
Abstract
Thiamine is an essential vitamin that is necessary to maintain the functional integrity of cells in the brain. Its deficiency is the underlying cause of Wernicke’s encephalopathy (WE), a disorder primarily associated with, but not limited to, chronic alcoholism. Thiamine deficiency leads to the development of impaired energy metabolism due to mitochondrial dysfunction in focal regions of the brain resulting in cerebral vulnerability. The consequences of this include oxidative stress, excitotoxicity, inflammatory responses, decreased neurogenesis, blood–brain barrier disruption, lactic acidosis and a reduction in astrocyte functional integrity involving a loss of glutamate transporters and other astrocyte-specific proteins which together contribute in a major way to the resulting neurodegeneration. Exactly how these factors acting in concert lead to the demise of neurons is unclear. In this review we reassess their relative importance in the light of more recent findings and discuss therapeutic possibilities that may provide hope for the future for individuals with WE.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Understanding the pathophysiology of a disease process is crucial for effecting its successful treatment. Thiamine deficiency (TD) represents a unique platform for the investigation of neurodegeneration. Lack of this essential vitamin results in a myriad of problems, mostly driven by the resulting impaired metabolic function of the cell, which in the brain can lead to Wernicke’s encephalopathy (WE), the neurological component of the Wernicke–Korsakoff syndrome (WKS), and a condition most commonly encountered in chronic alcoholics. In WE, initial dysfunction to key parts of the limbic system which can rapidly progress to structural damage results in serious and often life-threatening complications for those affected by this disorder. Although administration of thiamine remains the remedy for many of the metabolic-related problems associated with WE, the propensity of these individuals to return to their alcohol-associated habits leading to recurrent bouts of TD represents a major obstacle to successful treatment of the problem. Evidence suggest that TD occurs in chronic alcoholics at a frequency of at least 25–31 % [1–3] and up to 80 % [4, 5]. Significantly, reports suggest that WE occurs in chronic alcoholics at a frequency of ~35 %, and in the population as a whole, the figure is ~1.5 % [6]. When combined with the high percentage of adult cases of WE that are diagnosed at post-mortem of approximately 80 % [7] and 60 % for pediatric cases of WE [8], one can begin to appreciate the importance of, and urgency for, understanding the pathophysiology of this illness and establishing therapeutic strategies that are likely to have a major positive impact.
Pathophysiologic Changes in TD
Thiamine is an important cofactor [9, 10] involved in processes associated with the metabolism of lipids, glucose, amino acids, and neurotransmitters [11]. The active form of thiamine is phosphorylated to thiamine diphosphate [12] that is required for the proper functioning of four major enzyme systems, (1) pyruvate dehydrogenase (EC 1.2.4.1) complex which connects glycolysis with the tricarboxylic acid (TCA) cycle, (2) α-ketoglutarate dehydrogenase (EC 1.2.4.2) (α-KGDH) complex, a key rate-limiting enzyme of the TCA cycle involved in mitochondrial energy metabolism, (3) transketolase (EC 2.2.1.1), which plays an important role in nucleic acid and lipid biosynthesis through the pentose phosphate shunt, and (4) branched-chain α-keto-acid dehydrogenase (EC 1.2.4.4) complex, which is involved in the metabolism of branched-chain amino acids.
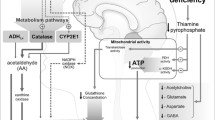
Since thiamine is required for decarboxylation in glucose metabolism, TD leads to impaired oxidative metabolism [13] associated with decreased ATP synthesis in the brain [14]. This decreased energy production along with resulting neuronal depolarization [15] are consequences of TD that have the capability on their own to lead to subsequent glutamate-mediated excitotoxicity and neurodegeneration [16, 17]. The impaired oxidative metabolism in TD leads to major changes in cerebral function that include the utilization of glucose [101], alterations in neurotransmitters [18, 19], oxidative/nitrosative stress [20, 21], lactic acidosis and decreased brain pH [22], excitotoxicity [16, 23, 24], inflammation [25], endoplasmic reticulum (ER) stress and apoptosis [20, 26], and dysfunction of the blood–brain barrier (BBB) which also constitutes a major part of the neurovascular unit that is involved in the control of regional blood flow [27–29]. Some of these important changes are shown in Fig. 1. The BBB also constitutes the physical barrier that regulates movement of blood nutrients into brain from the systemic circulation, thus playing an additional important role in preserving the integrity of the cerebral microenvironment [30]. Damage to the BBB can lead to altered blood flow [31] with subsequent neuronal loss and neurodegenerative events [32].

