerebellar Purkinje cells (Blackinton et al. 2007). PINK1 mRNA expression is abundantly found in neurons, but not in glial cells, whereas immunoreactivity of the protein was also shown in astrocytes (Wilhelmus et al. 2012). PINK1 is considered as key regulator of mitochondrial homeostasis and mitochondrial quality control on molecular and organellar levels.
Drp1 can be phosphorylated by PINK1, which in turn results in reduced mitochondrial scission possibly via decreased GTPase activity of Drp1. Drp1 overexpression exaggerates PINK1 deficiency phenotypes, which are characterized by enhanced mitochondrial fragmentation and increased amounts of dephosphorylated Drp1. Interestingly, Drp1 and fission protein 1 (Fis1) expression influence PINK1 expression possibly to provide a regulatory feedback mechanism (Mai et al. 2010; Taguchi et al. 2007; Sandebring et al. 2009). Parkin interacts with and subsequently ubiquitinates Drp1 for promoting its proteasome-dependent degradation. Pathogenic mutation or knockdown of parkin inhibits the ubiquitination and degradation of Drp1, leading to increased levels of Drp1 for mitochondrial fragmentation (Wang et al. 2011). DJ-1 exerts its cytoprotective function to oxidative stress either directly or by the regulation of antioxidative gene induction. Early studies by Canet-Aviles et al. (2004) in cells showed that cytosolic DJ-1 translocates to the outer mitochondrial membrane in response to oxidative stress and compromises Fis1-mediated mitochondrial fission by promoting its proteasomal degradation (Zhang et al. 2012). Shuttling to mitochondria is driven by the oxidization of the cysteine residue 106, which scavenges hydrogen peroxide and leads to a pI shift in DJ-1 to a more acidic point (Kinumi et al. 2004; Taira et al. 2004; Andres-Mateos et al. 2007). UV irradiation induces the SUMOylation of DJ-1, which is then oxidized and recruited to mitochondria. The K130 SUMOylation site of DJ-1 is located in close proximity to His126, which functions together with C106 as catalytic diad in a C-terminally cleaved and activity-enhanced fragment of DJ-1 (Chen et al. 2010b), further emphasizing the importance of SUMOylation for DJ-1 activity.
Taken together, the presented data suggest that Drp1 itself is modified and regulated by SUMOylation. Oxidative stress promotes the expression of SUMO and the translocation of Drp1, resulting in enhanced mitochondrial fission. Cytoprotective DJ-1 and parkin counteract SUMOylation and inhibit Drp1- and Fis1-mediated fragmentation (Fig. 1).
Full-length PINK1 is localized to mitochondria and partially imported through the outer and inner membranes in that the kinase domain faces the cytoplasm (Zhou et al. 2008). PINK1 is constitutively proteolyzed at the mitochondrial membrane of healthy mitochondria, keeping the levels of PINK1 low. The cleavage is catalyzed by the mitochondrial processing peptidase (MPP) and PARL/AFG3L2 (Meissner et al. 2011; Greene et al. 2012), leading to the formation of a 52-kDa fragment that can be further degraded by the proteasome (Lin and Kang 2008). Mitochondrial membrane depolarization leads to the accumulation of unprocessed PINK1 due to impaired mitochondrial import and reduced proteasomal degradation (Zhou et al. 2008; Narendra et al. 2010; Becker et al. 2012; Greene et al. 2012). The stabilized PINK1 directly phosphorylates parkin, which is required for efficient mitochondrial translocation of parkin (Shiba-Fukushima et al. 2012). The E3 ubiquitin ligase parkin catalyzes ubiquitination of outer mitochondrial membrane proteins. Subsequently, damaged mitochondria are transported to the perinuclear region and undergo mitophagy (Fig. 1). Intriguingly, SUMO-1 modification of parkin enhances its autoubiquitination and drives parkin translocation into the nucleus (Um and Chung 2006). By this means, SUMOylation of parkin might also diminish the pool of parkin that is available for the mitochondrial recruitment. PINK1-dependent phosphorylation of other mitochondrial proteins such as the mitochondrial chaperone TRAP1/Hsp75 or HtrA2, a mitochondrial serine protease encoded by PARK13, is likewise implicated in the protection against oxidative stress (Pridgeon et al. 2007; Plun-Favreau et al. 2007).
A surprising number of SUMO-1 conjugates were observed in mitochondrial fractions, hypothesizing that SUMO may play a central role in regulating mitochondrial processes. Several proteins are recruited to mitochondria in order to fulfill their specific function. SUMOylation could be a posttranslational switch that allows the dynamic and reversible regulation of mitochondrial homeostasis and function in response to various cellular stressors.
Response to Cellular Stress
Response to cellular stress can be regulated in several ways within the cell. This section highlights SUMO-regulated signal transduction events that are involved in cell survival and apoptosis. Signal transduction cascades can be initiated by extracellular stimuli and intrinsic factors. Extracellular signals such as growth factors, hormones, cytokines, and toxins must be transmitted via receptor mediation or by crossing the plasma membrane. Nuclear receptors such as androgen receptor, peroxisome proliferator-activated receptor (PPAR), and retinoid X receptor α (RXRα) represent a specific class of proteins that directly bind to DNA response elements and regulate the expression of specific genes. Intrinsic factors such as caspases and proapoptotic and antiapoptotic proteins play an important role in the regulation of apoptosis.
Rearranged during transfection (RET) belongs to the family of receptor tyrosine kinases that are expressed on the cell surface. Binding of glial cell-derived neurotrophic factor (GDNF) to RET is neuroprotective for dopaminergic neurons. The signal is intracellularly transduced through the activation of the phosphatidylinositide 3-kinase (PI3K) and the Akt/protein kinase B pathway. The PI3K class IA phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3). The phosphatase PTEN (phosphatase and tensin homolog) hydrolyzes PIP3 to PIP2, thus acting as the catalytic antagonist of PI3K and inhibitor of the Akt pathway (Stambolic et al. 1998). The activity of PTEN requires its association with the inner face of the plasma membrane via electrostatic interactions. SUMO-1 modification of PTEN at K266 within the CBR loop facilitates the cooperative binding between PTEN and the plasma membrane (Huang et al. 2012). This SUMO effect might not only be relevant in tumorigenesis but also have a broad impact on cell survival in general. DJ-1 is a key negative regulator of PTEN function. DJ-1 overexpression, which is found in various carcinoma samples, leads to hyperphosphorylation of Akt/protein kinase B and increased cell survival, whereas DJ-1 knockdown has opposite effects (Kim et al. 2005; Davidson et al. 2008). Additionally, DJ-1 prevents the down-regulation of RET mRNA and RET receptor by hypoxia-inducible factor 1α (HIF-1α) (Foti et al. 2010). Recently, SUMOylation of Akt itself was shown to be essential for its activation (Li et al. 2013), demonstrating a variety of regulatory mechanisms through SUMOylation.
Activation of the nuclear factor-kappaB (NF-κB) signaling pathway is widely connected to cell survival. NF-κB activity is tightly regulated by several factors including posttranslational modifications such as phosphorylation, acetylation, and ubiquitination. The transcription factor NF-κB, in its most abundant form a heterodimer of the p50/p65 (RelA) subunits, is retained in a cytosolic complex with IκBα. Phosphorylation and ubiquitination of IκBα release NF-κB from the complex and enable NF-κB translocation into the nucleus and transcriptional regulation. The phosphorylation of IκBα is catalyzed by the IκB kinase (IKK), a complex of the two catalytic subunits IKKα and IKKβ and the regulatory subunit IKKγ/NEMO (NF-κB essential modulator).
Growing evidence suggests that several proteins in the NF-κB pathway are subject to SUMOylation (Girdwood et al. 2003). One of the first proteins that were shown to be competitively modified by ubiquitin and SUMO is IκBα. IκBα is conjugated to SUMO on the same lysine residue K21 that also serves as ubiquitin target. In contrast to ubiquitination, SUMOylation neither requires prior phosphorylation nor results in IκBα proteasomal degradation (Desterro et al. 1998). NF-κB activity is inhibited by the SUMO E3 ligases protein inhibitor of activated signal transducers and activators of transcription (PIAS) 1 and 3 that bind to the C-terminal transactivation domain and N-terminal DNA binding domain of the RelA subunit, respectively (Jang et al. 2004). The RelA subunit of NF-κB is, in addition, covalently SUMOylated by PIAS3. SUMO-1 modification of NEMO is critical for stress-mediated NF-κB activation although the underlying mechanisms remain unclear. The SUMO E3 ligase PIASγ seems to mediate NEMO SUMOylation with substrate specificity for SUMO-1, but not for SUMO-2 and SUMO-3, thereby contributing to NF-κB activation (Mabb et al. 2006).
The PD-associated proteins DJ-1, parkin, and ubiquitin C-terminal hydrolase L1 (UCH-L1) are modulators of NF-κB activity. The deubiquitinating enzyme cellular zinc finger anti-NF-kappaB (Cezanne) inhibits NF-κB translocation by stabilizing the inhibitor IκBα. DJ-1 interacts with Cezanne, inhibits its deubiquitination activity, promotes NF-κB nuclear translocation, and enhances cell survival (McNally et al. 2011). Parkin mediates neuroprotection through activation of NF-κB signaling (Henn et al. 2007). UCH-L1 inhibits NF-κB activation possibly through reversal of IκBα ubiquitination (Takami et al. 2007). SUMOylation of members of the NF-кB pathway (e.g., IкBα) and binding of SUMO E3 ligases to nuclear NF-кB/RelA seem to prevent full NF-кB activation and cell survival. SUMOylation of NEMO though contributes to NF-кB activation. It remains to be determined whether and how SUMOylation of these proteins might provide a general mechanism to regulate cell survival depending on upstream signals or intrinsic factors.
Daxx was identified in 1997 as Fas-death-domain-associated protein that activates the JNK pathway and potentiates Fas-mediated apoptosis (Yang et al. 1997; Torii et al. 1999). Later studies showed that Daxx is involved in UV-induced cell death (Khelifi et al. 2005). Daxx possesses both cytoplasmic and nuclear functions. Cytosolic localization of Daxx is required for the binding and activation of the proapoptotic factor apoptosis signal-regulating kinase 1 (ASK-1) (Chang et al. 1998). DJ-1 binding to Daxx blocks the nucleocytoplasmic translocation of Daxx. Retention of Daxx in the nucleus prevents it from binding to ASK-1 (Junn et al. 2005). Although recent findings focus on nuclear functions of Daxx, the role of SUMO-interacting motifs (SIM) for cytosolic functions of Daxx will be of additional interest.
Hypoxia and the formation of reactive oxygen species (ROS) stabilize HIF-1α. Under physiological conditions, HIF-1α is constitutively ubiquitinated and degraded by the proteasome, keeping the levels of HIF-1α low. Oxidative stress blocks the degradation of HIF-1α, which in turn shuttles into the nucleus and induces the transcription of proapoptotic genes. Hypoxic stress promotes the SUMOylation of HIF-1α. SUMOylated HIF-1α binds to the ubiquitin ligase von Hippel-Lindau (VHL) and undergoes ubiquitination and degradation. In this case, SUMOylation serves as a direct signal for ubiquitin-dependent degradation. The SUMO isopeptidase SENP1 removes SUMO from HIF-1α and enhances its stability. Hypoxia-induced transcription of HIF-1α-dependent genes such as vascular endothelial growth factor, erythropoietin, and glucose transporter-1 is markedly reduced in SENP1-deficient mice, indicating that SENP1 contributes to the regulation of the hypoxic stress response (Cheng et al. 2007). In one report, Seliger et al. (2007) suggested that the PD-associated protein UCH-L1 may counteract VHL-driven HIF-1α ubiquitination, stabilizing the factor for subsequent transcriptional activity.
Oxidative stress within a certain range has been shown to induce global protein modification by SUMO-2/SUMO-3 rather than SUMO-1 (Bossis and Melchior 2006). Furthermore, the free pool of SUMO-2/SUMO-3 ready for conjugation to target proteins is much bigger than the amount of free SUMO-1. This led to the hypothesis that cells may use SUMO-2/SUMO-3 modification as a protective response to various cellular stress stimuli. Only a few proteins have been identified as exclusive targets for specific SUMO isoforms so far. Functional differences between SUMO paralogs remain elusive. Taken together, SUMO-mediated regulation in response to cellular stress appears veritable. The limited number of studies and the fragmentary knowledge do not allow a general concept of SUMO under stress.
SUMO: Nuclear Functions and Gene Regulation
Gene regulation is a complex interplay between various regulatory factors. It requires the availability of specific transcription factors, by either surface-receptor-mediated signaling and nuclear translocation or internalization of nuclear receptor proteins or shuttling of other proteins into the nucleus. Histones play a key role in the accessibility of DNA regions, a prerequisite for transcriptional regulation. Histones undergo posttranslational modification, especially acetylation. The addition of acetyl to the ε-amino group of lysine residues neutralizes the positive charge of histones and decreases the interaction with the negative-charged DNA. Highly acetylated histones form more accessible chromatin, which in general is associated with active transcription. The action of the responsible histone acetyltransferases (HATs) is counteracted by HDACs. Deacetylation of histones allows the negative-charged DNA to wrap more tightly.
