
Abstract
The Na+ and Ca2+-permeable melastatin related transient receptor potential 2 (TRPM2) channels can be gated either by ADP-ribose (ADPR) in concert with Ca2+ or by hydrogen peroxide (H2O2), an experimental model for oxidative stress, binding to the channel’s enzymatic Nudix domain. Since the mechanisms that lead to TRPM2 gating in response to ADPR and H2O2 are not understood in neuronal cells, I summarized previous findings and important recent advances in the understanding of Ca2+ influx via TRPM2 channels in different neuronal cell types and disease processes. Considering that TRPM2 is activated by oxidative stress, mediated cell death and inflammation, and is highly expressed in brain, the channel has been investigated in the context of central nervous system. TRPM2 plays a role in H2O2 and amyloid β-peptide induced striatal cell death. Genetic variants of the TRPM2 gene confer a risk of developing Western Pacific amyotropic lateral sclerosis and parkinsonism-dementia complex and bipolar disorders. TRPM2 also contributes to traumatic brain injury processes such as oxidative stress, inflammation and neuronal death. There are a limited number of TRPM2 channel blockers and they seem to be cell specific. For example, ADPR-induced Ca2+ influx in rat hippocampal cells was not blocked by N-(p-amylcinnomoyl)anthralic acid (ACA), the IP3 receptor inhibitor 2-aminoethoxydiphenyl borate or PLC inhibitor flufenamic acid (FFA). However, the Ca2+ entry in rat primary striatal cells was blocked by ACA and FFA. In conclusion TRPM2 channels in neuronal cells can be gated by either ADPR or H2O2. It seems to that the exact relationship between TRPM2 channels activation and neuronal cell death still remains to be determined.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Of all the cell types in the body, neuronal cells may be among the most vulnerable to oxidative stress. These cells are continuously exposed to reactive oxygen species (ROS) generated via the auto-oxidation of polyunsaturated fatty acids [1]. A variety of neurodegenerative disease states have been also associated with oxidative stress [2]. Oxidative stress is thought to be mediated by excessive exposure of cells to reactive oxygen or nitrogen species, which can be generated by ischemia, radiation, seizure, trauma etc. Exposure to oxidative stress induces both apoptotic-like delayed neural death and necrosis in cultured neurons, mediated by a rise in intracellular Ca2+ concentration ([Ca2+]i), mitochondrial dysfunction, and activation of poly[adenosine diphosphate ribose (ADPR)] polymerase (PARP) due to DNA damage [2]. In this regard, one potentially important calcium influx pathway may be activation Na+ and Ca2+ permeable Transient Receptor Potential (TRP) channels. Of particular interest is a member of the melastatin subfamily (TRPM2, it is also known as LTRPC2) that is ROS and ADPR gated [3, 4]. TRPM2 channels are widely expressed in brain, and TRPM2 channels have been implicated in neuronal damage induced by oxidants, amyloid β-peptide and tumor necrosis factor alpha (TNF-α) [2, 4]. Therefore, they are regarded as a potential therapeutic target in some neurological disorders including Alzheimer’s disease (AD), bipolar disorders, Parkinson disease (PD) and traumatic brain injury.
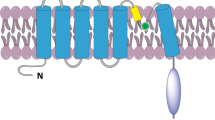
TRPM2, TRPM6, and TRPM7 share a feature unique among known channels, by having a functional enzyme moiety in the C-terminal domain. In TRPM2, this is a type of Nudix hydrolase (NUDT9-H) that can bind to and hydrolyze ADPR, although not as effectively as other known Nudix ADPR-hydrolases (Fig. 1) [5]. Binding of ADPR to NUDT9-H activates the channel, allowing the passage of cations down their electrochemical gradient. Because TRPM2 is a plasma membrane channel, Ca2+ and Na+ will flow into the cell when TRPM2 opens. ADPR is the most potent physiological activator of TRPM2, but other less potent activators have been proposed [6, 7]. These include nicotinamide adenine dinucleotide (NAD+) [4], oxidants such as H2O2 [4], and cyclic ADPR (cADPR) [5]. NAD+ and H2O2 increase intracellular levels of ADPR and could thus gate TRPM2 either directly [5–7] or indirectly [8, 9].