Some important processes involved in neurodegeneration in thiamine deficiency (TD) and potential therapeutic approaches. TD leads to impaired mitochondrial function that is associated with decreased activity of thiamine-dependent enzymes. This produces decreased energy production in the cell and lowered redox function resulting in net production of oxidative stress in this organelle. Increased endoplasmic reticulum (ER) stress is also a consequence of this reduced mitochondrial function. The impaired mitochondrial function results in development of an inflammatory process e.g. in microglial cells, and reactive oxygen species-driven oxidative stress that extends outside the mitochondria and into other parts of the cell, leading to decreased neurogenesis in neural stem/progenitor cells. Loss of mitochondrial function is likely to produce functional impairment of neurons directly, leading to overstimulation and potential excitotoxic conditions within and between these cells (not shown in this figure, oxidative stress and inflammation probably affect neuronal functional integrity as well). Impaired mitochondrial function, inflammation and oxidative stress also target astrocytes leading to increased lactate production and loss of astrocyte-specific gene expression producing a downregulation of the glutamate transporters GLT-1 and GLAST that results in increased extracellular glutamate levels and excitotoxicity. Since astrocytes also play a major role in establishing and maintaining BBB integrity, targeting of these cells in TD is likely to be a major contributing factor in the disruption of this barrier under these conditions. In addition, alterations in levels of the astrocyte water channel protein aquaporin (AQP)-4 are probably a major cause of the cerebral edema observed in TD and WE. Astrocyte targeting also results in disruption of astrocyte–neuronal trafficking of metabolites, thus further increasing neuronal dysfunction and the risk of cell death. Collectively, these resulting changes destabilize and impair neuronal function, ultimately leading to neurodegeneration. Future therapeutic strategies to be considered include novel glutamate receptor antagonists (e.g. flupirtine) antioxidants, along with anti-inflammatory agents (e.g. NSAIDS, minocycline, NP031112), β-lactam antibiotics (e.g. ceftriaxone), stem cells, and the use of nanomedicine-based procedures that could monitor or directly impact vulnerable brain regions (e.g. “ceria”)
Recent studies have indicated that astrocytes are a major source of lactate production in brain [33], with the increased lactic acidosis in TD being associated with decreased pH in focal regions of the brain [34]. This lactate production is a likely major contributor to the development of the selective lesions inherent in this disorder. Reports have also established that cerebral vulnerability is associated with the presence of edema in cases of WE [35, 36]. Previous findings indicate that TD leads to swelling of astrocytes in association with altered levels of aquaporin-4 (AQP-4) [37], a major water channel protein that is localized predominantly in these cells in brain (Fig. 1). As swelling is an important feature of TD [38–40], and acidosis is associated with cellular edema [41–43], it is probable that astrocytes play a significant role in the observed brain edema in WE. Lactic acidosis may play an important role in these changes in AQP-4 levels, with recent evidence in favor of this contention in which lactic acid increases AQP-4 protein expression in astrocytes [44]. In addition, previous studies have established the importance of metabolite trafficking between astrocytes and neurons, and the important role that astrocytes play in supplying neurons with new carbon skeletons necessary for maintenance of activity of their TCA cycle and efficient oxidative metabolism [45]. Disruption of such trafficking by impairment of astrocyte function due to targeting of these cells in TD is therefore likely to lead to increased vulnerability of neurons to dysfunction and the possibility of cell death.
TD-induced focal damage occurs in selectively vulnerable areas of the brain such as the thalamus, mammillary bodies, inferior colliculus of the midbrain, and vestibular nuclei and inferior olivary complex of the brainstem [46]. In WKS, damage to the diencephalic–hippocampal circuitry including thalamic nuclei and mammillary bodies causes the chronic amnesic syndrome “diencephalic amnesia” typical of Korsakoff psychosis. The anterior thalamic nuclei (ATN) are important for processing learning and memory events, and isolated damage to ATN produces a persistent amnestic syndrome [47], similar to that observed in WKS manifestations.
Clinical Aspects of TD
TD is considered to be a nutritional, metabolic, enzymatic, and neuroinflammatory disease, affecting both the nervous and cardiovascular systems. Beriberi, affecting primarily the peripheral nervous system, is classified into neurological (dry beriberi) leading to selective neurodegeneration, and cardiovascular (wet beriberi) associated with heart failure [48–50]. However neurological problems occur in both forms of the illness [51, 52]. In dry beriberi, which is predominately a neurological disorder, there is sensory–motor peripheral neuropathy that is more distal in nature, while in the wet type in addition to the presence of peripheral neuropathy, there are edema-related manifestations that include congestive heart failure, cardiomegaly and tachycardia [51, 53].
TD commonly occurs in patients with other co-morbidities such as alcoholism, liver and gastrointestinal diseases, head trauma, and seizures, but WE is the most serious complication of TD. In TD there is lactic acidosis with increased lactate levels in brain that can be detected in peripheral blood [54]. However, other uncommon clinical features may include stupor, hypotension and tachycardia, hypothermia, bilateral visual changes and papilloedema, convulsions, deafness, hallucinations and behavioural change, hyperthermia, hypertonia and spastic paresis, choreic dyskinesias and coma at the late-stage [55], which provides a basis for potential misdiagnosis of the illness.
WE commonly presents with an abrupt onset of a triad of neurological symptoms that consists of ophthalmoplegia, gait ataxia and confusion [56, 57]. However in many cases, these classic characteristics are not all present, leading to potential complications in the diagnosis. Modified criteria for clinical diagnosis of WE patients have been proposed that would include the presence of two out of four signs; dietary deficiencies, oculomotor abnormalities, cerebellar dysfunction, and either altered mental state or mild memory impairment [58]. Although there is no specific investigation to diagnose WE [59], magnetic resonance imaging is an important tool for visualizing the localization of lesions and in confirmation (or ruling out) of the illness [55, 60, 61].
Glutamate-Mediated Excitotoxicity
Over a number of years, strong evidence has been established for the existence of a glutamate-mediated excitotoxic event in TD. Earlier studies had shown reduced incorporation of [14C]-glucose into glutamate [62] consistent with reduced activities of KGDH, resulting in a decrease in energy status in TD animals [14]. In addition, treatment with the noncompetitive N-methyl-d-aspartate (NMDA) glutamate receptor antagonist MK-801 was shown to lead to a reduction in the extent of neuronal damage in thiamine-deficient rats [63]. Identification of increased interstitial glutamate concentration was then reported that was limited to vulnerable brain regions in TD [23, 24], thus providing the first direct evidence for glutamate-mediated excitotoxicity, along with the presence of excitotoxic-like lesions in damaged areas of the brain [64]. This was also consistent with description of the ultrastructural appearance of the affected thalamus being similar to that seen in excitotoxic-mediated necrosis [65, 66].