The nuclear receptors PPAR family of transcription factors, their co-activators (e.g., PPARγ co-activator 1α (PGC-1α) or RXRα), and their co-repressors (e.g., nuclear receptor co-repressor (NCoR)) are key gene regulators in mitochondrial biogenesis, energy metabolism, and apoptosis (Wadosky and Willis 2012). SUMOylation of the PPAR isoforms α, γ1, and γ2 blocks their transcriptional activities through the recruitment of co-repressors (e.g., NCoR). SUMO thereby inhibits PPAR-mediated apoptosis (Pourcet et al. 2010; Ohshima et al. 2004; Yamashita et al. 2004; Chung et al. 2011).
SUMOylation also suppresses the transcriptional activity of RXRα, which is reversed through specific deSUMOylation of RXRα by SENP6 (also named SUMO-specific isopeptidase (SUSP)1) (Choi et al. 2006).
The oncogene mouse double minute 2 homolog (MDM2) is a RING type E3 ubiquitin ligase, which mediates autoubiquitination as well as the ubiquitination of other substrates, including the p53 tumor suppressor protein. The p53 tumor suppressor has been identified as a key effector protein in familial as well as sporadic cases of PD (Alves da Costa and Checler 2011). p53-mediated death of dopaminergic neurons was shown in a zebrafish model (Bretaud et al. 2007). p53 is tightly regulated by MDM2 in two ways. First, polyubiquitination of p53 by MDM2 targets p53 to proteasomal degradation (Honda et al. 1997; Haupt et al. 1997; Kubbutat et al. 1997). Second, interaction of MDM2 with the p53 transcription activation domain blocks transcriptional activities of p53 (Chen et al. 1995).
MDM2 is SUMO-1-modified by the SUMO E3 ligases PIAS1 and PIASxβ. Upon SUMOylation, the E3 ligase activity of MDM2 is shifted from self-ubiquitination toward substrate ubiquitination (Buschmann et al. 2001). Consistently, SUSP4 promotes MDM2 self-ubiquitination and positively regulates p53 (Lee et al. 2006).
Interestingly, p53 itself undergoes SUMO-1 and SUMO-2/SUMO-3 modification that is believed to increase p53 transcriptional activities (Gostissa et al. 1999; Rodriguez et al. 1999; Stindt et al. 2011). Likewise, a SUMOylation-defective mutant of Drosophila p53 showed markedly less activity than the wild type (Mauri et al. 2008). Modifications of mammalian p53 are catalyzed by the SUMO ligase activity of MDM2 (Chen and Chen 2003; Stindt et al. 2011). Alternate reading frame (ARF) tumor suppressor stimulates MDM2-mediated SUMOylation but blocks ubiquitination of p53 by MDM2.
Homeodomain-interacting protein kinase 2 (HIPK2), a member of the nuclear serine/threonine kinases, directly phosphorylates and activates p53. HIPK2 itself can be modified by SUMOylation and acetylation. HIPK2 localizes with p53 into nuclear bodies and shows only low basal acetylation. SUMOylation of HIPK2 enhances its association with HDAC3 and subsequent HIPK2 deacetylation. ROS trigger the deSUMOylation of HIPK2 that is paralleled by HIPK2 acetylation and significantly enhance cell survival (de la Vega et al. 2012). Consistently, HIPK2-null mice showed a loss of about 40 % of dopaminergic neurons in the substantia nigra and Parkinson-like motor symptoms (Zhang et al. 2007). In this context, it is interesting that SUMOylation mediates the recruitment of the HDAC6 and HDAC2, leading to SUMO-dependent transcriptional repression (Girdwood et al. 2003; Yang and Sharrocks 2004). Parkin is reversibly recruited to the centrosome by direct binding with HDAC6 through multiple interaction domains (Jiang et al. 2008).
The first DJ-1-interacting protein identified was PIASx-α, also known as androgen-receptor-interacting protein 3, which suppresses androgen receptor reporter activity (Takahashi et al. 2001). DJ-1 restores the activity of the androgen receptor reporter by sequestering its negative regulator PIASx-α (Tillman et al. 2007). Due to the nature of PIASx-α as SUMO E3 ligase, it remains to be determined whether SUMOylation might have an impact on androgen receptor activity or not. Interestingly, androgen response elements are present in the promoter region of tyrosine hydroxylase (TH), the rate-limiting enzyme for catecholamine biosynthesis (Jeong et al. 2006). The influence of SUMOylation on the TH promoter will be discussed below. Similarly, DJ-1 antagonizes the inhibitory effect of the DJ-1 binding protein (DJBP) on the androgen receptor by abrogation of the DJBP–HDAC complex (Niki et al. 2003). DJ-1 mediates the induction of the glutamate cysteine ligase, the rate-limiting enzyme for the biosynthesis of glutathione, an antioxidant that prevents damage caused by ROS (Zhou and Freed 2005).
SUMO has diverse effects on transcriptional activity. In many cases, SUMOylation represses transcription either by modifying transcription factors or by the recruitment of HDACs. But there are also some exceptions to the rule with SUMO-mediated transactivation. In the case of p53, SUMO seems to promote p53-mediated cell death.
SUMO and Aggregation
Brain tissues from PD patients show characteristic cytoplasmic eosinophilic inclusions, the LBs, and dense structures in neuronal processes, the Lewy neurites. LBs are considered the hallmark of idiopathic PD and DLB, but they are also detectable in cases of Pick’s disease, corticobasal degeneration, tauopathies, and multiple-system atrophy. LBs are particularly abundant in the substantia nigra but also present in the cortex, amygdala, and locus coeruleus (Braak et al. 2003).
Although α-synuclein is considered the primary structural component of LBs, immunoreactivity against a variety of other proteins such as ubiquitin, β-amyloid, synphilin-1 (Wakabayashi et al. 2000), neurofilaments, lipofuscin, and neuromelanin was observed in the deposits. LBs seem to be a consequence of excess protein misfolding, failure of intracellular clearance/recycling systems, protein accumulation, and segregation. Despite marked abnormalities, neurons with inclusions may survive for years and preserve sufficient functional integrity prior to cell death.
It is still a matter of debate whether fibrils and aggregates are contributors to α-synuclein neurotoxicity or not. Analog questions were stated in tauopathies or aggregopathies regarding the role of key proteins such as Tau, amyloid precursor protein, huntingtin, and ataxin. It was hypothesized that aggregates and fibrils are harmful due to occlusion of the cell, blockage of the proteasome, and segregation of protein, thereby diminishing the pool of functional protein or nucleation of aggregation in a prion-like manner. On the other hand, aggregates were considered as beneficial for cells in terms of protection from aberrant misfolded protein, neutralization of toxic intermediate species, or deposition in order to preserve cellular function (Gosavi et al. 2002; Masliah et al. 2000; Sharon et al. 2003). There is growing evidence that inclusions do contain not only ubiquitinated species but also SUMOylated species. With certainty, SUMOylation can enhance the solubility of a protein as shown in numerous studies.
A recent study investigated the direct effect of SUMOylation on the aggregation propensity of α-synuclein in vitro. SUMOylation of a small portion of α-synuclein was sufficient to prevent α-synuclein from aggregation. In vivo experiments suggested that SUMO-deficient α-synuclein is more prone to aggregate and exaggerates α-synuclein-mediated toxicity, implicating a detrimental role of α-synuclein aggregation (Krumova et al. 2011). Seemingly contradictory conclusions were drawn from another study claiming that proteasomal dysfunction results in the aggregation of SUMOylated species (Kim et al. 2011). However, the presented data do not necessarily exclude each other.
Several studies investigated the relationship between α-synuclein and other PD-associated proteins such as parkin, DJ-1, UCH-L1, and synphilin-1. Intriguingly, many of these proteins are involved in ubiquitination processes and their action may converge at several junctions. Interference between ubiquitination and SUMOylation, although manifested in rare cases only, might be a general concept for cytoprotection.
Parkin and DJ-1 seem be protective against α-synuclein toxicity on TH-positive neurons and α-synuclein fibrillation (Shendelman et al. 2004; Petrucelli et al. 2002). Interaction of α-synuclein and synphilin-1 via their C-termini promotes the formation of cytosolic inclusions (Engelender et al. 1999; Kawamata et al. 2001). Hsp70 inhibits α-synuclein fibril formation (Dedmon et al. 2005). Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex (the so-called PPD complex) that promotes unfolded protein degradation (Xiong et al. 2009). The E3 ubiquitin ligase tumor necrosis factor receptor-associated factor 6 (TRAF6) binds and ubiquitinates misfolded mutant DJ-1 and α-synuclein and promotes their accumulation into cytoplasmic aggregates. Parkin mediates the ubiquitination and targeting of misfolded DJ-1 into aggresomes (Olzmann et al. 2007b). Parkin ubiquitination of the α-synuclein-interacting protein synphilin-1 (Kawamata et al. 2001; Chung et al. 2001) may be involved in the formation of LB inclusions associated with PD (Lim et al. 2005). Excess α-synuclein worsens the disease in mice lacking the neuronal deubiquitinating enzyme UCH-L1 (Shimshek et al. 2012).
Finally, an exploratory quantitative proteomic study has isolated potential SUMO substrates that are specifically up-regulated in response to proteasome inhibition, a common feature in brain aging and neurodegeneration (Abbas et al. 1999).
Regulation of Subcellular Localization and Translocation
To fulfill their specific function, proteins need to be translocated to or localized at their correct site of action. Therefore, the regulation of the subcellular localization has a major impact on the protein’s function. Additionally, the function of a protein might be dependent on the regulation of potential interaction partners.
Several studies implicate that SUMOylation is able to drive the subcellular localization, translocation, or interaction of proteins. Molecular consequences of SUMOylation, such as creating a new binding site for interaction, inhibiting interaction, and inducing a conformational change, have been described previously (Geiss-Friedlander and Melchior 2007). The initial discovery of SUMOylation as a posttranslational modification was shown for RanGAP1, a protein that, depending on its SUMOylation status, shuttles from the cytosol to the nuclear pore complex and interacts with the Ran-binding protein 2 (RanBP2) (Mahajan et al. 1997; Matunis et al. 1996).
RanBP2, a member of the nucleoporin family with SUMO E3 ligase activity (Pichler et al. 2002), is localized to the cytoplasmic fibers of the nuclear pore complex and involved in the regulation of nucleocytoplasmic shuttling of proteins. RanBP2 is a substrate of parkin, leading to ubiquitination and proteasomal degradation of RanBP2 (Um et al. 2006). RanBP2 binds to both parkin and HDAC4. HDAC4 belongs to class II HDACs (also HDAC5, 6, 7, 9, and 10) that are able to shuttle between the cytoplasm and nucleus through the nuclear pore complex. HDAC4 is a substrate of RanBP2 and conjugated to SUMO-1 at a single lysine residue. SUMOylation seems to be important for the full activity of HDAC4 (Kirsh et al. 2002), implicating consequences for histone deacetylation and transcriptional repression. Parkin can control the levels of SUMOylated and functional HDAC4 as a consequence of RanBP2 ubiquitination and degradation. Similarly, parkin may also regulate the SUMOylation or translocation of other RanBP2 substrates. Considering additionally the SUMO-dependent shuttling of parkin itself into the nucleus, SUMOylation might be one molecular mechanism for parkin-mediated transcriptional control.
TRAF- and TNF-receptor-associated protein (TTRAP) was identified as a novel effector protein for signal transduction by distinct members of the TNF receptor family (Pype et al. 2000). TTRAP inhibits the transcriptional activation of NF-кB, is involved in DNA repair, and may associate with PML nuclear bodies. The TTRAP sequence contains a SIM that is required for its binding to SUMO-2/SUMO-3 and TTRAP nucleolar localization (Hecker et al. 2006; Vilotti et al. 2012). Thus, SUMOylation controls TTRAP nuclear activity in response to cellular stressors. TTRAP was identified as a novel DJ-1 interactor with a stronger binding to the PD-associated DJ-1 mutants M261 and L166P over DJ-1 wild type (Zucchelli et al. 2009).