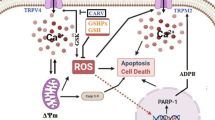
Cells regulate intracellular Ca2+ levels lightly and excessive Ca2+ loads can lead to inappropriate activation of process that are normally operate at low levels, causing metabolic derangements and eventual cell death. For example, excesses elevations in intracellular Ca2+ due to ischemia, radiation, trauma etc. may activate enzymatic degradation, induce formation of reactive oxygen species (ROS) or disrupt normal mitochondrial function leading to oxidative stress and bioenergetic failure. Voltage gated Ca2+ channels (VGCC) and transient potential melastatin 2 (TRPM2)-mediated Ca2+ entry triggers a neurotoxic signal cascade involving the activation of neuronal nitric oxide (NO) synthase (nNOS), formation of the toxic ROS and NO and activation of the pro-apoptotic protein poly[ADP-ribose, (ADPR)] polymerase (PARP-1). The TRPM2 channels are containing C (Nudix box) and N sections. Nodix box domain has ADP-ribose pyrophosphatase activity. The TRPM2 channels can be gated by H2O2 and ADP-Ribose via activation ADP-ribose pyrophosphatase [1, 8]. Antioxidants regulate Ca2+ influx into cytosol by inhibition of ROS. Sustained depolarization of mitochondrial membranes and enhanced ROS production activates TRPM2 and VGCC and Ca2+ influx increases by activation of TRPM2 via ROS [67]. The molecular pathway may be a cause of neurological symptoms and represents a fruitful subject for further study
Several chemicals have been reported to act, as TRPM2 agonists. However, they cannot be easily used for experimental purposes. For example, imidazole, derivates, miconazole and clotrimazole, inhibit TRPM2 in an irreversible manner. Flufenamic acid (FFA) is also a TRPM2 channel blocker. FFA is a nonsteroidal anti-inflammatory drug, is not easily dissolved in aqueous solution which makes it difficult to prepare solutions containing the high concentrations of FFA necessary to inhibit TRPM2. ADPR-induced TRPM2 currents are blocked by FFA [10]. N-(p-amylcinnomoyl)anthranilic acid (ACA) blocks TRPM2 channels blocker although it is relatively nonspecific, inhibiting both phospholipase A2 as well as TRPM2 channels [11]. 2-aminoethyldiphenyl borate (2-APB) blocks 1,4,5-trisphosphate (InsP3) receptors as well as store-operated channels, and it is also a TRPM2 channel blocker in TRPM2-expressing cell lines and rat pancreatic islet cells [12]. All of the TRPM2 channel antagonists, with the exception of 2-APB, either gradually or irreversibly block ADPR-induced currents.
In the current review I summarized previous findings and important recent advances in the understanding of Ca2+ influx via TRPM2 channels in different neuronal cell types and in disease processes. I discussed the possible use of nonspecific TRPM2 channels blockers in neuronal cells.
TRP Superfamily
The trp gene was identified through genetic studies of a mutation in the Drosophilia visual transduction system. The term of “TRP” is derived from “transient receptor potential”, because photoreceptors with the trp gene mutant fail to generate the Ca2+-dependent “sustained” phase of receptor potential and therefore fail to show subsequent Ca2+-dependent adaptation to light [13]. There are 30 mammalian TRP channels, grouped into six subfamilies. Members of the TRP channels superfamily include TRP canonical (TRPC) subfamily consisting of 7, TRP vanilloid (TRPV) subfamily consisting of 6, TRP melastatin (TRPM) subfamily consisting of 8, TRP polycystein (TRPP) subfamily consisting of 3, TRP mucolipin (ML) subfamily consisting of 3, and TRP ankyrin (TRPA) subfamily consisting of only one member. Some members of this superfamily are poorly characterized, but these channels are attracting increasing interest because of their apparent involvement in several human diseases. TRP channels have a basic structure similar to voltage-gated potassium channels, with homo- or hetero-tetrameric arrangements around a central ion conducting pore formed from a sequence between the 5th and 6th transmembrane domains [14]. The N-termini of TRPV and TRPC, but not those of TRPM channels, contain multiple ankyrin binding repeats. The C-terminal part of 6th segment in TRPC and TRPM channels includes the ‘TRP C and N domains’, a conserved stretch of 25 amino acids starting with the nearly invariant ‘TRP box’ that is missing in TRPV channels. In addition, all TRP channels have multiple regulatory protein interaction sites. Multiple protein kinase A (PKA) and C (PKC) putative phosphorylation sites have been identified and partially tested for function [15]. Phosphatidylinositide 3-kinase SH2-recognition domains have also been identified in several TRP channels [15, 16].
TRP channels contribute to changes in [Ca2+]i concentrations by acting as Ca2+ entry channels in the plasma membrane directly or by changing membrane potential, modulating the driving force for other Ca2+ entry channels. All functionally characterized TRP channels are permeable to Ca2+ with the exceptions of TRPM4 and TRPM5, which are only permeable to monovalent cations. Two mammalian TRPs, TRPV5 and TRPV6, are highly Ca2+ permeable. TRPM6 channels are highly selective to Mg2+ [16]. TRPM7 is also a cation selective ion channel that is highly permeable to both Ca2+ and Mg2+, but it can also conduct essential or toxic metals. At least three TRP channels, TRPV1, TRPML1, and TRPP3, are highly permeable to H+ ions [17].