More recently, loss of the astrocytic glutamate transporters Glutamate Transporter 1 (GLT-1) and Glutamate/Aspartate Transporter (GLAST) has been reported to be localized to vulnerable brain regions in TD [16] (Fig. 1). These astrocytic transporters provide the major spatial buffering of extracellular glutamate levels in brain [67]. Along with loss of the GLAST protein in an astrocyte model of TD [68] and the recent findings of a considerable downregulation of GLT-1 and GLAST in human post-mortem cases of WE [69], these reports have together provided considerable credence for implication of excitotoxicity as a major cause of the histological lesions observed in TD and WE. In addition, studies indicate that levels of complexin I and II, presynaptic terminal proteins that play an important role in the regulation of neurotransmitter release [70, 71] are downregulated in vulnerable brain regions in TD [72]. Given that complexins play an important regulatory role in the release of glutamate and GABA at the synaptic cleft, altered expression of these proteins may reflect either enhanced or reduced release of glutamate and/or GABA by neurons. This effect on complexin levels is reversible with antioxidants such as N-acetylcysteine [72], suggesting an involvement of oxidative stress. Further studies are required to establish the exact significance of these changes in the levels of both proteins in TD. Recently, studies have identified the presence of complexin II but not complexin I in astrocytes [73], with complexin II being known to be associated with excitatory synapses [74]. Since recent studies have indicated that astrocytes contain similar cellular machinery to that of the presynaptic terminal for exocytotic release of neurotransmitter and release glutamate in a vesicular manner similar to that at the synaptic cleft [75, 76], it is possible these cells release glutamate during TD in a manner similar to that of neurons that may accentuate the rise of extracellular glutamate concentration and contribute to excitotoxic cell death.
Role of Oxidative Stress in TD
Physiologically, the brain is more sensitive to oxidative stress due to its high oxygen consumption where neurons use more oxygen than they produce from the mitochondria [77]. Oxidative stress occurs due to imbalance between synthesis and elimination of reactive oxygen species (ROS) as a result of failure of the redox mechanisms of the cell. The increased net production of ROS such as superoxide anion (O2 −) and nitric oxide (NO) forming reactive nitrogen species (nitrosative stress) lead to activation of NMDA receptors [78] and increased extracellular glutamate levels resulting in excitotoxicity, BBB disruption, activation of microglia, and induction of apoptosis [77], all features of TD [21]. Excessive NO production can also cause disruption of the BBB [79], allowing the passage of multiple vascular factors from the systemic circulation into the brain that can cause cell death [80]. Peroxynitrite, ONOO− which is formed from NO and O2 −, is a strong neurotoxic oxidizing and nitrating agent and thus can play a role in neuronal loss and tissue damage in neurodegenerative diseases [17, 77, 81].
In TD, the reduction in thiamine-dependent enzyme activity, particularly that of the α-KGDH complex, leads to mitochondrial dysfunction and hence decreased TCA cycle activity in endothelial cells, astrocytes and microglia that result in oxidative stress due to induction of both endothelial and inducible forms of NOS (eNOS and iNOS respectively) and the production of ROS, along with increased production of cytokines through microglial activation [79, 82, 83] (Fig. 1). Interestingly, TD also causes stress of the ER which results from excessive accumulation of unfolded or misfolded proteins in the ER lumen leading to overexpression of four ER stress markers, glucose-regulated protein 78, growth arrest and DNA damage-inducible protein 153 (CCAAT/enhancer binding protein homologous protein), phosphorylation of eukaryotic initiation factor 2α and cleavage of caspase-12 [84]. Nitrosative stress can occur as well, inducing neurodegeneration in a similar way to that produced by oxidative stress [85, 86].
Inflammatory Processes in TD
The inflammatory process that develops in TD includes a series of events that can be summarised as follows: alteration in glial cell morphology, which leads to swelling and the appearance of phagocytic vacuoles, and increased microglial reactivity, which results in the upregulation of inflammatory genes, transcripts and transcription factors, and production of pro-inflammatory molecules [87, 88].
During TD, alterations in glial cell morphology occur in both astrocytes and oligodendrocytes, with glial swelling of both cytoplasmic and nuclear compartments being the first ultrastructural abnormality to occur [37, 38]. Generally, the initial swelling of glial cells develops into two types of cerebral edema, cytotoxic (mostly common in ischemic and hypoxic states) and vasogenic edema (mostly common in trauma, tumors, and cerebrovascular insults). In TD, the type of edema that is encountered is initially of the cytotoxic type [38]. However, it is also possible that astrocytic dysfunction plays an important role in altering the BBB with consequent vasogenic edema in TD leading to brain tissue swelling [89], a finding previously confirmed using neuroimaging in cases of WE and in experimental models of TD [80, 90, 91].
Microglia are the most immunoresponsive cells in the CNS, being the principal resident immune cells and the primary mediators of neuroinflammation [92, 93] which put them at the first line of neuroprotection against exogenous harmful triggers such as viruses and bacteria [94, 95]. Thus, they are similar to peripheral macrophages [93]. Evidence suggests that microglial activation is one of the initial TD-induced cellular events to occur [25]. However, in TD the activated microglia lead to a sequelae of events themselves that contribute to the overall neurological disorder [96]. This includes the production of pro-inflammatory cytokines such as interleukin-1β (IL-1β), and IL-6, and tumor necrosis factor-α (TNF-α) [97], and upregulation of inflammatory genes, transcripts and transcription factors in thalamus and inferior colliculus [98]. It is conceivable that at different stages of their activation, microglia wander between neuroprotection and neurotoxicity, and thus dual opposed responses within the brain [99]. However, this inflammatory response may exert a major influence on astrocyte function (Fig. 1). Pro-inflammatory cytokines IL-1β and TNF-α can also induce transcription factors CCAAT/enhancer binding protein β and δ in astrocytes that cause progression of the inflammatory response to the surrounding area through a second peak of activation [100].