The multifunctional protein Daxx acts as a transcriptional co-regulator and modulator of apoptosis. Daxx has a nuclear export signal (NES)-like motif and two nuclear localization signal (NLS) motifs, one in the center and one near the carboxy terminus. The C-terminal NLS ranging from amino acid 627 to 634 includes the two major covalent SUMOylation sites K630 and K631 (Jang et al. 2002; Yeung et al. 2008). In addition, Daxx has two SUMO interacting motifs, one at the very N-terminus and one at the very C-terminus, named SIM-N and SIM-C, respectively. The two SIMs feature differences in affinities and binding modes to SUMO probably due to differences in charged residues that flank the identical hydrophobic core sequences (I–I–V–L) of the two SIMs. SIM-N shows a higher affinity toward SUMO than SIM-C and binds SUMO-1 predominantly in parallel orientation, whereas SIM-C interconverted between parallel and antiparallel binding to SUMO-1. Furthermore, SIM-N binds intramolecularly to an adjacent α-helical bundle, suggesting an autoregulatory mechanism for Daxx (Lin et al. 2006; Santiago et al. 2009; Escobar-Cabrera et al. 2011). SUMO interacting motifs in the protein Daxx are important for the subcellular localization and function of Daxx (Chen et al. 2006). Nuclear localization of Daxx and its interaction with p65 inhibit the acetylation of p65, which is required for the full activation of NF-κB-mediated gene expression (Croxton et al. 2006; Park et al. 2007; Rothgiesser et al. 2010). Interaction of Daxx with DJ-1 prevents the nucleocytoplasmic translocation of Daxx and sequesters Daxx in the nucleus, thereby blocking cytosolic functions of Daxx (e.g., in apoptosis). Similarly, the subcellular localization and gene expression of the Drosophila homolog of Daxx, the Daxx-like protein (DLP), is regulated by DJ-1β, a Drosophila homolog of DJ-1. DJ-1β protects against oxidative stress and UV-induced apoptosis, whereas loss-of-function DJ-1β mutants or overexpression of DLP induces apoptosis via the c-Jun N-terminal kinase (JNK)/Drosophila fork head box subgroup O (dFOXO) pathway (Hwang et al. 2013). Daxx associates via SIMs with several SUMOylated transcription factors and acts as transcriptional co-repressor of antiapoptotic genes (e.g., via the recruitment of HDACs) (Chang et al. 2005; Lin et al. 2006; Kuo et al. 2005; Mizuta and Kuroda 2004). PML oncogenic domains (PODs), also called PML nuclear bodies (PML-NBs) or nuclear domain 10 (ND10), are discrete interchromosomal accumulations of several proteins, including PML and Sp100. Daxx can be recruited to PODs, a process that requires SUMO modification of PML and a SIM within Daxx (Ishov et al. 1999; Lin et al. 2006). A SUMO-defective PML mutant that cannot be SUMOylated is no longer able to interact with Daxx (Lehembre et al. 2001; Li et al. 2000). Sequestration of Daxx to PODs can attenuate the transcriptional co-repressor activity/transrepression (Betarbet et al. 2000; Chang et al. 2005; Lin et al. 2006).
Under stress conditions such as UV irradiation, Daxx seems to be recruited to PIAS1 foci and interact with PIAS1-SUMOylated substrates via its C-terminal SIM. Thereby, Daxx appears to act as proapoptotic downstream effector of PIAS1. The SUMO E3 ligase activity of PIAS, but not SUMOylation of Daxx itself, seems to be necessary for the recruitment of Daxx- and PIAS-mediated UV-induced apoptosis (Sudharsan and Azuma 2012).
UV irradiation leads to SUMOylation and subsequent oxidization of DJ-1, followed by translocation to the nucleus. Interestingly, disease-associated mutant DJ-1L166P shows improper SUMOylation and is more sensitive to UV-induced apoptosis. Besides that, DJ-1L166P recovers in insoluble fractions and shows partial degradation by the proteasome (Shinbo et al. 2006). The DJ-1 mutant is strongly associated with the chaperones Hsp70 and CHIP compared to DJ-1wt (Li et al. 2005). BAG1 (Bcl-2-associated athanogene 1), a cochaperone of HSP70 and DJ-1 interacting protein, is able to compensate for mutant DJ-1L166P by restoring DJ-1 subcellular distribution, dimer formation, and chaperone activity and by ameliorating cell survival (Deeg et al. 2010).
Interaction Networks of Posttranslational Modifications
Proteins can be subject to various posttranslational modifications. Some of them target the same amino acid residues within a protein, but a cross-talk between the modifications is not necessarily restricted to direct mechanisms. The discovery of SUMO opened the doors to speculations that ubiquitination and SUMOylation act competitively regarding the targeted lysine residue and the fate of a protein. Indeed, some studies showed that proteins such as NF-кB, yeast PCNA, and SHMT1 were competitively modified by ubiquitin or SUMO (Desterro et al. 1998; Hoege et al. 2002; Anderson et al. 2012). Surprisingly, SUMOylation was identified as a targeting signal for ubiquitination and ubiquitin-dependent degradation (Uzunova et al. 2007; Cheng et al. 2007). The ubiquitinating enzymes were designated as STUbLs (Prudden et al. 2007). Several E3 ligases such as topoisomerase I binding arginine/serine-rich (TOPORS), MDM2, tripartite motif containing protein 27 (TRIM27), and ubiquitin-like with PHD and ring finger domains 2 (UHRF2) have dual functions as ubiquitin and SUMO E3 ligases, depending on the specific substrate (Weger et al. 2005; Chen and Chen 2003; Chu and Yang 2011; Oh and Chung 2013). A common perception especially in neurodegeneration suggested that failure of the proteasomal system leads to the accumulation of ubiquitinated species that might harm the cell, whereas SUMOylation may enhance protein solubility. However, inhibition of the proteasomal function also results in the accumulation of SUMOylated proteins, indicating that the interplay between SUMOylation, ubiquitination, and the UPS appears to be more complex than anticipated (Tatham et al. 2011; Kim et al. 2011).
Lysine residues of transcription factors can be posttranslationally modified by acetylation, a process known to modulate protein-protein interaction, enhance DNA binding, and regulate transactivation. SUMOylation of these transcription factors seems to play opposing roles as shown for p65. In addition, several enzymes of the HATs and HDAC families were shown to be modified by SUMO, thereby regulating their localization and activity (David et al. 2002; Kirsh et al. 2002; Girdwood et al. 2003; Cheng et al. 2004). Finally, acetylation has been suggested to enhance SUMOylation of histone H4 (Shiio and Eisenman 2003). Site-specific acetylation of SUMO paralogs (SUMO-1 at lysine 37 and SUMO-2 at lysine 33) was suggested as a regulatory principle in the control of SUMO–SIM interactions. Acetylation of SUMO prevents binding to several SIM-containing proteins such as PML, Daxx, and PIAS, thereby modulating SUMO-dependent transcriptional repression (Ullmann et al. 2012). A recent study reported that in vitro N-terminally acetylated α-synuclein primarily presents as monomeric protein and does not form dimers, tetramers, or higher molecular weight species. N-acetylated α-synuclein forms similar fibrils as the non-modified form but shows lower fibril assembly rates (Kang et al. 2012).
Phosphorylation attained interest through the identification of phosphorylation-dependent SUMOylation motifs (PDSMs) in proteins such as heat shock factors (HSFs) and MEF2A (Hietakangas et al. 2006). Similarly, the phosphorylation of proteins might be dependent on their SUMOylation status (Yao et al. 2011). Phosphorylation of serine residues flanking SIM-C of Daxx increased the local negative charge and thereby facilitated electrostatic interactions with a conserved lysine residue in the basic interface of SUMO. Moreover, SIM phosphorylation markedly enhanced the binding affinity of SUMO-1 over SUMO-2/SUMO-3, thus enabling SUMO-paralog-specific selectivity (Chang et al. 2011).
In conclusion, posttranslational modifications are likely to orchestrate the harmonic interplay of proteins and may act as molecular switches that determine the specific function of proteins at a given time and place. This applies to the physiological situation and may be even more important in diseases. Conversely, disharmony of interaction networks might account for cellular insult and cell death.
Vulnerability of Dopaminergic Cells
Many efforts were made to elucidate the cause for the specific vulnerability of dopaminergic neurons in PD. Moreover, similar to other neurodegenerative diseases, the idiopathic pathology initiates in a small region of the brain and spreads according to a predestined sequence to other areas (Braak et al. 2003). Dopamine neuron loss and depletion of the neurotransmitter dopamine are hallmarks of PD. However, many PD symptoms only manifest when at least 55–60 % of the dopaminergic neurons are gone. Dopamine is synthesized from the amino acid tyrosine, which is converted to L-dihydroxyphenylalanine (L-DOPA) by the enzyme TH, the rate-limiting enzyme for catecholamine biosynthesis. L-DOPA is further converted to dopamine by the enzyme DOPA decarboxylase (or aromatic amino acid decarboxylase), which is found in the cytoplasm. After neurotransmitter release, dopamine can be taken up from the synaptic cleft via the dopamine active transporter DAT. Dopamine can be metabolized by the monoamine oxidase, an enzyme located in the outer mitochondrial membrane.
TH is depleted in PD, and there is evidence of an increase in oxidative and inflammatory nigral environment (Jeong et al. 2006). Although rare, mutations of PD-associated proteins were helpful to unravel potential pathological mechanisms in the disease. Loss of function might lead to selective neurodegeneration of nigrostriatal dopaminergic neurons. PET neuroimaging in ante mortem studies demonstrated severe dopamine depletion and reduced dopamine uptake in homozygous DJ-1 mutation carriers (Dekker et al. 2004). DJ-1 is one candidate protein that seems to prevent the specific and slowly progressive loss of nigrostriatal dopamine neuron function, while the underlying molecular mechanisms still remain to be elucidated.
A very interesting study of Zhong et al. (2006) showed that DJ-1 transcriptionally up-regulates the human TH, whereas pyrimidine tract-binding protein-associated splicing factor (PSF) acts as a transcriptional repressor (Fig. 2). PSF is SUMOylated at K338, which facilitates the recruitment of HDAC1 and enables transcriptional repression. SUMO mutation of PSF as well as the presence of DJ-1 that prevents the posttranslational modification of PSF interrupts the interaction between PSF and HDAC1 and relieves the transcriptional repression of the TH promoter. Down-regulation of DJ-1 results in lower acetylation levels of TH-promoter-bound histones H2A, H2B, H3, and H4. A similar involvement was shown for the transcriptional up-regulation of MnSOD (Zhong and Xu 2008).

SUMOylation regulates transcriptional activity. TH is the rate-limiting enzyme for catecholamine synthesis. Both DJ-1 and PSF can bind to the TH promoter. SUMOylation of PSF facilitates the recruitment of HDAC1 and enables transcriptional repression of the TH promoter by PSF. DJ-1 prevents the SUMO modification of PSF and thereby interrupts the interaction between PSF and HDAC1. Subsequently, the transcriptional repression of the TH promoter is relieved. PSF: pyrimidine tract-binding protein-associated splicing factor; HDAC1 histone deacetylase 1, SUMO small ubiquitin-related modifier, TH tyrosine hydroxylase
The synaptic calcium-/calmodulin-dependent serine protein kinase (CASK) belongs to the membrane-associated guanylate kinase protein family (Hata et al. 1996). CASK functions as a multidomain scaffolding protein and interacts among others with several cell surface proteins, including amyloid precursor protein, neurexins, and syndecans. It regulates calcium/calmodulin serine/threonine kinase II (CaMKII), a central molecule in mechanisms of synaptic plasticity and memory (Malik et al. 2013). Conjugation of SUMO-1 to CASK promotes the dissociation of CASK from the membrane and reduces the interaction between CASK and protein 4.1. The function of protein 4.1 is to bind spectrin, a postsynaptic density protein required for synapse formation (Sytnyk et al. 2006). The interaction between CASK and protein 4.1 is required for CASK function in spinogenesis. Overexpression of a CASK–SUMO-1 fusion construct, which mimics CASK SUMOylation, impairs spine formation (Chao et al. 2008). CASK interacts with parkin but is not a substrate for parkin-mediated ubiquitination. The authors speculated that CASK may be involved in trafficking parkin to the appropriate subcellular compartments (Fallon et al. 2002).
Ras homolog enriched in striatum (Rhes) is a small GTP binding protein whose expression is highly enriched in striatum. Signaling by 7 transmembrane receptors through heterotrimeric G proteins is inhibited by Rhes. Rhes is involved in cAMP/protein kinase A signaling pathway. Rhes is required for a correct dopamine-mediated GTP binding, a function mainly associated with the stimulation of dopamine D2 receptors, indicating that Rhes is an important modulator of dopaminergic transmission in the striatum (Errico et al. 2008). Rhes is a physiological regulator of SUMOylation. Rhes binds directly to both E1 and Ubc9, enhancing cross-SUMOylation as well as thioester transfer from E1 to Ubc9 and is therefore considered to act as E3 ligase for the attachment of SUMO (Subramaniam et al. 2010). Incidentally, Rhes induces the SUMOylation of mutant huntingtin, which leads to enhanced cytotoxicity (Subramaniam et al. 2009).
Parkin-associated endothelin-receptor-like receptor (Pael-R)/GPR37 is a G-protein-coupled orphan receptor that is highly expressed in dopaminergic neurons in the substantia nigra and accumulates in LBs (Murakami et al. 2004). The membrane protein Pael-R is a substrate for parkin. Accumulation of Pael-R in the ER results in ER stress-induced death of dopaminergic neurons. Correct folding of Pael-R protein is difficult, and approximately half of newly synthesized Pael-R becomes misfolded and is ubiquitinated by parkin and degraded via the ubiquitin–proteasome system (Imai et al. 2001; Kitao et al. 2007).