TRPM Subfamily
The TRPM2 subfamily consist of eight members, which are sub-divided into three subgroups on the basis of sequence homology; TRPM1/TRPM3, TRPM4/TRPM5 and TRPM6/7, with TRPM2/TRPM8 being distinct proteins from the other groups (Fig. 2) [1].

The TRPM2 subfamily consist of 8 members, which are sub-divided into four subgroups on the basis of sequence homology; TRPM1/TRPM3, TRPM4/TRPM5, TRPM6/7 and TRPM2/TRPM8
Three members of this subfamily carry a functional enzyme in their COOH-termini: TRPM2 contains a functional NUDT9 homology domain, exhibiting ADPR pyrophophatase activity (Fig. 1), whereas both TRPM6 and TRPM7 contain a functional COOH-terminal serine/threonine kinase. Except for TRPM1 which is not as yet functionally characterized, all TRPM channels are cation channels, although the Ca2+ permeability is diverse, ranging from highly permeable (TRPM6/TRPM7 and splice variants of TRPM3) to Ca2+ impermeable (TRPM4 and TRPM5). TRPM4 and TRPM5 are heat-sensitive, Ca2+ activated channels [17]. TRPM2 is activated by ADPR, H2O2 and heat. TRPM2 and TRPM7 are involved in oxidative stress-induced cell death [4]. Interestingly, TRPM2 and TRPM7 have been reported to play pathological roles in the brain [18]. TRPM8 is the infamous cold receptor [19]. TRPM3 channels are, much like TRPM6 and TRPM7, regulated by intracellular Mg2+ levels. TRPM3 channels show considerable constitutive activity, and TRPM3 activation was also reported after cell swelling and by sphingosine [20]. TRPM7 channels are also activated by oxidative stress, ADPR and both.
TRPM2 Channels
The founding member of the TRPM family is melastatin (TRPM1), so named because it was first described in connection with metastatic melanomas. TRPM2 was formerly known as TRPC2 and LTRPC2. TRPM2 channels were first identified in 1998 [21] and subsequently recognized as a member of the TRPM family [14]. All eight TRPM family members have been linked to disease, either by functional studies in mouse models or by genetic evidence [17]. TRPM2 channels are nonselective cation channels. They are highly expressed in brain [4], where they are preferentially localized in microglia cells, the host macrophages of the central nervous system. A cation current with TRPM2-like properties has been described in rat striatal neurons [22].
Molecular Mechanisms on the Activation of TRPM2 in Neurons
Three extracellular signals are known to active TRPM2; oxidative stress, ADPR/NAD+ metabolism and TNF-α [4]. The channel may also be temperature sensitive with body temperature acting as ‘an endogenous co-activator’ for TRPM2 [14].
Oxidative Stress
Oxidative stress is defined as an imbalance between higher cellular levels and ROS, e.g., superoxide and hydroxyl radicals and cellular antioxidant defense. Generation of ROS is ubiquitous since ROS are generated during aerobic metabolism, i.e., mitochondrial oxidation and phagocytosis. In order to scavenge ROS, various defense systems exist in the brain [2].
ROS act as subcellular messengers when they are generated excessively or when enzymatic and non-enzymatic defense systems are impaired [1], and can regulate such complex processes as mitogenic signal transduction, gene expression, and cell proliferation. While many intra- and extracellular molecules may participate in neuronal injury and cell apoptosis, accumulation of oxidative stress due to excessive generation of ROS appears to be a potential factor for cell damage and death [2].
Role of Oxidative Stress on Activation of TRPM2
Oxidative stress, for which application of H2O2 is an experimental paradigm, induces TRPM2 currents and an increase in [Ca2+]i concentrations in various cell types transfected with TRPM2 [6, 7], as well as in pancreatic β-cells [9], neutrophil granulocytes [5], and U937 monocytes [19, 23]. However, the exact mediator for H2O2-induced TRPM2 channel activation remains to be identified. The ROS sensitivity of TRPM2 could be mediated by NAD+ [4] or ADPR [4, 24] released from mitochondria or through protein oxidation [25]. H2O2 and cADPR have been proposed to potentiate the effect of ADPR at lower concentrations and to gate the TRPM2 channel directly at higher concentrations [26].