Neuroinflammation can also lead to disruption of the BBB with decreased cerebral blood flow, a condition that may contribute to neuronal cell loss and progression of neurodegenerative maladies such as Alzheimer’s disease (AD). This is collectively due to the effect of pro-inflammatory cytokines, secretion of endothelin-1 that suppresses blood flow, and overexpression of vascular genes causing increased synthesis of proteins leading to hypercontractile vascular smooth muscle state and gene expression changes that regulate Ca2+ homeostasis [32]. Such changes may also contribute to reported changes in local cerebral blood flow in vulnerable brain regions during TD [101].
Future Therapeutic Strategies in TD
Thiamine is crucial to many metabolic processes in the brain. The finding that WE occurs in the population at a frequency of about 1.5 % [6] makes this disorder an important health care issue. WE is a medical emergency that needs to be treated with high doses of intravenous vitamin B in order to hinder the development of the chronic and debilitating condition of Korsakoff psychosis. Future non-conventional therapies in TD will depend largely on the use of strategies that can either prevent or inhibit mechanisms involved in the neurodegenerative process occurring in this illness (see Fig. 1), particularly in light of recurrent TD, a common occurrence in cases of WE.
A move towards the use of agents that exert an anti-inflammatory influence such as non-steroidal anti-inflammatory drugs (NSAIDS) is presently being taken to investigate their potential benefit in several neurodegenerative maladies including Parkinson’s disease (PD) [87, 102], and which may also be beneficial in treating WE. Previous studies have shown promising results for the treatment of TD using the anti-inflammatory drug minocycline, which also possesses anti-oxidant and anti-apoptotic characteristics, in which it delayed the onset of major neurological impairment in rats by over 24 h [96]. In addition, studies have reported that treatment with the β-lactam antibiotic ceftriaxone was able to prevent loss of the GLT-1b splice-variant in animals with TD and improve glutamate uptake into astrocytes under TD conditions [103]. Ceftriaxone has previously been shown to have the ability to upregulate GLT-1 [104]. Furthermore, the use of NP031112, a compound that protects against inflammation and excitotoxic-mediated cell death is being considered as a possible therapeutic strategy in brain disorders, and may also be beneficial in treating WE cases [105].
Since TD results in both necrosis and apoptotic cell death [26], small anti-apoptotic drugs which are more able to cross the BBB and hence can directly affect the brain also represent another group of compounds for which a possible therapeutic avenue may exist [106]. The potential for use of glutamate receptor antagonists should also be considered, and has been used in the past with beneficial effects in both neurodegenerative disease states [107] and in experimental TD [63]. Flupirtine, which is a centrally-acting, non-opioid analgesic with muscle relaxant properties, but which interestingly possesses NMDA antagonist properties could also prove beneficial in treating TD.
Recently, the potential therapeutic benefit of stem cells in treating a wide range of illnesses including neurodegenerative diseases has gained considerable momentum [108]. These cells which have the ability for both indefinite self-renewal and differentiation into one or more specialized mature cell types hold the potential for successfully treating and even curing many disease states in which loss of a particular cell type or function is a major factor. These cells are currently being used in clinical trials to treat illnesses such as PD, stroke, Huntington’s disease (HD) and amyotrophic lateral sclerosis [108]. In the past, the adult brain was considered to be non-neurogenic and non-regenerative in nature. Nowadays, the concept of neurogenesis and the existence of neural stem cells (NSCs) have been widely adopted in the scientific field [109]. NSCs give rise to differentiated neurons or glial cells in specific regions within an adult brain that favour neurogenesis; most notably in the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone of the hippocampal dentate gyrus [110, 111]. Neurogenesis can be increased as in AD [112] but with decreased survival of newly born adult neurons, decreased neurogenesis also occurs in PD [113] and is increased in HD [114], and is aberrant in the epileptic rat dentate gyrus [115]. Following a stroke, migration of the newly born neuroblasts occurs from SVZ to the border of the vascular insult leading to simultaneous enhancement of post-stroke neurogenesis and angiogenesis [116, 117]. In the case of TD, neurogenesis is reduced in both major neurogenic areas of the brain [118–120]. Identifying the underlying basis of this suppressed production of neurons may facilitate the development of therapeutic approaches that will allow repair of the damage in vulnerable regions of the brain in this disorder. During TD, loss of mitochondrial function is a major feature, and studies have demonstrated that irradiation of animals leads to decreased neurogenesis, but co-treatment with thiamine can prevent this effect [121], suggesting that impaired neurogenesis may involve a loss of mitochondrial functional integrity. Thus, improved mitochondrial function may be an important therapeutic tool for maintaining neuroblast production and survival in the face of a damaging insult to the brain. Interestingly, some neurodegenerative disease states such as AD display evidence of decreased thiamine status, e.g. low plasma thiamine levels along with decreased activities of α-KGDH complex and transketolase [122, 123], suggesting that thiamine may have a positive role to play in these illnesses. Use of thiamine as a therapeutic approach in such cases may therefore increase mitochondrial function in neuroblasts, thus promoting their survival.
Other potential therapeutic approaches involve the use of nanoparticles (i.e. diameter <100 nm), e.g. cerium oxide, or “ceria” to reverse oxidative stress that is encountered in many neurological conditions by their prolonged antioxidant properties that can lead to improvement of neurological function [124]. Such nanomaterials can also be used in the diagnosis of neurodegenerative diseases, however caution is required at this stage as our knowledge in this field of nanomedicine is still in its infantile stages [125].
Conclusions
TD produces a complex pathophysiology that is multifactorial in nature. While significant progress has been made over the last 10 years in terms of our understanding of this illness, a clearer picture is still required in order to effectively treat and potentially reverse the neuropsychiatric problems associated with WKS including the chronic debilitating Korsakoff psychosis. Given the pace of developing technology, however, the next 10 years should prove engaging.