PD Models and Neurotoxins
Milestones in PD were the identification of neurotoxins such as MPTP, 6-hydroxydopamine (6-OHDA), rotenone, and N,N′-dimethyl-4,4′-bipyridinium dichloride (trade name paraquat), which induce Parkinson-like syndromes. Neurotoxins are widely used in animal studies to selectively mimic Parkinsonian mechanisms in dopaminergic neurons. These models serve to identify key players in pathological mechanisms and to investigate the specific role of PD-associated proteins.
MPTP appeared as a lipophilic by-product of an analog of the narcotic meperidine (Davis et al. 1979; Langston et al. 1983). The toxicity of MPTP is due to its metabolite MPP+, formed in glial cells in the reaction catalyzed by the mitochondrial monoamine oxidase B. When taken up by dopaminergic neurons, MPP+ accumulates in mitochondria and inhibits complex I (Sayre 1989). Cellular damage caused by MPP+ is primarily due to inhibition of NAD-linked mitochondrial respiration (Javitch et al. 1985) and energy depletion and secondarily by ROS production and oxidative insult (Cleeter et al. 1992). DJ-1 knockout mice do not show overt neurodegeneration under basal conditions (Andres-Mateos et al. 2007; R. H. Kim et al. 2005). However, additional MPTP treatment greatly enhanced the loss of nigral dopaminergic neurons concomitant with striatal denervation. Protective effects of overexpressed DJ-1 are noteworthy and not restricted to dopaminergic neurotoxicity and seem to act more generally against oxidative stress (R. H. Kim et al. 2005).
6-OHDA shares structural similarities with dopamine and noradrenaline and therefore can be taken up by both the dopamine (DAT) and the noradrenaline transporters (NET). 6-OHDA poorly crosses the blood–brain barrier and has to be stereotactically injected into the substantia nigra, the medial forebrain bundle (nigrostriatal tract), or the striatum.
The neurotoxin rotenone is naturally occurring in plant roots and was used as an insecticide, piscicide, and pesticide. Unlike MPP+ or 6-OHDA, rotenone is extremely lipophilic, by which it freely crosses the blood–brain barrier and biological membranes, thus reaching the brain rapidly. Rotenone inhibits mitochondrial complex I and causes oxidative insult via the formation of intracellular ROS. Strikingly, in vivo rotenone administrations to rodents have mimicked typical histopathological features of PD, including the formation of LBs (Sherer et al. 2003). Heikkila et al. (1985) reported a substantial depletion of striatal dopamine and its metabolites after acute stereotaxic injection of rotenone into the median forebrain bundle of rats. In a Drosophila model, chronic exposure to sublethal doses of rotenone is accompanied by the selective loss of dopaminergic neurons and locomotor deficits (Sherer et al. 2002). SUMO immunoreactivity was found within and surrounding glial inclusion bodies in atypical PD such as multiple-system atrophy and progressive supranuclear palsy. SUMO-1 showed punctate co-localization with the lysosomal marker cathepsin D in affected brain regions. Association of SUMO-1 with lysosomes was also detected in glial cells bearing α-synuclein aggregates in a rotenone-lesioned rat model, suggesting a role for SUMO-1 in lysosome function (Wong et al. 2013). Increased levels of SUMO-1 and α-synuclein-specific species ranging from 12 to 190 kDa were observed in the lesioned brain hemisphere in a unilateral rotenone-lesioned mouse model, with 6–12-month-old mice showing proportionately greater increases in SUMO-1 than mice older than 21 months (Weetman et al. 2013).
Today, paraquat is among the most commonly used herbicides worldwide. It is possibly taken up into the brain by the neutral amino acid transport system and then transported into cells in an Na+-dependent manner (Shimizu et al. 2001). Paraquat is an inhibitor of complex I that undergoes redox cycling, being reduced and oxidized to produce superoxide, a major ROS (Bus and Gibson 1984). Paraquat-induced toxicity has been linked to Parkinson-like neurodegenerative phenotype in mice, rats, and primates (Betarbet et al. 2000). Drosophila melanogaster as a model organism has proven to be invaluable to dissect molecular determinants of PD-associated protein function (Botella et al. 2009). As in knockout mice and zebrafish, DJ-1 deletions in D. melanogaster did not lead to dopaminergic neurodegeneration under basal conditions. However, DJ-1 knockout flies were specifically sensitized to neurotoxin-mediated oxidative stress and have been very informative for the elucidation of the role of DJ-1 in the antioxidative defense mechanisms implicated in PD (Meulener et al. 2005; Menzies et al. 2005).
Many Pieces: Few Conjunctions
Hundreds of SUMOylated substrates were identified since the discovery of SUMO as a ubiquitin-like posttranslational modification. Several mechanisms were described and a variety of diverse SUMO functions were revealed that ultimately distinguish this modification from ubiquitination.
However, our knowledge is only in part. Single pieces of the whole were investigated, but more is needed to fit them together. In some cases, bridging parts are still missing. For this reason, it is important to search for other SUMO targets or SUMOylating enzymes. Due to the subtle and highly dynamic character of SUMOylation, the identification of new targets turned out to be difficult. Basic approaches are essential and were already introduced in order to identify endogenous SUMO substrates (Tirard et al. 2012; Becker et al. 2013; Lamoliatte et al. 2013). In some cases, it is known that SUMOylation is “somehow” influencing cellular processes, but the exact mechanisms are difficult to comprehend. Therefore, SUMOylated proteins need to be further characterized in terms of SUMOylation sites and molecular consequences. Here, the use of SUMO-defective mutants might be helpful. Although the pathology in PD and other neurodegenerative diseases is rather complex, the development of simple models and organisms may be advantageous to dissect molecular pathways. Likewise, these models can serve to specifically target enzymes of the SUMO pathway such as E3 ligases and SUMO isopeptidases in therapeutical approaches (Yang et al. 2013). To find the missing pieces to the puzzle might require thinking “outside the box”. In earlier times, aggregates were considered as a principal disease-causing species, whereas at present, oligomer species take the center stage. LBs were recognized as defining features of PD and DLB but lack in rare cases of PD. The long-held dogma that α-synuclein, but not β-synuclein, is toxic to cells needs to be questioned in a similar way (Taschenberger et al. 2013).
Conclusion
α-Synuclein, DJ-1, and parkin, three representatives of typically PD-associated proteins, are modified by SUMO. The consequences of SUMOylation for each protein seem to be quite diverse. SUMOylation of α-synuclein might modulate the solubility and toxicity of the protein although possible mechanisms remain elusive due to an indistinct α-synuclein function. The multifunctional character of the DJ-1 protein might be causal for the diverse aspects of DJ-1 SUMOylation. SUMOylation appears to be supportive for DJ-1 activity and cytoprotective function as a molecular chaperone and transcriptional regulator. SUMOylation of parkin shifts the equilibrium toward autoubiquitination and nuclear shuttling of parkin. Whether SUMOylation is intended to block parkin function as a major player in mitochondrial quality control or to support parkin function in transcriptional regulation remains to be answered.
The impact of SUMOylation on protein aggregation is controversially discussed. Moreover, aggregation might be beneficial or detrimental for the cells, as suggested for α-synuclein or huntingtin, respectively (Krumova et al. 2011; Steffan et al. 2004). Lastly, SUMO interferes with ubiquitin and the UPS. This suggests that SUMO may act as regulatory switch in concert with ubiquitin depending on the cellular context.
Apart from its direct modification of disease-associated proteins, SUMO is involved in many other cellular processes. In general, cellular stress results in increased levels of SUMO and SUMOylated species. Therefore, SUMOylation might be involved in stress response as well as mitochondrial homeostasis. SUMO increased mitochondrial fragmentation, whereas rather protective players such as DJ-1, PINK1, and parkin counteracted fission.
SUMO is an important regulator of transcriptional activity in several ways. In the cytosol, SUMO interfered with several members of signaling pathways such as HIF-1α, IKK/NEMO, IкBα, and Daxx, resulting in degradation, stabilization, or translocation of the targeted proteins. Nuclear regulation of SUMO included the modulation of transcription factor activity or recruitment of HDACs. The consequences were shown to be very diverse, either promoting cell survival or inducing apoptosis. Therefore, SUMOylation might provide regulatory mechanisms for the fine-tuning of cellular processes in order to enable a dynamic response to various stress stimuli. The ability of SUMO to modulate transcriptional activity applied also to regulatory elements of the TH promoter. Therefore, research on SUMO targets and function is of particular interest to answer to the vulnerability of dopaminergic cells in PD.
References
Abbas, N., Lucking, C. B., Ricard, S., Durr, A., Bonifati, V., De Michele, G., et al. (1999). A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Human Molecular Genetics, 8(4), 567–574.
Akhtar, M. W., Sunico, C. R., Nakamura, T., & Lipton, S. A. (2012). Redox regulation of protein function via cysteine S-Nitrosylation and its relevance to neurodegenerative diseases. International Journal of Cell Biology, 2012, 463756.
Alves da Costa, C., & Checler, F. (2011). Apoptosis in Parkinson’s disease: Is p53 the missing link between genetic and sporadic Parkinsonism? Cellular Signalling, 23(6), 963–968.
Anderson, D. D., Eom, J. Y., & Stover, P. J. (2012). Competition between sumoylation and ubiquitination of serine hydroxymethyltransferase 1 determines its nuclear localization and its accumulation in the nucleus. The Journal of Biological Chemistry, 287(7), 4790–4799.
Andres-Mateos, E., Perier, C., Zhang, L., Blanchard-Fillion, B., Greco, T. M., Thomas, B., et al. (2007). DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proceedings of the National Academy of Sciences of the United States of America, 104(37), 14807–14812.
Bandopadhyay, R., Kingsbury, A. E., Cookson, M. R., Reid, A. R., Evans, I. M., Hope, A. D., et al. (2004). The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain: A Journal of Neurology, 127(Pt 2), 420–430.
Bandopadhyay, R., Kingsbury, A. E., Muqit, M. M., Harvey, K., Reid, A. R., Kilford, L., et al. (2005). Synphilin-1 and parkin show overlapping expression patterns in human brain and form aggresomes in response to proteasomal inhibition. Neurobiology of Disease, 20(2), 401–411.
Becker, J., Barysch, S. V., Karaca, S., Dittner, C., Hsiao, H. H., Berriel Diaz, M., et al. (2013). Detecting endogenous SUMO targets in mammalian cells and tissues. Nature Structural & Molecular Biology, 20(4), 525–531.
Becker, D., Richter, J., Tocilescu, M. A., Przedborski, S., & Voos, W. (2012). Pink1 kinase and its membrane potential (Deltapsi)-dependent cleavage product both localize to outer mitochondrial membrane by unique targeting mode. The Journal of Biological Chemistry, 287(27), 22969–22987.
Beilina, A., Van Der Brug, M., Ahmad, R., Kesavapany, S., Miller, D. W., Petsko, G. A., et al. (2005). Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proceedings of the National Academy of Sciences of the United States of America, 102(16), 5703–5708.
Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V., & Greenamyre, J. T. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nature Neuroscience, 3(12), 1301–1306.
Beyer, K. (2006). Alpha-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathologica, 112(3), 237–251.
Beyer, K., Domingo-Sabat, M., Humbert, J., Carrato, C., Ferrer, I., & Ariza, A. (2008). Differential expression of alpha-synuclein, parkin, and synphilin-1 isoforms in Lewy body disease. Neurogenetics, 9(3), 163–172.
Blackinton, J. G., Anvret, A., Beilina, A., Olson, L., Cookson, M. R., & Galter, D. (2007). Expression of PINK1 mRNA in human and rodent brain and in Parkinson’s disease. Brain Research, 1184, 10–16.
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science, 299(5604), 256–259.
Bossis, G., & Melchior, F. (2006). Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Molecular Cell, 21(3), 349–357.
Botella, J. A., Bayersdorfer, F., Gmeiner, F., & Schneuwly, S. (2009). Modelling Parkinson’s disease in Drosophila. NeuroMolecular Medicine, 11(4), 268–280.
Braak, H., Del Tredici, K., Rub, U., de Vos, R. A., Jansen Steur, E. N., & Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of Aging, 24(2), 197–211.
Bretaud, S., Allen, C., Ingham, P. W., & Bandmann, O. (2007). p53-dependent neuronal cell death in a DJ-1-deficient zebrafish model of Parkinson’s disease. Journal of Neurochemistry, 100(6), 1626–1635.
Bus, J. S., & Gibson, J. E. (1984). Paraquat: Model for oxidant-initiated toxicity. Environmental Health Perspectives, 55, 37–46.
Buschmann, T., Lerner, D., Lee, C. G., & Ronai, Z. (2001). The Mdm-2 amino terminus is required for Mdm2 binding and SUMO-1 conjugation by the E2 SUMO-1 conjugating enzyme Ubc9. The Journal of Biological Chemistry, 276(44), 40389–40395.
Canet-Aviles, R. M., Wilson, M. A., Miller, D. W., Ahmad, R., McLendon, C., Bandyopadhyay, S., et al. (2004). The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proceedings of the National Academy of Sciences of the United States of America, 101(24), 9103–9108.