It was reported that oxidative stress-induced TRPM2 activation is triggered via the production of ADPR from mitochondria [1]. N domain of TRPM2 has ADPR pyrophosphates enzyme activity and the enzyme activated by oxidative stress and ADPR. ADPR is synthesized from NAD of mitochondria and nucleolus via different three ways [1, 8]. In the nucleus, ADPR generation is attributed to a pathway involving poly(ADPR) polymerase-1 (PARP-1), and may be initiated by DNA damage because of different factors such as oxidative stress and radiation. Free ADPR is generated following the degradation of poly(ADPR) by poly(ADPR) glycohydrolase (PARG) and ADP-ribosyl protein lyase activities [27]. Thus, it may be hypothesized that free-ADPR, which activates TRPM2, is produced by the activation of PARP-1 and PARG. However, electrophysiological studies have shown that PARP inhibitors don’t interfere with activation of TRPM2 by ADPR [8, 28].
Role of ADPR/NAD+ Products on Activation of TRPM2
Ca2+ influx via TRPM2 is thought to occur via production of ADPR. ADPR may arise from a mitochondrial source or alternatively via activation of poly(ADPR). ADPR may also arise from a mitochondrial source [25].
NAD+ has been reported to stimulate TRPM2 [4, 6]. TRPM2 contains a characteristic structural feature known as a Nudix domain in its C-terminal cytosolic tail [23]. A Nudix domain is a consensus region that is known to be present in a class of pyrophosphates that degrade nucleoside diphosphates (Fig. 1) [1]. Indeed, the nudix domain of TRPM2 cleaves ADPR, a breakdown product of NAD+ and cyclic ADPR, the latter being an intracellular second messenger that stimulates Ca2+ release by ryanodine receptors [4]. While ADPR is hydrolyzed by TRPM2, it also activates TRPM2 and induces TRPM2 currents when infused by patch pipettes [4, 23]. In two recent studies, we observed activation of TRPM2 by NAD+, which supports the idea that this substance activates TRPM2 directly [9, 10]. A more recent study [29] reported that extracellular cyclic-ADP-ribose (cADPR) induced temperature-sensitive [Ca2+]i concentration increases by Ca2+ influx through TRPM2 channels in rodent NG108-15 neuronal cells.
Previously published data indicated that TRPM2 can be gated by H2O2 and it has been suggested that this occurs independently of ADPR [1, 6, 7, 24]. Perraud et al. [23] demonstrated that reducing the ADPR concentration within mitochondria largely suppresses H2O2-mediated activation of TRPM2, suggesting that the gating mechanism of H2O2 is primarily based on its ability to release ADPR from mitochondria, which then proceeds to active TRPM2. However, Kolisek et al. [30] recently demonstrated that H2O2 may act as a direct stimulus for TRPM2 activation, as it can initiate the release of ADPR from mitochondria and at the same time function as a potentiating cofactor of ADPR. In our recent study, we observed that the H2O2-mediated activation of TRPM2 appears to result from a direct gating mechanism, because TRPM2 activation by H2O2 was relatively rapid in whole cell recordings, and the compound triggered single-channel activity in excised membrane patches [6]. After adding H2O2 to the bath after recording in the inside out configuration, the patches were washed by intracellular buffer and then H2O2 was given again. The channel was activated again by a second administration of H2O2 although no intracellular components should be present. From this, we concluded that H2O2-induced, single channel activity observed in excised membrane patches is likely caused by a direct gating mechanism.
Methodological problems are likely responsible for differing reports on the activation of TRPM2 by NAD+, ADPR and H2O2. For examples, in a previous study [24] using HEK293 cells as an expression system, we demonstrated characteristic currents through the splice variant of TRPM-ΔC induced by H2O2. In later experiments using a CHO expression system, we could stimulate wild-type TRPM2 channels with NAD+ but we could not stimulate them with H2O2 with sufficient consistency [30].
Second problem leading to differing results arises from the use of different cell types to study TRPM channel activation by H2O2, ADPR and its metabolites. On the other hand, channel activation by H2O2 appears to represent a cell-specific process in cells with endogenous expression of TRPM2. For example, the TRPM2 channel is activated by H2O2 in HEK293 cells [4, 24], CHO cells [31], CRI-GI rat insulinoma cell lines [9], rat primary striatal cultures [32] but not in human neutrophil granulocytes [5]. Kolisek et al. [30] in experiments with HEK293 cells found that cADPR stimulated TRPM2 channels. Later, this report was supported by the results of Gasser et al. [33] who reported that TRPM2 channels in Jurkat cells were gated by cADPR. However, a study from Aachen did not support the results for neutrophils granulocytes [5] where TRPM2 channels were not activated by cADPR.