Abbreviations
- AD:
-
Alzheimer’s disease
- ATN:
-
Anterior thalamic nuclei
- BBB:
-
Blood–brain barrier
- CNS:
-
Central nervous system
- ER:
-
Endoplasmic reticulum
- HD:
-
Huntington’s disease
- α-KGDH:
-
Alpha-ketoglutarate dehydrogenase
- IL-1β:
-
Interleukin-1 beta
- NO:
-
Nitric oxide
- NOS:
-
NO synthase
- NSCs:
-
Neural stem cells
- PD:
-
Parkinson’s disease
- ROS:
-
Reactive oxygen species
- SGZ:
-
Subgranular zone
- O2 − :
-
Superoxide anion
- SVZ:
-
Subventricular zone
- TCA:
-
Tricarboxylic acid cycle
- TD:
-
Thiamine deficiency
- TNF-α:
-
Tumor necrosis factor-alpha
- WE:
-
Wernicke’s encephalopathy
- WKS:
-
Wernicke–Korsakoff syndrome
References
Baines M (1978) Detection and incidence of B and C vitamin deficiency in alcohol related illness. Ann Clin Biochem 15:307–312
Camilo ME, Morgan MY, Sherlock S (1981) Erythrocyte transketolase activity in alcoholic liver disease. Scand J Gastroenterol 16:273–279
Lévy S, Hervé C, Delacoux E, Erlinger S (2002) Thiamine deficiency in hepatitis C virus and alcohol-related liver diseases. Dig Dis Sci 47:543–548
Thomson AD, Jeyasingham MD, Pratt OE, Shaw GK (1987) Nutrition and alcoholic encephalopathies. Acta Med Scand Suppl 717:55–65
Morgan MY (1999) Nutritional aspects of liver and biliary disease. In: Bircher J, Benhamou JP, McIntyre N, Rizzetto M, Rode’s J (eds) Oxford textbook of clinical hepatology. Oxford Medical Publication, Oxford, pp 1923–1981
Cook CC, Hallwood PM, Thomson AD (1998) B vitamin deficiency and neuropsychiatric syndromes in alcohol misuse. Alcohol Alcohol 33:317–336
Harper CG, Giles M, Finlay-Jones R (1986) Clinical signs in the Wernicke–Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry 49:341–345
Vasconcelos MM, Silva KP, Vidal G, Silva AF, Domingues RC, Berditchevsky CR (1999) Early diagnosis of pediatric Wernicke’s encephalopathy. Pediatr Neurol 20:289–294
Breslow R (1958) On the mechanism of thiamine action. IV. Evidence from studies on model systems. J Am Chem Soc 80:3719–3726
Kern D, Kern G, Neef H, Tittmann K, Killenberg-Jabs M, Wikner C, Schneider G, Hübner G (1997) How thiamine diphosphate is activated in enzymes. Sci 275:67–70
Chisolm-Straker M, Cherkas D (2013) Altered and unstable: wet beriberi, a clinical review. J Emerg Med 45:341–344
Kraut J, Reed HJ (1962) The crystal structure of thiamine hydrochloride (vitamin B1). Acta Crystallogr 15:747–757
Witte KK, Clark AL, Cleland JG (2001) Chronic heart failure and micronutrients. J Am Coll Cardiol 37:1765–1774
Aikawa H, Watanabe IS, Furuse T, Iwasaki Y, Satoyoshi E, Sumi T, Moroji T (1984) Low energy levels in thiamine-deficient encephalopathy. J Neuropathol Exp Neurol 43:276–287
Hazell AS, Hakim AM, Senterman MK, Hogan MJ (1998) Regional activation of L-type voltage-sensitive calcium channels in experimental thiamine deficiency. J Neurosci Res 52:742–749
Hazell AS, Rao KV, Danbolt NC, Pow DV, Butterworth RF (2001) Selective down-regulation of the astrocyte glutamate transporters GLT-1 and GLAST within the medial thalamus in experimental Wernicke’s encephalopathy. J Neurochem 78:560–568
Beal MF (1996) Mitochondria, free radicals, and neurodegeneration. Curr Opin Neurobiol 6:661–666
Butterworth RF (1982) Neurotransmitter function in thiamine-deficiency encephalopathy. Neurochem Int 4:449–464
Van Woert MH, Plaitakis A, Hwang EC, Berl S (1979) Effect of thiamine deficiency on brain serotonin turnover. Brain Res 179:103–110
Martin PR, Singleton CK, Hiller-Sturmhöfel S (2003) The role of thiamine deficiency in alcoholic brain disease. Alcohol Res Health 27:134–142
Jhala SS, Hazell AS (2011) Modeling neurodegenerative disease pathophysiology in thiamine deficiency: consequences of impaired oxidative metabolism. Neurochem Int 58:248–260
Navarro D, Zwingmann C, Hazell AS, Butterworth RF (2005) Brain lactate synthesis in thiamine deficiency: a re-evaluation using 1H-13C nuclear magnetic resonance spectroscopy. J Neurosci Res 79:33–41
Hazell AS, Butterworth RF, Hakim AM (1993) Cerebral vulnerability is associated with selective increase in extracellular glutamate concentration in experimental thiamine deficiency. J Neurochem 61:1155–1158
Langlais PJ, Zhang SX (1993) Extracellular glutamate is increased in thalamus during thiamine deficiency-induced lesions and is blocked by MK-801. J Neurochem 61:2175–2182
Todd KG, Butterworth RF (1999) Early microglial response in experimental thiamine deficiency: an immunohistochemical analysis. Glia 25:190–198
Matsushima K, MacManus JP, Hakim AM (1997) Apoptosis is restricted to the thalamus in thiamine deficient rats. NeuroReport 8:867–870
Iadecola C (2004) Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci 5:347–360
Schipke CG, Kettenmann H (2004) Astrocyte responses to neuronal activity. Glia 47:226–232
Abbott NJ, Rönnbäck L, Hansson E (2006) Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 7:41–53
Quaegebeur A, Segura I, Carmeliet P (2010) Pericytes: blood–brain barrier safeguards against neurodegeneration? Neuron 68:321–323
Hakim AM, Pappius HM (1981) The effect of thiamine deficiency on local cerebral glucose utilization. Ann Neurol 9:334–339