Chandra, S., Fornai, F., Kwon, H. B., Yazdani, U., Atasoy, D., Liu, X., et al. (2004). Double-knockout mice for alpha- and beta-synucleins: Effect on synaptic functions. Proceedings of the National Academy of Sciences of the United States of America, 101(41), 14966–14971.
Chandra, S., Gallardo, G., Fernandez-Chacon, R., Schluter, O. M., & Sudhof, T. C. (2005). Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell, 123(3), 383–396.
Chang, C. C., Lin, D. Y., Fang, H. I., Chen, R. H., & Shih, H. M. (2005). Daxx mediates the small ubiquitin-like modifier-dependent transcriptional repression of Smad4. The Journal of Biological Chemistry, 280(11), 10164–10173.
Chang, C. C., Naik, M. T., Huang, Y. S., Jeng, J. C., Liao, P. H., Kuo, H. Y., et al. (2011). Structural and functional roles of Daxx SIM phosphorylation in SUMO paralog-selective binding and apoptosis modulation. Molecular Cell, 42(1), 62–74.
Chang, H. Y., Nishitoh, H., Yang, X., Ichijo, H., & Baltimore, D. (1998). Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science, 281(5384), 1860–1863.
Chao, H. W., Hong, C. J., Huang, T. N., Lin, Y. L., & Hsueh, Y. P. (2008). SUMOylation of the MAGUK protein CASK regulates dendritic spinogenesis. The Journal of Cell Biology, 182(1), 141–155.
Chartier-Harlin, M. C., Kachergus, J., Roumier, C., Mouroux, V., Douay, X., Lincoln, S., et al. (2004). Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet, 364(9440), 1167–1169.
Chen, H., & Chan, D. C. (2009). Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Human molecular genetics, 18(R2), R169–176.
Chen, L., & Chen, J. (2003). MDM2-ARF complex regulates p53 sumoylation. Oncogene, 22(34), 5348–5357.
Chen, D., Gao, F., Li, B., Wang, H., Xu, Y., Zhu, C., et al. (2010a). Parkin mono-ubiquitinates Bcl-2 and regulates autophagy. The Journal of Biological Chemistry, 285(49), 38214–38223.
Chen, J., Li, L., & Chin, L. S. (2010b). Parkinson disease protein DJ-1 converts from a zymogen to a protease by carboxyl-terminal cleavage. Human Molecular Genetics, 19(12), 2395–2408.
Chen, J., Lin, J., & Levine, A. J. (1995). Regulation of transcription functions of the p53 tumor suppressor by the mdm-2 oncogene. Molecular Medicine, 1(2), 142–152.
Chen, A., Wang, P. Y., Yang, Y. C., Huang, Y. H., Yeh, J. J., Chou, Y. H., et al. (2006). SUMO regulates the cytoplasmonuclear transport of its target protein Daxx. Journal of Cellular Biochemistry, 98(4), 895–911.
Cheng, J., Kang, X., Zhang, S., & Yeh, E. T. (2007). SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell, 131(3), 584–595.
Cheng, J., Wang, D., Wang, Z., & Yeh, E. T. (2004). SENP1 enhances androgen receptor-dependent transcription through desumoylation of histone deacetylase 1. Molecular and Cellular Biology, 24(13), 6021–6028.
Choi, S. J., Chung, S. S., Rho, E. J., Lee, H. W., Lee, M. H., Choi, H. S., et al. (2006). Negative modulation of RXRalpha transcriptional activity by small ubiquitin-related modifier (SUMO) modification and its reversal by SUMO-specific protease SUSP1. The Journal of Biological Chemistry, 281(41), 30669–30677.
Choi, P., Ostrerova-Golts, N., Sparkman, D., Cochran, E., Lee, J. M., & Wolozin, B. (2000). Parkin is metabolized by the ubiquitin/proteosome system. NeuroReport, 11(12), 2635–2638.
Chu, Y., & Yang, X. (2011). SUMO E3 ligase activity of TRIM proteins. Oncogene, 30(9), 1108–1116.
Chung, S. S., Ahn, B. Y., Kim, M., Kho, J. H., Jung, H. S., & Park, K. S. (2011). SUMO modification selectively regulates transcriptional activity of peroxisome-proliferator-activated receptor gamma in C2C12 myotubes. The Biochemical Journal, 433(1), 155–161.
Chung, K. K., Zhang, Y., Lim, K. L., Tanaka, Y., Huang, H., Gao, J., et al. (2001). Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: Implications for Lewy-body formation in Parkinson disease. Nature Medicine, 7(10), 1144–1150.
Cleeter, M. W., Cooper, J. M., & Schapira, A. H. (1992). Irreversible inhibition of mitochondrial complex I by 1-methyl-4-phenylpyridinium: Evidence for free radical involvement. Journal of Neurochemistry, 58(2), 786–789.
Cookson, M. R., Lockhart, P. J., McLendon, C., O’Farrell, C., Schlossmacher, M., & Farrer, M. J. (2003). RING finger 1 mutations in Parkin produce altered localization of the protein. Human Molecular Genetics, 12(22), 2957–2965.
Croxton, R., Puto, L. A., de Belle, I., Thomas, M., Torii, S., Hanaii, F., et al. (2006). Daxx represses expression of a subset of antiapoptotic genes regulated by nuclear factor-kappaB. Cancer Research, 66(18), 9026–9035.
Cuervo, A. M., Wong, E. S., & Martinez-Vicente, M. (2010). Protein degradation, aggregation, and misfolding. Movement Disorders, 25(Suppl 1), S49–S54.
Dagata, V., & Cavallaro, S. (2004). Parkin transcript variants in rat and human brain. Neurochemical Research, 29(9), 1715–1724.
David, G., Neptune, M. A., & DePinho, R. A. (2002). SUMO-1 modification of histone deacetylase 1 (HDAC1) modulates its biological activities. The Journal of Biological Chemistry, 277(26), 23658–23663.
Davidson, B., Hadar, R., Schlossberg, A., Sternlicht, T., Slipicevic, A., Skrede, M., et al. (2008). Expression and clinical role of DJ-1, a negative regulator of PTEN, in ovarian carcinoma. Human Pathology, 39(1), 87–95.
Davis, G. C., Williams, A. C., Markey, S. P., Ebert, M. H., Caine, E. D., Reichert, C. M., et al. (1979). Chronic Parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Research, 1(3), 249–254.
de la Vega, L., Grishina, I., Moreno, R., Kruger, M., Braun, T., & Schmitz, M. L. (2012). A redox-regulated SUMO/acetylation switch of HIPK2 controls the survival threshold to oxidative stress. Molecular Cell, 46(4), 472–483.
Dedmon, M. M., Christodoulou, J., Wilson, M. R., & Dobson, C. M. (2005). Heat shock protein 70 inhibits alpha-synuclein fibril formation via preferential binding to prefibrillar species. The Journal of Biological Chemistry, 280(15), 14733–14740.
Deeg, S., Gralle, M., Sroka, K., Bahr, M., Wouters, F. S., & Kermer, P. (2010). BAG1 restores formation of functional DJ-1 L166P dimers and DJ-1 chaperone activity. The Journal of Cell Biology, 188(4), 505–513.
Dekker, M. C., Eshuis, S. A., Maguire, R. P., Veenma-van der Duijn, L., Pruim, J., Snijders, P. J., et al. (2004). PET neuroimaging and mutations in the DJ-1 gene. Journal of Neural Transmission, 111(12), 1575–1581.
Desterro, J. M., Rodriguez, M. S., & Hay, R. T. (1998). SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Molecular Cell, 2(2), 233–239.
Dorval, V., & Fraser, P. E. (2006). Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. The Journal of Biological Chemistry, 281(15), 9919–9924.
Doss-Pepe, E. W., Chen, L., & Madura, K. (2005). Alpha-synuclein and parkin contribute to the assembly of ubiquitin lysine 63-linked multiubiquitin chains. The Journal of Biological Chemistry, 280(17), 16619–16624.
Elfferich, P., Verleun-Mooijman, M. C., Maat-Kievit, J. A., van de Warrenburg, B. P., Abdo, W. F., Eshuis, S. A., et al. (2011). Breakpoint mapping of 13 large parkin deletions/duplications reveals an exon 4 deletion and an exon 7 duplication as founder mutations. Neurogenetics, 12(4), 263–271.
Engelender, S., Kaminsky, Z., Guo, X., Sharp, A. H., Amaravi, R. K., Kleiderlein, J. J., et al. (1999). Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nature Genetics, 22(1), 110–114.
Errico, F., Santini, E., Migliarini, S., Borgkvist, A., Centonze, D., Nasti, V., et al. (2008). The GTP-binding protein Rhes modulates dopamine signalling in striatal medium spiny neurons. Molecular and Cellular Neurosciences, 37(2), 335–345.
Escobar-Cabrera, E., Okon, M., Lau, D. K., Dart, C. F., Bonvin, A. M., & McIntosh, L. P. (2011). Characterizing the N- and C-terminal Small ubiquitin-like modifier (SUMO)-interacting motifs of the scaffold protein DAXX. The Journal of Biological Chemistry, 286(22), 19816–19829.
Fallon, L., Moreau, F., Croft, B. G., Labib, N., Gu, W. J., & Fon, E. A. (2002). Parkin and CASK/LIN-2 associate via a PDZ-mediated interaction and are co-localized in lipid rafts and postsynaptic densities in brain. The Journal of Biological Chemistry, 277(1), 486–491.
Farrer, M., Gwinn-Hardy, K., Muenter, M., DeVrieze, F. W., Crook, R., Perez-Tur, J., et al. (1999). A chromosome 4p haplotype segregating with Parkinson’s disease and postural tremor. Human Molecular Genetics, 8(1), 81–85.
Figueroa-Romero, C., Iniguez-Lluhi, J. A., Stadler, J., Chang, C. R., Arnoult, D., Keller, P. J., et al. (2009). SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, 23(11), 3917–3927.
Foti, R., Zucchelli, S., Biagioli, M., Roncaglia, P., Vilotti, S., Calligaris, R., et al. (2010). Parkinson disease-associated DJ-1 is required for the expression of the glial cell line-derived neurotrophic factor receptor RET in human neuroblastoma cells. The Journal of Biological Chemistry, 285(24), 18565–18574.
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nature Cell Biology, 4(2), 160–164.
Galter, D., Westerlund, M., Belin, A. C., & Olson, L. (2007). DJ-1 and UCH-L1 gene activity patterns in the brains of controls, Parkinson and schizophrenia patients and in rodents. Physiology & Behavior, 92(1–2), 46–53.
Geisler, S., Holmstrom, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature Cell Biology, 12(2), 119–131.
Geiss-Friedlander, R., & Melchior, F. (2007). Concepts in sumoylation: A decade on. Nature Reviews Molecular Cell Biology, 8(12), 947–956.
Giasson, B. I., Duda, J. E., Murray, I. V., Chen, Q., Souza, J. M., Hurtig, H. I., et al. (2000). Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science, 290(5493), 985–989.
Girdwood, D., Bumpass, D., Vaughan, O. A., Thain, A., Anderson, L. A., Snowden, A. W., et al. (2003). P300 transcriptional repression is mediated by SUMO modification. Molecular Cell, 11(4), 1043–1054.
Goedert, M., Spillantini, M. G., Del Tredici, K., & Braak, H. (2013). 100 years of Lewy pathology. Nature Reviews. Neurology, 9(1), 13–24.
Gorner, K., Holtorf, E., Waak, J., Pham, T. T., Vogt-Weisenhorn, D. M., Wurst, W., et al. (2007). Structural determinants of the C-terminal helix-kink-helix motif essential for protein stability and survival promoting activity of DJ-1. The Journal of Biological Chemistry, 282(18), 13680–13691.
Gosavi, N., Lee, H. J., Lee, J. S., Patel, S., & Lee, S. J. (2002). Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. The Journal of Biological Chemistry, 277(50), 48984–48992.
Gostissa, M., Hengstermann, A., Fogal, V., Sandy, P., Schwarz, S. E., Scheffner, M., et al. (1999). Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. The EMBO Journal, 18(22), 6462–6471.
Greene, A. W., Grenier, K., Aguileta, M. A., Muise, S., Farazifard, R., Haque, M. E., et al. (2012). Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Reports, 13(4), 378–385.
Harder, Z., Zunino, R., & McBride, H. (2004). Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Current Biology: CB, 14(4), 340–345.
Hata, Y., Butz, S., & Sudhof, T. C. (1996). CASK: A novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 16(8), 2488–2494.
Hattori, N., Kitada, T., Matsumine, H., Asakawa, S., Yamamura, Y., Yoshino, H., et al. (1998). Molecular genetic analysis of a novel Parkin gene in Japanese families with autosomal recessive juvenile parkinsonism: Evidence for variable homozygous deletions in the Parkin gene in affected individuals. Annals of Neurology, 44(6), 935–941.
Haupt, Y., Maya, R., Kazaz, A., & Oren, M. (1997). Mdm2 promotes the rapid degradation of p53. Nature, 387(6630), 296–299.
Hauser, D. N., & Hastings, T. G. (2013). Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic parkinsonism. Neurobiology of disease, 51, 35–42.