Studies of TRPM2 Knockout Mice
The first TRPM2 knockout (TRPM2-KO) mouse study was published in 2008 [19], and additional studies using this model have continued. For example, an important advance in understanding the biological role of TRPM2 is provided by a study on TRPM2-KO mice [19]. The H2O2-activated signaling cascade involved in the production of inflammatory protein-2 cytokine is impaired in monocytes from TRPM2-KO mice. Recently, Lange et al. [34] reported that in TRPM2-KO mice, TRPM2 functions as a Ca2+-release channel activated by intracellular ADPR in a lysosomal compartment in addition to its role as a plasma membrane channel. They showed that both functions of TRPM2 are critically linked to hydrogen peroxide-induced beta cell death. Additionally, extracellular ADPR production by the ectoenzyme CD38 from its substrates NAD+ or cADPR causes IP3-dependent Ca2+ release via P2Y and adenosine receptors. Lange et al. [34] concluded that ADPR and TRPM2 represent multimodal signaling elements regulating Ca2+ mobilization in β-cells through membrane depolarization, Ca2+ influx, and release of Ca2+ from intracellular stores.
Role of TRPM2 Channels in Neuronal Cells
Northern blotting and quantitative PCR techniques indicted that TRPM2 is broadly expressed in the central nervous system (CNS). However, as this evidence was derived from homogenized tissue samples, it does not allow expression in neurons to be distinguished from that in glial cells. The importance of making such a distinction is highlighted by a recent study that failed to identify TRPM2 transcripts or functional channels in cerebellar granule cells and astrocytes [35]. Rather, TRPM2 was detected in microglial cells leading to the suggestion that the CNS distribution of TRPM2 corresponds to its expression in non-neuronal cells (Table 1) [36, 37].
Role of Oxidative Stress on TRPM2 Channels in Microglia and Astroglia Cells
The NADPH-oxidase in phagocytic cells such as microglia is an electron transport system that catalyzes the reduction of oxygen to superoxide radical. Under physiological conditions, the system contributes to the elimination of pathogens but under chronic inflammatory conditions such as scleroderma and liver fibrosis it is through to induce neurodegeneration by the massive accumulation of superoxide radical [2]. Monocyte superoxide, in high glucose media, is released by the NADPH-oxidase but not by the mitochondrial respiratory chain, and antioxidant such as α-tocopherol inhibits superoxide release via inhibition of PKC-α. The assembly of a functional NADPH-oxidase complex at the plasma membrane depends on the phosphorylation and subsequent translocation of several cytosolic subunits (p40phox, p47phox, p67phox, and Rac1/2) [38]. In microglia cells, antioxidants inactivates PKC via phosphatase-mediated pathway (PP1 or PP2A) and, as a consequence, block the phosphorylation-dependent translocation of p67phox to the plasma membrane. As a result, the production of superoxide radical by the microglial NADPH-oxidase system is substantially inhibited, offering a partial explanation for the beneficial effect of antioxidants such as α-tocopherol on a variety of neurodegenerative diseases [2].
Like other activated macrophages, microglia remove bacteria and cellular debris and produce a diverse range mediators of the inflammatory cascade including arachidonic acid derivatives and H2O2. Thus microglia cells are a key factor in the immune defense and tissue repair in the central nervous system. Kraft et al. [35] described a novel calcium influx pathway in microglial cells coupled to hydrogen peroxide and ADPR and provided evidence that this pathway involved TRPM2 although they failed to detect TRPM2 in cultured cerebellar granule neurons. Recently, Ohana et al. [39] investigated expression of putative Ca2+-permeable TRPM2 channels in rat cultured microglial cells by quantitative real-time RT-PCR. They detected transcripts in the rat cultured microglial cells for TRPM2 genes.
Although the role of astroglia in the progression of neurodegenerative disease is still relatively unknown, their importance in regulating the normal and abnormal neuronal environment is attracting increasing attention. Through Ca2+ signaling cascades, astroglia control gene expression, neuronal differentiation, and programmed cell death, which are all integral to developmental and degenerative processes. Under conditions of oxidative stress, glial cells provide energy support for neurons, exert a protective function by scavenging and detoxifying ROS, and direct neuronal resistance or vulnerability to degeneration through Ca2+-dependent secretion of trophic or inflammatory factors [40]. There are few reports on TRPM2 in astroglia. Bond and Greenfield [41] reported that the additive effects of L-VGCC blockade and TRPM2 inhibition during oxidative stress significantly enhanced recovery from protein synthesis suppression and repressed subsequent compensatory protein over-expression. These results indicated that Ca2+ signaling is integral to astroglial transcriptional and translational responses to oxidative stress.