Zlokovic BV (2008) The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201
Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ (2007) Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55:1251–1262
Hakim AM (1984) The induction and reversibility of cerebral acidosis in thiamine deficiency. Ann Neurol 16:673–679
Yokote K, Miyagi K, Kuzuhara S, Yamanouchi H, Yamada H (1991) Wernicke encephalopathy: follow-up study by CT and MR. J Comput Assist Tomogr 15:835–838
Bergui M, Bradac GB, Zhong JJ, Barbero PA, Durelli L (2001) Diffusion-weighted MR in reversible Wernicke encephalopathy. Neuroradiology 43:969–972
Chan H, Butterworth RF, Hazell AS (2004) Astrocytes respond to thiamine deficiency-induced swelling by downregulating aquaporin-4 levels. Neurosci Lett 366:231–234
Collins GH (1967) Glial cell changes in the brain stem of thiamine-deficient rats. Am J Pathol 50:791–814
Robertson DM, Wasan SM, Skinner DB (1968) Ultrastructural features of early brain stem lesions of thiamine-deficient rats. Am J Pathol 52:1081–1097
Watanabe I, Kanabe S (1978) Early edematous lesion of pyrithiamine induced acute thiamine deficient encephalopathy in the mouse. J Neuropathol Exp Neurol 37:401–413
Myers RE (1979) Lactic acid accumulation as cause of brain edema and cerebral necrosis resulting from oxygen deprivation. In: Korobkin R, Guilleminault G (eds) Advances in perinatal neurology. Spectrum, New York, pp 85–114
Kalimo H, Rehncrona S, Söderfeldt B, Olsson Y, Siesjö BK (1981) Brain lactic acidosis and ischemic cell damage: 2. Histopathology. J Cereb Blood Flow Metab 1:313–327
Jenkins LW, Becher DP, Coburn TH (1984) A quantitative analysis of glial swelling and ischemic neuronal injury following complete cerebral ischemia. In: Go TG, Baethmann A (eds) Recent progress in the study and therapy of brain edema. Plenum, New York, pp 523–537
Morishima T, Aoyama M, Iida Y, Yamamoto N, Hirate H, Arima H, Fujita Y, Sasano H, Tsuda T, Katsuya H, Asai K, Sobue K (2008) Lactic acid increases aquaporin 4 expression on the cell membrane of cultured rat astrocytes. Neurosci Res 61:18–26
Hertz L, Dringen R, Schousboe A, Robinson SR (1999) Astrocytes: glutamate producers for neurons. J Neurosci Res 57:417–428
Troncoso JC, Johnston MV, Hess KM, Griffin JW, Price DL (1981) Model of Wernicke’s encephalopathy. Arch Neurol 38:350–354
Nardone R, Höller Y, Storti M, Christova M, Tezzon F, Golaszewski S, Trinka E, Brigo F (2013) Thiamine deficiency induced neurochemical, neuroanatomical, and neuropsychological alterations: a reappraisal. Sci World J. Article ID 309143
Platt BS, Lu GD (1936) Chemical and clinical findings in beri-beri with special reference to vitamin B1 deficiency. QJM 5:355–374
Jones RH (1959) Beriberi heart disease. Circulation 19:275–283
McIntyre N, Stanley NN (1971) Cardiac beriberi: two modes of presentation. Br Med J 3:567–569
Motherway C (1999) Acute pernicious (sho-shin) beri-beri: a report of three cases. Crit Care Resusc 1:69–73
Amato AA, Dumitru D (2001) Acquired neuropathies. In: Dumitru D, Amato AA, Zwarts M (eds) Electrodiagnostic medicine, 2nd edn. Elsevier, New York, pp 937–1041
Butterworth RF (2003) Thiamin deficiency and brain disorders. Nutr Res Rev 16:277–284
Engbers JG, Molhoek GP, Arntzenius AC (1984) Shoshin beriberi: a rare diagnostic problem. Br Heart J 51:581–582
Sechi G, Serra A (2007) Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol 6:442–455
Wernicke C (1881) Lehrbuch der Gehirnkrankheiten für Aerzte und Studierende, vol II. Theodor Fischer, Kassel, pp 229–242
Victor M, Adams R, Collins G (1989) The Wernicke–Korsakoff syndrome and related neurologic disorders due to alcoholism and malnutrition. F.A. Davies, Philadelphia
Caine D, Halliday GM, Kril JJ, Harper CG (1997) Operational criteria for the classification of Chronic alcoholics: identification of Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry 62:51–60
Thomson AD, Cook CC, Touquet R, Henry JA (2002) The Royal College of Physicians report on alcohol: guidelines for managing Wernicke’s encephalopathy in the accident and Emergency Department. Alcohol Alcohol 37:513–521
Chu K, Kang DW, Kim HJ, Lee YS, Park SH (2002) Diffusion-weighted imaging abnormalities in Wernicke encephalopathy: reversible cytotoxic edema? Arch Neurol 59:123–127
Sullivan EV, Pfefferbaum A (2009) Neuroimaging of the Wernicke–Korsakoff syndrome. Alcohol Alcohol 44:155–165
Gaitonde MK (1975) Conversion of [U-14C]threonine into 14C-labelled amino acids in the brain of thiamin-deficient rats. Biochem J 150:285–295
Langlais PJ, Mair RG (1990) Protective effects of the glutamate antagonist MK-801 on pyrithiamine-induced lesions and amino acid changes in rat brain. J Neurosci 10:1664–1674
Armstrong-James M, Ross DT, Chen F, Ebner FF (1988) The effect of thiamine deficiency on the structure and physiology of the rat forebrain. Metab Brain Dis 3:91–124
Watanabe I (1978) Pyrithiamine-induced acute thiamine-deficient encephalopathy in the mouse. Exp Mol Pathol 28:381–394