Hecker, C. M., Rabiller, M., Haglund, K., Bayer, P., & Dikic, I. (2006). Specification of SUMO1- and SUMO2-interacting motifs. The Journal of Biological Chemistry, 281(23), 16117–16127.
Heikkila, R. E., Nicklas, W. J., Vyas, I., & Duvoisin, R. C. (1985). Dopaminergic toxicity of rotenone and the 1-methyl-4-phenylpyridinium ion after their stereotaxic administration to rats: Implication for the mechanism of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity. Neuroscience Letters, 62(3), 389–394.
Henn, I. H., Bouman, L., Schlehe, J. S., Schlierf, A., Schramm, J. E., Wegener, E., et al. (2007). Parkin mediates neuroprotection through activation of IkappaB kinase/nuclear factor-kappaB signaling. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 27(8), 1868–1878.
Hietakangas, V., Anckar, J., Blomster, H. A., Fujimoto, M., Palvimo, J. J., Nakai, A., et al. (2006). PDSM, a motif for phosphorylation-dependent SUMO modification. Proceedings of the National Academy of Sciences of the United States of America, 103(1), 45–50.
Hod, Y., Pentyala, S. N., Whyard, T. C., & El-Maghrabi, M. R. (1999). Identification and characterization of a novel protein that regulates RNA-protein interaction. Journal of Cellular Biochemistry, 72(3), 435–444.
Hoege, C., Pfander, B., Moldovan, G. L., Pyrowolakis, G., & Jentsch, S. (2002). RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature, 419(6903), 135–141.
Honda, R., Tanaka, H., & Yasuda, H. (1997). Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Letters, 420(1), 25–27.
Huang, J., Yan, J., Zhang, J., Zhu, S., Wang, Y., Shi, T., et al. (2012). SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nature Communications, 3, 911.
Hwang, S., Song, S., Hong, Y. K., Choi, G., Suh, Y. S., Han, S. Y., et al. (2013). Drosophila DJ-1 decreases neural sensitivity to stress by negatively regulating Daxx-like protein through dFOXO. PLoS Genetics, 9(4), e1003412.
Imai, Y., Soda, M., Inoue, H., Hattori, N., Mizuno, Y., & Takahashi, R. (2001). An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell, 105(7), 891–902.
Ishov, A. M., Sotnikov, A. G., Negorev, D., Vladimirova, O. V., Neff, N., Kamitani, T., et al. (1999). PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. The Journal of Cell Biology, 147(2), 221–234.
Jakes, R., Spillantini, M. G., & Goedert, M. (1994). Identification of two distinct synucleins from human brain. FEBS Letters, 345(1), 27–32.
Jang, M. S., Ryu, S. W., & Kim, E. (2002). Modification of Daxx by small ubiquitin-related modifier-1. Biochemical and Biophysical Research Communications, 295(2), 495–500.
Jang, H. D., Yoon, K., Shin, Y. J., Kim, J., & Lee, S. Y. (2004). PIAS3 suppresses NF-kappaB-mediated transcription by interacting with the p65/RelA subunit. The Journal of Biological Chemistry, 279(23), 24873–24880.
Javitch, J. A., D’Amato, R. J., Strittmatter, S. M., & Snyder, S. H. (1985). Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: Uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proceedings of the National Academy of Sciences of the United States of America, 82(7), 2173–2177.
Jeong, H., Kim, M. S., Kwon, J., Kim, K. S., & Seol, W. (2006). Regulation of the transcriptional activity of the tyrosine hydroxylase gene by androgen receptor. Neuroscience Letters, 396(1), 57–61.
Jiang, Q., Ren, Y., & Feng, J. (2008). Direct binding with histone deacetylase 6 mediates the reversible recruitment of parkin to the centrosome. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 28(48), 12993–13002.
Junn, E., Taniguchi, H., Jeong, B. S., Zhao, X., Ichijo, H., & Mouradian, M. M. (2005). Interaction of DJ-1 with Daxx inhibits apoptosis signal-regulating kinase 1 activity and cell death. Proceedings of the National Academy of Sciences of the United States of America, 102(27), 9691–9696.
Kalia, L. V., Kalia, S. K., McLean, P. J., Lozano, A. M., & Lang, A. E. (2013). alpha-Synuclein oligomers and clinical implications for Parkinson disease. Annals of Neurology, 73(2), 155–169.
Kang, L., Moriarty, G. M., Woods, L. A., Ashcroft, A. E., Radford, S. E., & Baum, J. (2012). N-terminal acetylation of alpha-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Science: A Publication of the Protein Society, 21(7), 911–917.
Kawamata, H., McLean, P. J., Sharma, N., & Hyman, B. T. (2001). Interaction of alpha-synuclein and synphilin-1: Effect of Parkinson’s disease-associated mutations. Journal of Neurochemistry, 77(3), 929–934.
Khelifi, A. F., D’Alcontres, M. S., & Salomoni, P. (2005). Daxx is required for stress-induced cell death and JNK activation. Cell Death and Differentiation, 12(7), 724–733.
Kim, Y. M., Jang, W. H., Quezado, M. M., Oh, Y., Chung, K. C., Junn, E., et al. (2011). Proteasome inhibition induces alpha-synuclein SUMOylation and aggregate formation. Journal of the Neurological Sciences, 307(1–2), 157–161.
Kim, R. H., Peters, M., Jang, Y., Shi, W., Pintilie, M., Fletcher, G. C., et al. (2005). DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell, 7(3), 263–273.
Kim, J. M., Shin, H. I., Cha, S. S., Lee, C. S., Hong, B. S., Lim, S., et al. (2012). DJ-1 promotes angiogenesis and osteogenesis by activating FGF receptor-1 signaling. Nature Communications, 3, 1296.
Kinumi, T., Kimata, J., Taira, T., Ariga, H., & Niki, E. (2004). Cysteine-106 of DJ-1 is the most sensitive cysteine residue to hydrogen peroxide-mediated oxidation in vivo in human umbilical vein endothelial cells. Biochemical and Biophysical Research Communications, 317(3), 722–728.
Kirsh, O., Seeler, J. S., Pichler, A., Gast, A., Muller, S., Miska, E., et al. (2002). The SUMO E3 ligase RanBP2 promotes modification of the HDAC4 deacetylase. The EMBO Journal, 21(11), 2682–2691.
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature, 392(6676), 605–608.
Kitada, T., Asakawa, S., Minoshima, S., Mizuno, Y., & Shimizu, N. (2000). Molecular cloning, gene expression, and identification of a splicing variant of the mouse parkin gene. Mammalian Genome: Official Journal of the International Mammalian Genome Society, 11(6), 417–421.
Kitao, Y., Imai, Y., Ozawa, K., Kataoka, A., Ikeda, T., Soda, M., et al. (2007). Pael receptor induces death of dopaminergic neurons in the substantia nigra via endoplasmic reticulum stress and dopamine toxicity, which is enhanced under condition of parkin inactivation. Human Molecular Genetics, 16(1), 50–60.
Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., et al. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nature Genetics, 18(2), 106–108.
Krumova, P., Meulmeester, E., Garrido, M., Tirard, M., Hsiao, H. H., Bossis, G., et al. (2011). Sumoylation inhibits alpha-synuclein aggregation and toxicity. The Journal of Cell Biology, 194(1), 49–60.
Kubbutat, M. H., Jones, S. N., & Vousden, K. H. (1997). Regulation of p53 stability by Mdm2. Nature, 387(6630), 299–303.
Kubo, S. I., Kitami, T., Noda, S., Shimura, H., Uchiyama, Y., Asakawa, S., et al. (2001). Parkin is associated with cellular vesicles. Journal of Neurochemistry, 78(1), 42–54.
Kumaran, R., Vandrovcova, J., Luk, C., Sharma, S., Renton, A., Wood, N. W., et al. (2009). Differential DJ-1 gene expression in Parkinson’s disease. Neurobiology of Disease, 36(2), 393–400.
Kuo, H. Y., Chang, C. C., Jeng, J. C., Hu, H. M., Lin, D. Y., Maul, G. G., et al. (2005). SUMO modification negatively modulates the transcriptional activity of CREB-binding protein via the recruitment of Daxx. Proceedings of the National Academy of Sciences of the United States of America, 102(47), 16973–16978.
Lamoliatte, F., Bonneil, E., Durette, C., Caron-Lizotte, O., Wildemann, D., Zerweck, J., et al. (2013). Targeted identification of SUMOylation sites in human proteins using affinity enrichment and paralog-specific reporter ions. Molecular & cellular proteomics: MCP
Langston, J. W., Ballard, P., Tetrud, J. W., & Irwin, I. (1983). Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science, 219(4587), 979–980.
Lee, M. H., Lee, S. W., Lee, E. J., Choi, S. J., Chung, S. S., Lee, J. I., et al. (2006). SUMO-specific protease SUSP4 positively regulates p53 by promoting Mdm2 self-ubiquitination. Nature Cell Biology, 8(12), 1424–1431.
Lehembre, F., Muller, S., Pandolfi, P. P., & Dejean, A. (2001). Regulation of Pax3 transcriptional activity by SUMO-1-modified PML. Oncogene, 20(1), 1–9.
Leroy, E., Anastasopoulos, D., Konitsiotis, S., Lavedan, C., & Polymeropoulos, M. H. (1998). Deletions in the Parkin gene and genetic heterogeneity in a Greek family with early onset Parkinson’s disease. Human Genetics, 103(4), 424–427.
Li, H., Leo, C., Zhu, J., Wu, X., O’Neil, J., Park, E. J., et al. (2000). Sequestration and inhibition of Daxx-mediated transcriptional repression by PML. Molecular and Cellular Biology, 20(5), 1784–1796.
Li, H. M., Niki, T., Taira, T., Iguchi-Ariga, S. M., & Ariga, H. (2005). Association of DJ-1 with chaperones and enhanced association and colocalization with mitochondrial Hsp70 by oxidative stress. Free Radical Research, 39(10), 1091–1099.
Li, R., Wei, J., Jiang, C., Liu, D., Deng, L., Zhang, K., et al. (2013). Akt SUMOylation regulates cell proliferation and tumorigenesis. Cancer research.
Lim, K. L., Chew, K. C., Tan, J. M., Wang, C., Chung, K. K., Zhang, Y., et al. (2005). Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 25(8), 2002–2009.
Lin, D. Y., Huang, Y. S., Jeng, J. C., Kuo, H. Y., Chang, C. C., Chao, T. T., et al. (2006). Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Molecular Cell, 24(3), 341–354.
Lin, W., & Kang, U. J. (2008). Characterization of PINK1 processing, stability, and subcellular localization. Journal of Neurochemistry, 106(1), 464–474.
Mabb, A. M., Wuerzberger-Davis, S. M., & Miyamoto, S. (2006). PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress. Nature Cell Biology, 8(9), 986–993.
Mahajan, R., Delphin, C., Guan, T., Gerace, L., & Melchior, F. (1997). A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell, 88(1), 97–107.
Mai, S., Klinkenberg, M., Auburger, G., Bereiter-Hahn, J., & Jendrach, M. (2010). Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. Journal of Cell Science, 123(Pt 6), 917–926.
Malik, B. R., Gillespie, J. M., & Hodge, J. J. (2013). CASK and CaMKII function in the mushroom body alpha’/beta’ neurons during Drosophila memory formation. Frontiers in Neural Circuits, 7, 52.
Maroteaux, L., Campanelli, J. T., & Scheller, R. H. (1988). Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 8(8), 2804–2815.
Masliah, E., Rockenstein, E., Veinbergs, I., Mallory, M., Hashimoto, M., Takeda, A., et al. (2000). Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science, 287(5456), 1265–1269.
Matunis, M. J., Coutavas, E., & Blobel, G. (1996). A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. The Journal of Cell Biology, 135(6 Pt 1), 1457–1470.
Mauri, F., McNamee, L. M., Lunardi, A., Chiacchiera, F., Del Sal, G., Brodsky, M. H., et al. (2008). Modification of Drosophila p53 by SUMO modulates its transactivation and pro-apoptotic functions. The Journal of Biological Chemistry, 283(30), 20848–20856.
McNally, R. S., Davis, B. K., Clements, C. M., Accavitti-Loper, M. A., Mak, T. W., & Ting, J. P. (2011). DJ-1 enhances cell survival through the binding of Cezanne, a negative regulator of NF-kappaB. The Journal of Biological Chemistry, 286(6), 4098–4106.
Meissner, C., Lorenz, H., Weihofen, A., Selkoe, D. J., & Lemberg, M. K. (2011). The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. Journal of Neurochemistry, 117(5), 856–867.
Menzies, F. M., Yenisetti, S. C., & Min, K. T. (2005). Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress. Current Biology: CB, 15(17), 1578–1582.
Meulener, M., Whitworth, A. J., Armstrong-Gold, C. E., Rizzu, P., Heutink, P., Wes, P. D., et al. (2005). Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Current Biology: CB, 15(17), 1572–1577.
Meulmeester, E., Kunze, M., Hsiao, H. H., Urlaub, H., & Melchior, F. (2008). Mechanism and consequences for paralog-specific sumoylation of ubiquitin-specific protease 25. Molecular Cell, 30(5), 610–619.