Role of Oxidative Stress on TRPM2 Channel in Hippocampal Neurons
As it was mentioned in previous section, TRPM2 is expressed in diverse cell types and despite convincing evidence for high expression in the mammalian brain, much of this signal was attributed to strong expression in non-neuronal cells. Thus, the existence of functional TRPM2 channels in neurons is controversial at best. Recently, Lipski et al. [42] reported that TRP or TRP-like channels including TRPM2 in hippocampal CA1 neurons are activated by cellular stress and contribute to ischemia-induced membrane depolarization, intracellular calcium accumulation and cell swelling. More recently, Olah et al. [37] reported that functional TRPM2 channels are highly expressed in pyramidal neurons of hippocampus, including those of CA1 interneurons in hippocampal slices. They also reported that ADPR alone is insufficient to gate TRPM2 in hippocampal neurons. They concluded that concomitant influx of Ca2+ through voltage dependent Ca2+ channels and or NMDAR is necessary to fully active TRPM2 channels. Recently, Bai and Lipski [43] investigated expression of TRPM2 and TRPV4 channels and their potential role in oxidative stress-induced cell damage in organotypic hippocampal slice cultures. In this study oxidative stress induced by H2O2 (600 μM) caused preferential damage of pyramidal neurons although oxidative stress induced with mercaptosuccinate (400 μM) or buthionine sulfoximine (4 μM) damaged astrocytes. Antioxidants (Trolox 500 μM, MitoE 2 μM) reduced both neuronal and astrocytic cell damage.
TRPM2 Channels and Dorsal Root Ganglion Neurons
There are several types of sensory neurons in the DRG, with responsiveness to different of external and internal stimuli. These stimuli, i.e. nociceptive, thermal or mechanical, activate different receptors and ion channels that are present in the nerve terminals at the sensory receptive fields and their expression in selective subsets of DRG neurons determines the response profile of non-selective cation channels that play important functions in sensory neurons. TRPM8 is the only TRPM channel with a clearly assigned function in DRG neurons. It is activated by innocuous cool stimuli and responds to menthol and icilin with intracellular Ca2+ elevations [16, 17]. Recently, TRPM2 channels were for the first time detected in DRG cells of mouse [44]. These investigators observed also that levels of TRPM2 channels in DRG cells were significantly higher in lumbar tissue than in thoracic tissue in the adult mouse although they did not detect the channels in the DRG cells of embryonic day (12) to 12 weeks age. Recently, my group was performed in whole cell patch clamp experiments in DRG cells of mouse and we observed that the TRPM2 channels in DRG cells of mouse were activated by either ADPR (1 mM) or rotenone (1 μM) (Submitted data).
Role of TRPM2 Channels in Oxidative Stress-Induced Neurological Diseases
TRPM2 is highly expressed in the brain, in both microglia and neuronal cells, but its biological role in these cells still needs to be understood. There are few studies on the role of TRPM2 cations channels in neurological diseases those few suggest that Ca2+ influx via TRPM2 is necessary for microglia and other phagocytes to mount effective inflammatory and clearance responses [35].
Alzheimer’s Disease, Oxidative Stress and TRPM2 Channels
Alzheimer’s disease (AD) is a form of dementia in which patients show neurodegeneration, complete loss of cognitive abilities and prematurely die. Central to the neurodegenerative process is the inability of neurons to properly regulate [Ca2+]i concentration. In AD, correlations between the pathological hallmarks of AD (amyloid plaques and neurofibrillary tangles) and perturbed cellular Ca2+ homeostasis have been established in studies of patients, as well as in animal and cell culture models of AD. Specifically, increased levels of amyloid β-peptide (Aβ) induce neurotoxic factors including ROS and cytokines, which impair cellular Ca2+ homeostasis and render neurons vulnerable to apoptosis and excitotoxicity [18].
Ca2+ influx via H2O2-activated TRPM2 induces cell death in rat insulinoma RIN-5F cells expressing TRPM2; TRPM2-specific antisense oligonucleotide significantly suppressed Ca2+ influx and cell death induced by H2O2 [4]. TRPM2-specific antisense almost completely abolished TNF-α-evoked [Ca2+]i concentration oscillation in RIN-5F cells, similar to the effect of omitting extracellular Ca2+, and also significantly suppressed TNF-α-induced death in RIN-5F cells [4]. These results suggest an important role of TRPM2 in TNF-α-activated Ca2+ channels that mediate cell death. Therefore, it is possible that TNF-α released from Aβ-activated microglia triggers neuronal cell death via TRPM2 activation in AD. In fact, TRPM2 expressed in rat cortical neurons is critically involved in H2O2-induced Ca2+ influx that causes neuronal cell death [45]. Importantly, Fonfria et al. [32] have suggested that activation of TRPM2, functionally expressed in primary cultures of rat striatum, contributes to Aβ- and oxidative stress-induced striatal cell death, thereby providing evidence that TRPM2 activity may contribute to neuronal cell death in pathophysiological circumstances in which ROS are generated abundantly.