Olney JW (1971) Glutamate-induced neuronal necrosis in the infant mouse hypothalamus. An electron microscopic study. J Neuropathol Exp Neurol 30:75–90
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65:1–105
Hazell AS, Pannunzio P, Rama Rao KV, Pow DV, Rambaldi A (2003) Thiamine deficiency results in downregulation of the GLAST glutamate transporter in cultured astrocytes. Glia 43:175–184
Hazell AS, Sheedy D, Oanea R, Aghourian M, Sun S, Jung JY, Wang D, Wang C (2010) Loss of astrocytic glutamate transporters in Wernicke encephalopathy. Glia 58:148–156
Pabst S, Hazzard JW, Antonin W, Südhof TC, Jahn R, Rizo J, Fasshauer D (2000) Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J Biol Chem 275:19808–19818
Tang J, Maximov A, Shin OH, Dai H, Rizo J, Südhof TC (2006) A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126:1175–1187
Hazell AS, Wang C (2005) Downregulation of complexin I and complexin II in the medial thalamus is blocked by N-acetylcysteine in experimental Wernicke’s encephalopathy. J Neurosci Res 79:200–207
Hazell AS, Wang D (2011) Identification of complexin II in astrocytes: a possible regulator of glutamate release in these cells. Biochem Biophys Res Commun 404:228–232
McMahon HT, Missler M, Li C, Südhof TC (1995) Complexins: cytosolic proteins that regulate SNAP receptor function. Cell 83:111–119
Parpura V, Fang Y, Basarsky T, Jahn R, Haydon PG (1995) Expression of synaptobrevin II, cellubrevin and syntaxin but not SNAP-25 in cultured astrocytes. FEBS Lett 377:489–492
Montana V, Ni Y, Sunjara V, Hua X, Parpura V (2004) Vesicular glutamate transporter-dependent glutamate release from astrocytes. J Neurosci 24:2633–2642
Emerit J, Edeas M, Bricaire F (2004) Neurodegenerative diseases and oxidative stress. Biomed Pharmacother 58:39–46
Schulz JB, Henshaw DR, Siwek D, Jenkins BG, Ferrante RJ, Cipolloni PB, Kowall NW, Rosen BR, Beal MF (1995) Involvement of free radicals in excitotoxicity in vivo. J Neurochem 64:2239–2247
Calingasan NY, Gibson GE (2000) Vascular endothelium is a site of free radical production and inflammation in areas of neuronal loss in thiamine-deficient brain. Ann NY Acad Sci 903:353–356
Calingasan NY, Baker H, Sheu KF, Gibson GE (1995) Blood–brain barrier abnormalities in vulnerable brain regions during thiamine deficiency. Exp Neurol 134:64–72
Torreilles F, Salman-Tabcheh S, Guérin M, Torreilles J (1999) Neurodegenerative disorders: the role of peroxynitrite. Brain Res Brain Res Rev 30:153–163
Desjardins P, Butterworth RF (2005) Role of mitochondrial dysfunction and oxidative stress in the pathogenesis of selective neuronal loss in Wernicke’s encephalopathy. Mol Neurobiol 31:17–25
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Wang X, Wang B, Fan Z, Shi X, Ke ZJ, Luo J (2007) Thiamine deficiency induces endoplasmic reticulum stress in neurons. Neuroscience 144:1045–1056
Gu Z, Nakamura T, Lipton SA (2010) Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Mol Neurobiol. 41(2–3):55–72
Fiedorowicz M, Grieb P (2012) Nitrooxidative stress and neurodegeneration. In: González-Quevedo A (ed) Brain damage—bridging between basic research and clinics. InTech, Rijeka, pp 139–162
Gao HM, Liu B, Zhang W, Hong JS (2003) Novel anti-inflammatory therapy for Parkinson’s disease. Trends Pharmacol Sci 24:395–401
Hazell AS, Butterworth RF (2009) Update of cell damage mechanisms in thiamine deficiency: focus on oxidative stress, excitotoxicity and inflammation. Alcohol Alcohol 44:141–147
Hazell AS (2009) Astrocytes are a major target in thiamine deficiency and Wernicke’s encephalopathy. Neurochem Int 55:129–135
Hong KS, Kang DW, Cho YJ, Hwang YJ, Hur G (2002) Diffusion-weighted magnetic resonance imaging in Wernicke’s encephalopathy. Acta Neurol Scand 105:132–134
Liu YT, Fuh JL, Lirng JF, Li AF, Ho DM, Wang SJ (2006) Correlation of magnetic resonance images with neuropathology in acute Wernicke’s encephalopathy. Clin Neurol Neurosurg 108:682–687
Marchetti B, Abbracchio MP (2005) To be or not to be (inflamed)—is that the question in anti-inflammatory drug therapy of neurodegenerative disorders? Trends Pharmacol Sci 26:517–525
Kohman RA, Rhodes JS (2013) Neurogenesis, inflammation and behavior. Brain Behav Immun 27:22–32
Graeber MB, Streit WJ (1990) Microglia: immune network in the CNS. Brain Pathol 1:2–5
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91:461–553
Wang D, Hazell AS (2010) Microglial activation is a major contributor to neurologic dysfunction in thiamine deficiency. Biochem Biophys Res Commun 402:123–128
Neri M, Cantatore S, Pomara C, Riezzo I, Bello S, Turillazzi E, Fineschi V (2011) Immunohistochemical expression of proinflammatory cytokines IL-1β, IL-6, TNF-α and involvement of COX-2, quantitatively confirmed by Western blot analysis, in Wernicke’s encephalopathy. Pathol Res Pract 207:652–658
Vemuganti R, Kalluri H, Yi JH, Bowen KK, Hazell AS (2006) Gene expression changes in thalamus and inferior colliculus associated with inflammation, cellular stress, metabolism and structural damage in thiamine deficiency. Eur J Neurosci 23:1172–1188