Michel, P. P., Toulorge, D., Guerreiro, S., & Hirsch, E. C. (2013). Specific needs of dopamine neurons for stimulation in order to survive: Implication for Parkinson disease. FASEB Journa: Official Publication of the Federation of American Societies for Experimental Biology.
Mizuta, H., & Kuroda, Y. (2004). Cloning and functional characterization of a rat Daxx that functions as a corepressor for the androgen receptor. Cell Biology International, 28(8–9), 609–614.
Morett, E., & Bork, P. (1999). A novel transactivation domain in parkin. Trends in Biochemical Sciences, 24(6), 229–231.
Mori, F., Tanji, K., Odagiri, S., Hattori, M., Hoshikawa, Y., Kono, C., et al. (2012). Ubiquitin-related proteins in neuronal and glial intranuclear inclusions in intranuclear inclusion body disease. Pathology International, 62(6), 407–411.
Mullett, S. J., Hamilton, R. L., & Hinkle, D. A. (2009). DJ-1 immunoreactivity in human brain astrocytes is dependent on infarct presence and infarct age. Neuropathology: Official Journal of the Japanese Society of Neuropathology, 29(2), 125–131.
Murakami, T., Shoji, M., Imai, Y., Inoue, H., Kawarabayashi, T., Matsubara, E., et al. (2004). Pael-R is accumulated in Lewy bodies of Parkinson’s disease. Annals of Neurology, 55(3), 439–442.
Nagakubo, D., Taira, T., Kitaura, H., Ikeda, M., Tamai, K., Iguchi-Ariga, S. M., et al. (1997). DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochemical and Biophysical Research Communications, 231(2), 509–513.
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biology, 8(1), e1000298.
Narendra, D., Tanaka, A., Suen, D. F., & Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of Cell Biology, 183(5), 795–803.
Neumann, M., Muller, V., Gorner, K., Kretzschmar, H. A., Haass, C., & Kahle, P. J. (2004). Pathological properties of the Parkinson’s disease-associated protein DJ-1 in alpha-synucleinopathies and tauopathies: Relevance for multiple system atrophy and Pick’s disease. Acta Neuropathologica, 107(6), 489–496.
Niki, T., Takahashi-Niki, K., Taira, T., Iguchi-Ariga, S. M., & Ariga, H. (2003). DJBP: A novel DJ-1-binding protein, negatively regulates the androgen receptor by recruiting histone deacetylase complex, and DJ-1 antagonizes this inhibition by abrogation of this complex. Molecular Cancer Research: MCR, 1(4), 247–261.
Oh, Y., & Chung, K. C. (2013). UHRF2, a ubiquitin E3 ligase, acts as a small ubiquitin-like modifier E3 ligase for zinc finger protein 131. The Journal of Biological Chemistry, 288(13), 9102–9111.
Oh, Y., Kim, Y. M., Mouradian, M. M., & Chung, K. C. (2011). Human Polycomb protein 2 promotes alpha-synuclein aggregate formation through covalent SUMOylation. Brain Research, 1381, 78–89.
Ohshima, T., Koga, H., & Shimotohno, K. (2004). Transcriptional activity of peroxisome proliferator-activated receptor gamma is modulated by SUMO-1 modification. The Journal of Biological Chemistry, 279(28), 29551–29557.
Olzmann, J. A., Bordelon, J. R., Muly, E. C., Rees, H. D., Levey, A. I., Li, L., et al. (2007a). Selective enrichment of DJ-1 protein in primate striatal neuronal processes: Implications for Parkinson’s disease. The Journal of Comparative Neurology, 500(3), 585–599.
Olzmann, J. A., Li, L., Chudaev, M. V., Chen, J., Perez, F. A., Palmiter, R. D., et al. (2007b). Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. The Journal of Cell Biology, 178(6), 1025–1038.
Park, J., Lee, J. H., La, M., Jang, M. J., Chae, G. W., Kim, S. B., et al. (2007). Inhibition of NF-kappaB acetylation and its transcriptional activity by Daxx. Journal of Molecular Biology, 368(2), 388–397.
Patel, V. P., & Chu, C. T. (2011). Nuclear transport, oxidative stress, and neurodegeneration. International Journal of Clinical and Experimental Pathology, 4(3), 215–229.
Petrucelli, L., O’Farrell, C., Lockhart, P. J., Baptista, M., Kehoe, K., Vink, L., et al. (2002). Parkin protects against the toxicity associated with mutant alpha-synuclein: Proteasome dysfunction selectively affects catecholaminergic neurons. Neuron, 36(6), 1007–1019.
Pichler, A., Gast, A., Seeler, J. S., Dejean, A., & Melchior, F. (2002). The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell, 108(1), 109–120.
Pilsl, A., & Winklhofer, K. F. (2012). Parkin, PINK1 and mitochondrial integrity: Emerging concepts of mitochondrial dysfunction in Parkinson’s disease. Acta Neuropathologica, 123(2), 173–188.
Plun-Favreau, H., Klupsch, K., Moisoi, N., Gandhi, S., Kjaer, S., Frith, D., et al. (2007). The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nature Cell Biology, 9(11), 1243–1252.
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science, 276(5321), 2045–2047.
Pountney, D. L., Chegini, F., Shen, X., Blumbergs, P. C., & Gai, W. P. (2005). SUMO-1 marks subdomains within glial cytoplasmic inclusions of multiple system atrophy. Neuroscience Letters, 381(1–2), 74–79.
Pountney, D. L., Huang, Y., Burns, R. J., Haan, E., Thompson, P. D., Blumbergs, P. C., et al. (2003). SUMO-1 marks the nuclear inclusions in familial neuronal intranuclear inclusion disease. Experimental Neurology, 184(1), 436–446.
Pountney, D. L., Raftery, M. J., Chegini, F., Blumbergs, P. C., & Gai, W. P. (2008). NSF, Unc-18-1, dynamin-1 and HSP90 are inclusion body components in neuronal intranuclear inclusion disease identified by anti-SUMO-1-immunocapture. Acta Neuropathologica, 116(6), 603–614.
Pourcet, B., Pineda-Torra, I., Derudas, B., Staels, B., & Glineur, C. (2010). SUMOylation of human peroxisome proliferator-activated receptor alpha inhibits its trans-activity through the recruitment of the nuclear corepressor NCoR. The Journal of Biological Chemistry, 285(9), 5983–5992.
Pridgeon, J. W., Olzmann, J. A., Chin, L. S., & Li, L. (2007). PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biology, 5(7), e172.
Prudden, J., Pebernard, S., Raffa, G., Slavin, D. A., Perry, J. J., Tainer, J. A., et al. (2007). SUMO-targeted ubiquitin ligases in genome stability. The EMBO Journal, 26(18), 4089–4101.
Pype, S., Declercq, W., Ibrahimi, A., Michiels, C., Van Rietschoten, J. G., Dewulf, N., et al. (2000). TTRAP, a novel protein that associates with CD40, tumor necrosis factor (TNF) receptor-75 and TNF receptor-associated factors (TRAFs), and that inhibits nuclear factor-kappa B activation. The Journal of Biological Chemistry, 275(24), 18586–18593.
Rizzu, P., Hinkle, D. A., Zhukareva, V., Bonifati, V., Severijnen, L. A., Martinez, D., et al. (2004). DJ-1 colocalizes with tau inclusions: A link between parkinsonism and dementia. Annals of Neurology, 55(1), 113–118.
Rodriguez, M. S., Desterro, J. M., Lain, S., Midgley, C. A., Lane, D. P., & Hay, R. T. (1999). SUMO-1 modification activates the transcriptional response of p53. The EMBO Journal, 18(22), 6455–6461.
Rothgiesser, K. M., Fey, M., & Hottiger, M. O. (2010). Acetylation of p65 at lysine 314 is important for late NF-kappaB-dependent gene expression. BMC Genomics, 11, 22.
Saitoh, H., & Hinchey, J. (2000). Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. The Journal of Biological Chemistry, 275(9), 6252–6258.
Sandebring, A., Thomas, K. J., Beilina, A., van der Brug, M., Cleland, M. M., Ahmad, R., et al. (2009). Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS One, 4(5), e5701.
Santiago, A., Godsey, A. C., Hossain, J., Zhao, L. Y., & Liao, D. (2009). Identification of two independent SUMO-interacting motifs in Daxx: Evolutionary conservation from Drosophila to humans and their biochemical functions. Cell Cycle, 8(1), 76–87.
Sayre, L. M. (1989). Biochemical mechanism of action of the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology Letters, 48(2), 121–149.
Schlossmacher, M. G., Frosch, M. P., Gai, W. P., Medina, M., Sharma, N., Forno, L., et al. (2002). Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. The American Journal of Pathology, 160(5), 1655–1667.
Schulz-Schaeffer, W. J. (2012). Neurodegeneration in Parkinson disease: Moving Lewy bodies out of focus. Neurology, 79(24), 2298–2299.
Seliger, B., Fedorushchenko, A., Brenner, W., Ackermann, A., Atkins, D., Hanash, S., et al. (2007). Ubiquitin COOH-terminal hydrolase 1: A biomarker of renal cell carcinoma associated with enhanced tumor cell proliferation and migration. Clinical Cancer Research, 13(1), 27–37.
Sharon, R., Bar-Joseph, I., Frosch, M. P., Walsh, D. M., Hamilton, J. A., & Selkoe, D. J. (2003). The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron, 37(4), 583–595.
Shaw, J. M., & Nunnari, J. (2002). Mitochondrial dynamics and division in budding yeast. Trends in Cell Biology, 12(4), 178–184.
Shendelman, S., Jonason, A., Martinat, C., Leete, T., & Abeliovich, A. (2004). DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biology, 2(11), e362.
Sherer, T. B., Betarbet, R., Stout, A. K., Lund, S., Baptista, M., Panov, A. V., et al. (2002). An in vitro model of Parkinson’s disease: Linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 22(16), 7006–7015.
Sherer, T. B., Kim, J. H., Betarbet, R., & Greenamyre, J. T. (2003). Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Experimental Neurology, 179(1), 9–16.
Shiba-Fukushima, K., Imai, Y., Yoshida, S., Ishihama, Y., Kanao, T., Sato, S., et al. (2012). PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Scientific Reports, 2, 1002.
Shiio, Y., & Eisenman, R. N. (2003). Histone sumoylation is associated with transcriptional repression. Proceedings of the National Academy of Sciences of the United States of America, 100(23), 13225–13230.
Shimizu, K., Ohtaki, K., Matsubara, K., Aoyama, K., Uezono, T., Saito, O., et al. (2001). Carrier-mediated processes in blood–brain barrier penetration and neural uptake of paraquat. Brain Research, 906(1–2), 135–142.
Shimshek, D. R., Schweizer, T., Schmid, P., & van der Putten, P. H. (2012). Excess alpha-synuclein worsens disease in mice lacking ubiquitin carboxy-terminal hydrolase L1. Scientific Reports, 2, 262.
Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S., et al. (2000). Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nature Genetics, 25(3), 302–305.
Shimura, H., Hattori, N., Kubo, S., Yoshikawa, M., Kitada, T., Matsumine, H., et al. (1999). Immunohistochemical and subcellular localization of Parkin protein: Absence of protein in autosomal recessive juvenile parkinsonism patients. Annals of Neurology, 45(5), 668–672.
Shinbo, Y., Niki, T., Taira, T., Ooe, H., Takahashi-Niki, K., Maita, C., et al. (2006). Proper SUMO-1 conjugation is essential to DJ-1 to exert its full activities. Cell Death and Differentiation, 13(1), 96–108.
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). alpha-Synuclein locus triplication causes Parkinson’s disease. Science, 302(5646), 841.
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., & Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature, 388(6645), 839–840.
Stambolic, V., Suzuki, A., de la Pompa, J. L., Brothers, G. M., Mirtsos, C., Sasaki, T., et al. (1998). Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell, 95(1), 29–39.
Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C., Slepko, N., et al. (2004). SUMO modification of Huntingtin and Huntington’s disease pathology. Science, 304(5667), 100–104.
Stichel, C. C., Augustin, M., Kuhn, K., Zhu, X. R., Engels, P., Ullmer, C., et al. (2000). Parkin expression in the adult mouse brain. The European Journal of Neuroscience, 12(12), 4181–4194.
Stindt, M. H., Carter, S., Vigneron, A. M., Ryan, K. M., & Vousden, K. H. (2011). MDM2 promotes SUMO-2/3 modification of p53 to modulate transcriptional activity. Cell Cycle, 10(18), 3176–3188.
Subramaniam, S., Mealer, R. G., Sixt, K. M., Barrow, R. K., Usiello, A., & Snyder, S. H. (2010). Rhes, a physiologic regulator of sumoylation, enhances cross-sumoylation between the basic sumoylation enzymes E1 and Ubc9. The Journal of Biological Chemistry, 285(27), 20428–20432.
Subramaniam, S., Sixt, K. M., Barrow, R., & Snyder, S. H. (2009). Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science, 324(5932), 1327–1330.