Ca2+ influx through TRPM2 then causes a positive feedback loop of ROS production, which kills the neuronal cells. In primary cortical neurons, knockdown of TRPM7 also results in knockdown of TRPM2, suggesting that expression of these two proteins in cortical neurons is co-ordinated in some manner. Yoshida et al. [46] reported that H2O2-induced Ca2+ responses mediated by TRPM7 were very small compared to those from TRPM2. Considering that TRPM2 and TRPM7 are activated by ROS and plays a critical role for the progression of anoxic cell death in a model of in vitro ischemic neuronal injury, it is then possible that Ca2+ overload via oxidative stress-activated TRPM7 may participate in the development of AD [18].
TRPM2 Channels, Western Pacific Amyotrophic Lateral Sclerosis and Parkinson Disease
Among neurodegenerative diseases, ALS and PD are ideal for studying the relative contributions of genes and environment in disease etiology because they occur in geographically separate foci among three genetically different, homogenous groups of people [47]. Intensive research conducted over the years led to the identification of two candidate environmental factors: (1) severely low levels of Ca2+ and Mg2+ in the soil and drinking water [48]; and (2) the putative neurotoxin-methylamino-l-alanine derived from the cycad plant, a traditional food source in Guam [49]. These findings led to the hypothesis that prolonged exposure to such an environment could be involved in the pathogenesis of Western Pacific ALS and PD [49]. Supporting this hypothesis are observations that afflicted individuals exhibit altered Ca2+ metabolism, and experimental reports of neuronal damage in experimental models of animals fed diets that mimic the mineral composition in the disease foci environment [50].
TRPM2 has been implicated in cell death induced by oxidants [4, 7, 24]. The presence of low Mg2+ and high transition metals in the Western Pacific ALS and PD foci create conditions of increased oxidative stress [47]. As a channel with high expression profile in microglia and neuronal cells that could sense and respond to oxidative stress, TRPM2 is an attractive candidate for a susceptibility gene for these disorders. Hermosura et al. [47] reported the presence of heterozygous TRPM2P1018L, a heterozygous variant of TRPM2 in the pathogenesis of Western Pacific ALS and PD. They observed also that P1018L forms functional channels that activate in response to H2O2 and ADPR. However, in the presence of physiological concentrations of extracellular Ca2+, P1018L channels inactivate quickly and are thus unable to allow sustained ion influx. In intact cells, this is manifested as an attenuated Ca2+ rise in response to H2O2. Defective Ca2+ handling is implicated in many diseases, including neurodegeneration. Of particular interest is a recent report that described another Pro-to-Leu substitution, this time in CalHM1, a putative Ca2+-permeable channel [51]. CalHM1P86L increases risk for Alzheimer’s disease. In functional studies, cells expressing CalHM1P86L proteins exhibited attenuated Ca2+ permeability, reduced cytosolic Ca2+ levels, and increased amyloid deposition. The similarity in the effects of CalHM1P86L and TRPM2P1018L suggests that attenuation of intracellular Ca2+ rises and its effects on downstream signaling pathways may contribute to the pathophysiological mechanism in neurodegenerative diseases [47].
One frequent studied animal model of PD, relevant to the ‘environmental toxin’ hypothesis of PD [52], is based on the use of rotenone, a naturally occurring isoflavonoid from the tropical plants Lonchocarpus and Derris [53]. Rotenone inhibits complex I of the mitochondrial electron chain reaction. It also decreases intracellular ATP levels and increases the production of ROS [54]. It also releases glutamate from presynaptic terminals’ leading to an additional increase in ROS production [55]. ROS gates TRPM2 channels [5]. Recently, Freestone et al. [54] indicated that TRPM2 or TRPM2-like channels are activated by ROS during exposure of substantia nigra pars compacta (SNc) neurons to rotenone. Their evidences were based on the following observations: rotenone induced a fast rise in [Ca2+]i concentration although the rise in [Ca2+]i concentration was reduced by N-(p-amylcinnamoyl) anthranilic acid (ACA); scavenging ROS by Trolox also decreased the rotenone-induced [Ca2+]i concentration increase.
TRPM2 Channels and Bipolar Disorders
While the C-terminal cytosolic tail mediates the interaction with ADPR, the role of the N-terminal tail that is also located within the cytosol has not been defined on a molecular level. A number of N-terminal truncated isoforms of TRPM2 have been identified and in some cases they regulate the function of the full-length channel [36]. In general, the N terminus is indispensable. Deletion of a stretch of 20 amino acid residues (Δ537–556), as has been found in the TRPM2-ΔN splice variant in neutrophils, abolishes any channel function [24]. This ΔN stretch comprises several structural elements which may explain why TRPM2-ΔN is dysfunctional. First, it contains an IQ-like sequence motif that represents a CaM binding domain [56]. Interestingly, a further IQ-like motif is found immediately downstream of the ΔN stretch. Second, the ΔN stretch contains two PxxP motifs that are characteristic for sites enabling interaction with other proteins [57]. For TRP channels the significance of endogenous PxxP motifs until now has only been investigated in the TRPC channel subfamily [58]. Although so far no proteins are known that form functional protein complexes with TRPM2, TRPM2 is unusually rich in PxxP motifs in comparison to other channels of the TRP superfamily. As a further remarkable property of the ΔN stretch, the conservative substitution of a single amino acid residue (D543E) was correlated with the presence of bipolar disorder in members of a family with a high incidence of this disease [59, 60]. Recently, it was reported that no functional role can be attributed to any of the structural motifs within the ΔN stretch, and that this sequence may act as a spacer segment for other functional sites in the N terminus [61].