Block ML, Hong JS (2005) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76:77–98
Cardinaux JR, Allaman I, Magistretti PJ (2000) Pro-inflammatory cytokines induce the transcription factors C/EBPbeta and C/EBPdelta in astrocytes. Glia 29:91–97
Hakim AM (1986) Effect of thiamine deficiency and its reversal on cerebral blood flow in the rat. Observations on the phenomena of hyperperfusion, “no reflow,” and delayed hypoperfusion. J Cereb Blood Flow Metab 6:79–85
Lleo A, Galea E, Sastre M (2007) Molecular targets of non-steroidal anti-inflammatory drugs in neurodegenerative diseases. Cell Mol Life Sci 64:1403–1418
Jhala SS, Wang D, Hazell AS (2011) Loss of the glutamate transporter splice-variant GLT-1b in inferior colliculus and its prevention by ceftriaxone in thiamine deficiency. Neurochem Int 58:558–563
Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB (2005) Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433:73–77
Luna-Medina R, Cortes-Canteli M, Sanchez-Galiano S, Morales-Garcia JA, Martinez A, Santos A, Perez-Castillo A (2007) NP031112, a thiadiazolidinone compound, prevents inflammation and neurodegeneration under excitotoxic conditions: potential therapeutic role in brain disorders. J Neurosci 27:5766–5776
Waldmeier PC (2003) Prospects for antiapoptotic drug therapy of neurodegenerative diseases. Prog Neuropsychopharmacol Biol Psychiatry 27:303–321
Papa SM, Chase TN (1996) Levodopa-induced dyskinesias improved by a glutamate antagonist in Parkinsonian monkeys. Ann Neurol 39:574–578
Lindvall O, Kokaia Z, Martinez-Serrano A (2004) Stem cell therapy for human neurodegenerative disorders-how to make it work. Nat Med 10(Suppl):S42–S50
Sierra A, Encinas JM, Maletic-Savatic M (2011) Adult human neurogenesis: from microscopy to magnetic resonance imaging. Front Neurosci 5:47
Gage FH (2000) Mammalian neural stem cells. Science 287:1433–1438
Alvarez-Buylla A, Garcia-Verdugo JM (2002) Neurogenesis in adult subventricular zone. J Neurosci 22:629–634
Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA (2004) Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci USA 101:343–347
Zhao M, Momma S, Delfani K, Carlen M, Cassidy RM, Johansson CB, Brismar H, Shupliakov O, Frisen J, Janson AM (2003) Evidence for neurogenesis in the adult mammalian substantia nigra. Proc Natl Acad Sci USA 100:7925–7930
Curtis MA, Penney EB, Pearson AG, van Roon-Mom WM, Butterworth NJ, Dragunow M, Connor B, Faull RL (2003) Increased cell proliferation and neurogenesis in the adult human Huntington’s disease brain. Proc Natl Acad Sci USA 100:9023–9027
Parent JM, Elliott RC, Pleasure SJ, Barbaro NM, Lowenstein DH (2006) Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann Neurol 59:81–91
Ohab JJ, Fleming S, Blesch A, Carmichael ST (2006) A neurovascular niche for neurogenesis after stroke. J Neurosci 26:13007–13016
Wang C, Liu F, Liu YY, Zhao CH, You Y, Wang L, Zhang J, Wei B, Ma T, Zhang Q, Zhang Y, Chen R, Song H, Yang Z (2011) Identification and characterization of neuroblasts in the subventricular zone and rostral migratory stream of the adult human brain. Cell Res 21:1534–1550
Zhao N, Zhong C, Wang Y, Zhao Y, Gong N, Zhou G, Xu T, Hong Z (2008) Impaired hippocampal neurogenesis is involved in cognitive dysfunction induced by thiamine deficiency at early pre-pathological lesion stage. Neurobiol Dis 29:176–185
Vetreno RP, Klintsova A, Savage LM (2011) Stage-dependent alterations of progenitor cell proliferation and neurogenesis in an animal model of Wernicke–Korsakoff syndrome. Brain Res 1391:132–146
Hazell AS, Wang D, Oanea R, Sun S, Aghourian M, Yong JJ (2014) Pyrithiamine-induced thiamine deficiency alters proliferation and neurogenesis in both neurogenic and vulnerable areas of the rat brain. Metab Brain Dis 29:145–152
Voloboueva LA, Lee SW, Emery JF, Palmer TD, Giffard RG (2010) Mitochondrial protection attenuates inflammation-induced impairment of neurogenesis in vitro and in vivo. J Neurosci 30:12242–12251
Gold M, Chen MF, Johnson K (1995) Plasma and red blood cell thiamine deficiency in patients with dementia of the Alzheimer’s type. Arch Neurol 52:1081–1086
Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, Perrino P (1988) Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Arch Neurol 45:836–840
Singh N, Cohen CA, Rzigalinski BA (2007) Treatment of neurodegenerative disorders with radical nanomedicine. Ann N Y Acad Sci 1122:219–230
Re F, Gregori M, Masserini M (2012) Nanotechnology for neurodegenerative disorders. Nanomedicine 8(Suppl 1):S51–S58
Acknowledgments
The senior author (A.S.H.) is supported by the Canadian Institutes of Health Research (CIHR).
Author information
Authors and Affiliations
Corresponding author
Additional information
Special Issue: In honor of Michael Norenberg.
Rights and permissions
About this article

Cite this article
Abdou, E., Hazell, A.S. Thiamine Deficiency: An Update of Pathophysiologic Mechanisms and Future Therapeutic Considerations. Neurochem Res 40, 353–361 (2015). https://doi.org/10.1007/s11064-014-1430-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-014-1430-z