Sudharsan, R., & Azuma, Y. (2012). The SUMO ligase PIAS1 regulates UV-induced apoptosis by recruiting Daxx to SUMOylated foci. Journal of Cell Science, 125(Pt 23), 5819–5829.
Sytnyk, V., Leshchyns’ka, I., Nikonenko, A. G., & Schachner, M. (2006). NCAM promotes assembly and activity-dependent remodeling of the postsynaptic signaling complex. The Journal of Cell Biology, 174(7), 1071–1085.
Taguchi, N., Ishihara, N., Jofuku, A., Oka, T., & Mihara, K. (2007). Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. The Journal of Biological Chemistry, 282(15), 11521–11529.
Taira, T., Saito, Y., Niki, T., Iguchi-Ariga, S. M., Takahashi, K., & Ariga, H. (2004). DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Reports, 5(2), 213–218.
Takahashi, K., Taira, T., Niki, T., Seino, C., Iguchi-Ariga, S. M., & Ariga, H. (2001). DJ-1 positively regulates the androgen receptor by impairing the binding of PIASx alpha to the receptor. The Journal of Biological Chemistry, 276(40), 37556–37563.
Takahashi-Fujigasaki, J., & Fujigasaki, H. (2006). Histone deacetylase (HDAC) 4 involvement in both Lewy and Marinesco bodies. Neuropathology and Applied Neurobiology, 32(5), 562–566.
Takami, Y., Nakagami, H., Morishita, R., Katsuya, T., Cui, T. X., Ichikawa, T., et al. (2007). Ubiquitin carboxyl-terminal hydrolase L1, a novel deubiquitinating enzyme in the vasculature, attenuates NF-kappaB activation. Arteriosclerosis, Thrombosis, and Vascular Biology, 27(10), 2184–2190.
Tao, X., & Tong, L. (2003). Crystal structure of human DJ-1, a protein associated with early onset Parkinson’s disease. The Journal of Biological Chemistry, 278(33), 31372–31379.
Taschenberger, G., Toloe, J., Tereshchenko, J., Akerboom, J., Wales, P., Benz, R., et al. (2013). ss-synuclein aggregates and induces neurodegeneration in dopaminergic neurons. Annals of neurology.
Tatham, M. H., Matic, I., Mann, M., & Hay, R. T. (2011). Comparative proteomic analysis identifies a role for SUMO in protein quality control. Science signaling, 4(178), rs4.
Terashima, T., Kawai, H., Fujitani, M., Maeda, K., & Yasuda, H. (2002). SUMO-1 co-localized with mutant atrophin-1 with expanded polyglutamines accelerates intranuclear aggregation and cell death. NeuroReport, 13(17), 2359–2364.
Tillman, J. E., Yuan, J., Gu, G., Fazli, L., Ghosh, R., Flynt, A. S., et al. (2007). DJ-1 binds androgen receptor directly and mediates its activity in hormonally treated prostate cancer cells. Cancer Research, 67(10), 4630–4637.
Tirard, M., Hsiao, H. H., Nikolov, M., Urlaub, H., Melchior, F., & Brose, N. (2012). In vivo localization and identification of SUMOylated proteins in the brain of His6-HA-SUMO1 knock-in mice. Proceedings of the National Academy of Sciences of the United States of America, 109(51), 21122–21127.
Tofaris, G. K., Razzaq, A., Ghetti, B., Lilley, K. S., & Spillantini, M. G. (2003). Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. The Journal of Biological Chemistry, 278(45), 44405–44411.
Torii, S., Egan, D. A., Evans, R. A., & Reed, J. C. (1999). Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs). The EMBO Journal, 18(21), 6037–6049.
Ueda, K., Fukushima, H., Masliah, E., Xia, Y., Iwai, A., Yoshimoto, M., et al. (1993). Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America, 90(23), 11282–11286.
Ullmann, R., Chien, C. D., Avantaggiati, M. L., & Muller, S. (2012). An acetylation switch regulates SUMO-dependent protein interaction networks. Molecular Cell, 46(6), 759–770.
Um, J. W., & Chung, K. C. (2006). Functional modulation of parkin through physical interaction with SUMO-1. Journal of Neuroscience Research, 84(7), 1543–1554.
Um, J. W., Min, D. S., Rhim, H., Kim, J., Paik, S. R., & Chung, K. C. (2006). Parkin ubiquitinates and promotes the degradation of RanBP2. The Journal of Biological Chemistry, 281(6), 3595–3603.
Uzunova, K., Gottsche, K., Miteva, M., Weisshaar, S. R., Glanemann, C., Schnellhardt, M., et al. (2007). Ubiquitin-dependent proteolytic control of SUMO conjugates. The Journal of Biological Chemistry, 282(47), 34167–34175.
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science, 304(5674), 1158–1160.
van de Warrenburg, B. P., Lammens, M., Lucking, C. B., Denefle, P., Wesseling, P., Booij, J., et al. (2001). Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology, 56(4), 555–557.
van Duijn, C. M., Dekker, M. C., Bonifati, V., Galjaard, R. J., Houwing-Duistermaat, J. J., Snijders, P. J., et al. (2001). Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. American Journal of Human Genetics, 69(3), 629–634.
Vilotti, S., Biagioli, M., Foti, R., Dal Ferro, M., Lavina, Z. S., Collavin, L., et al. (2012). The PML nuclear bodies-associated protein TTRAP regulates ribosome biogenesis in nucleolar cavities upon proteasome inhibition. Cell Death and Differentiation, 19(3), 488–500.
Wadosky, K. M., & Willis, M. S. (2012). The story so far: Post-translational regulation of peroxisome proliferator-activated receptors by ubiquitination and SUMOylation. American Journal of Physiology. Heart and Circulatory Physiology, 302(3), H515–H526.
Wakabayashi, K., Engelender, S., Yoshimoto, M., Tsuji, S., Ross, C. A., & Takahashi, H. (2000). Synphilin-1 is present in Lewy bodies in Parkinson’s disease. Annals of Neurology, 47(4), 521–523.
Wang, H., Song, P., Du, L., Tian, W., Yue, W., Liu, M., et al. (2011). Parkin ubiquitinates Drp1 for proteasome-dependent degradation: Implication of dysregulated mitochondrial dynamics in Parkinson disease. The Journal of Biological Chemistry, 286(13), 11649–11658.
Weetman, J., Wong, M. B., Sharry, S., Rcom-H’cheo-Gauthier, A., Gai, W. P., Meedeniya, A., et al. (2013). Increased SUMO-1 expression in the unilateral rotenone-lesioned mouse model of Parkinson’s disease. Neuroscience Letters.
Weger, S., Hammer, E., & Heilbronn, R. (2005). Topors acts as a SUMO-1 E3 ligase for p53 in vitro and in vivo. FEBS Letters, 579(22), 5007–5012.
Weinreb, P. H., Zhen, W., Poon, A. W., Conway, K. A., & Lansbury, P. T, Jr. (1996). NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry, 35(43), 13709–13715.
Wenzel, D. M., Lissounov, A., Brzovic, P. S., & Klevit, R. E. (2011). UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature, 474(7349), 105–108.
Wilhelmus, M. M., Nijland, P. G., Drukarch, B., de Vries, H. E., & van Horssen, J. (2012). Involvement and interplay of Parkin, PINK1, and DJ1 in neurodegenerative and neuroinflammatory disorders. Free Radical Biology & Medicine, 53(4), 983–992.
Wilkinson, K. A., & Henley, J. M. (2010). Mechanisms, regulation and consequences of protein SUMOylation. The Biochemical Journal, 428(2), 133–145.
Wilson, M. A., Collins, J. L., Hod, Y., Ringe, D., & Petsko, G. A. (2003). The 1.1-A resolution crystal structure of DJ-1, the protein mutated in autosomal recessive early onset Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America, 100(16), 9256–9261.
Wiltshire, K. M., Dunham, C., Reid, S., Auer, R. N., & Suchowersky, O. (2010). Neuronal Intranuclear Inclusion Disease presenting as juvenile Parkinsonism. The Canadian Journal of Neurological Sciences. Le Journal Canadien des Sciences Neurologiques, 37(2), 213–218.
Wong, M. B., Goodwin, J., Norazit, A., Meedeniya, A. C., Richter-Landsberg, C., Gai, W. P., et al. (2013). SUMO-1 is associated with a subset of lysosomes in glial protein aggregate diseases. Neurotoxicity Research, 23(1), 1–21.
Xiong, H., Wang, D., Chen, L., Choo, Y. S., Ma, H., Tang, C., et al. (2009). Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. The Journal of Clinical Investigation, 119(3), 650–660.
Yamashita, D., Yamaguchi, T., Shimizu, M., Nakata, N., Hirose, F., & Osumi, T. (2004). The transactivating function of peroxisome proliferator-activated receptor gamma is negatively regulated by SUMO conjugation in the amino-terminal domain. Genes to Cells: Devoted to Molecular & Cellular Mechanisms, 9(11), 1017–1029.
Yang, X., Khosravi-Far, R., Chang, H. Y., & Baltimore, D. (1997). Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell, 89(7), 1067–1076.
Yang, S. H., & Sharrocks, A. D. (2004). SUMO promotes HDAC-mediated transcriptional repression. Molecular Cell, 13(4), 611–617.
Yang, W., Wang, L., & Paschen, W. (2013). Development of a high-throughput screening assay for inhibitors of small ubiquitin-like modifier proteases. Journal of Biomolecular Screening, 18(5), 621–628.
Yao, Q., Li, H., Liu, B. Q., Huang, X. Y., & Guo, L. (2011). SUMOylation-regulated protein phosphorylation, evidence from quantitative phosphoproteomics analyses. The Journal of Biological Chemistry, 286(31), 27342–27349.
Yeung, P. L., Chen, L. Y., Tsai, S. C., Zhang, A., & Chen, J. D. (2008). Daxx contains two nuclear localization signals and interacts with importin alpha3. Journal of Cellular Biochemistry, 103(2), 456–470.
Youle, R. J., & van der Bliek, A. M. (2012). Mitochondrial fission, fusion, and stress. Science, 337(6098), 1062–1065.
Zarranz, J. J., Alegre, J., Gomez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, E46 K, of alpha-synuclein causes Parkinson and Lewy body dementia. Annals of Neurology, 55(2), 164–173.
Zhang, J., Pho, V., Bonasera, S. J., Holtzman, J., Tang, A. T., Hellmuth, J., et al. (2007). Essential function of HIPK2 in TGFbeta-dependent survival of midbrain dopamine neurons. Nature Neuroscience, 10(1), 77–86.
Zhang, L., Shimoji, M., Thomas, B., Moore, D. J., Yu, S. W., Marupudi, N. I., et al. (2005). Mitochondrial localization of the Parkinson’s disease related protein DJ-1: Implications for pathogenesis. Human Molecular Genetics, 14(14), 2063–2073.
Zhang, Q., Wu, J., Wu, R., Ma, J., Du, G., Jiao, R., et al. (2012). DJ-1 promotes the proteasomal degradation of Fis1: Implications of DJ-1 in neuronal protection. The Biochemical Journal, 447(2), 261–269.
Zhong, N., Kim, C. Y., Rizzu, P., Geula, C., Porter, D. R., Pothos, E. N., et al. (2006). DJ-1 transcriptionally up-regulates the human tyrosine hydroxylase by inhibiting the sumoylation of pyrimidine tract-binding protein-associated splicing factor. The Journal of Biological Chemistry, 281(30), 20940–20948.
Zhong, N., & Xu, J. (2008). Synergistic activation of the human MnSOD promoter by DJ-1 and PGC-1alpha: Regulation by SUMOylation and oxidation. Human Molecular Genetics, 17(21), 3357–3367.
Zhou, W., & Freed, C. R. (2005). DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T alpha-synuclein toxicity. The Journal of Biological Chemistry, 280(52), 43150–43158.
Zhou, C., Huang, Y., Shao, Y., May, J., Prou, D., Perier, C., et al. (2008). The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proceedings of the National Academy of Sciences of the United States of America, 105(33), 12022–12027.
Zucchelli, S., Vilotti, S., Calligaris, R., Lavina, Z. S., Biagioli, M., Foti, R., et al. (2009). Aggresome-forming TTRAP mediates pro-apoptotic properties of Parkinson’s disease-associated DJ-1 missense mutations. Cell Death and Differentiation, 16(3), 428–438.
Zungu, M., Schisler, J., & Willis, M. S. (2011). All the little pieces. -Regulation of mitochondrial fusion and fission by ubiquitin and small ubiquitin-like modifier and their potential relevance in the heart. Circulation Journal: Official Journal of the Japanese Circulation Society, 75(11), 2513–2521.
Zunino, R., Schauss, A., Rippstein, P., Andrade-Navarro, M., & McBride, H. M. (2007). The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. Journal of Cell Science, 120(Pt 7), 1178–1188.
Acknowledgments
The author was funded through the Cluster of Excellence and DFG Research Center Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eckermann, K. SUMO and Parkinson’s Disease. Neuromol Med 15, 737–759 (2013). https://doi.org/10.1007/s12017-013-8259-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-013-8259-5