Traumatic Brain Injury, Oxidative Stress and TRPM2 Channel
Traumatic brain injury is a leading cause of mortality and disability among the young population. Motor vehicle accidents accounts for the majority severe traumatic brain injury cases, while falls, sporting accidents and assault are responsible for most mild to moderate injuries. Traumatic brain injury contributes to fuelling a chronic central nervous system inflammation with increased formation of proinflammatory cytokines, enzymes and ROS [62]. ROS promote oxidative stress, which leads to neurodegeneration and ultimately results in programmed cell death. TRPM2 is implicated in inflammatory pathways, specifically as a key participant in monocyte chemokine production induced by H2O2 [19]. More recently, Cook et al. [63] investigated TRPM2 transcript and protein levels following experimental traumatic brain injury. They reported increases in TRPM2 mRNA and protein expression in cortical and hippocampal neurons of injured animals, and suggested a role for TRPM2 in traumatic brain injury pathophysiology.
Neuronal Cells and TRPM2 Channel Blockers
Phospholipase A(2) enzymes display a superfamily of structurally different enzymes classified in at least nine subfamilies by biochemical and structural properties. N-(p-amylcinnamoyl)anthranilic acid (ACA) is often used as a broad-spectrum inhibitor for the characterization of phospholipase A(2)-mediated pathways. Compounds like ACA and ACA-like structures have been described to block the receptor-induced release of arachidonic acid and subsequent signaling cascades in the pancreas and the cardiovascular system. Harteneck et al. [11] reported that ACA at 20 μM directly blocks several transient receptor potential (TRP) channels (TRPC6, TRPM2 and TRPM8).
Several TRPM2 blockers are shown in Table 2. Fenamates like flufenamic acid (FFA) are anti-inflammatory drugs known to alter ion fluxes through the plasma membrane. They are for instance potent blockers of cation and anion channels, and FFA at ≥50 μM is now commonly used to block currents through TRP channels and receptor-operated channels [31]. Two other substances, clotrimazole and econazole, have been described to inhibit TRPM2 currents at 3–30 μM concentrations [64]. Recently, Olah et al. [37] reported that the voltage-ramp-activated ADPR-primed currents in cultured neurons were strongly depressed by recombinant TRPM2 channel blockers including clotrimazole, FFA and ACA.
Ca2+ influx observed during rotetone application is due to activation of TRPM2 channels. Freestone et al. [54] indicated that TRPM2 or TRPM2-like channels are activated by ROS during exposure of SNc neurons to rotenone. They observed also that ACA and FFA did not change resting [Ca2+]i concentration or any other measured membrane property. ACA, however, caused a decrease in rotenone-induced [Ca2+]i concentration increase while FFA induced a tendency to decrease the [Ca2+]i concentration increase. Pretreatment with the antioxidant Trolox (500 μM for 10 min) also decreased the rotenone (200 nM)-induced [Ca2+]i concentration rise, consistent with the crucial role of a ROS-sensitive Ca2+ entry mechanisms.
Bai and Lipski [43] investigated the effects of three putative blockers of TRPM2 channels. Clotrimazole (20 μM), ACA (25 μM) and FFA (200 μM), in organotypic hippocampal slice cultures. They concluded that the three chemicals failed to protect pyramidal neurons from damage by exogenous H2O2 (600 μM) and increased damage of these neurons and astrocytes caused by oxidative stress induced with mercaptosuccinate (400 μM) or buthionine sulfoximine (4 μM). Antioxidants (Trolox 500 μ; MitoE 2 μM) reduced both neuronal and astrocytic cell damage. Recently, my group investigated antagonist effects of 2-APB and FFA in DRG cells of mouse by whole cell patch clamp experiments. Recently, we observed that ADPR (1 mM)-induced TRPM2 channel currents in the cells were inhibited by 2-APB (50 μM) and FFA (100 μM) (Submitted data).
Conclusions
The evidence for association of TRPM2 variants with neurological diseases such as Bipolar disorders suggested that genetic variations in TRPM2 may influence susceptibility to the diseases. In addition, the identification of TRPM2 as a key component of the neurological Ca2+ entry pathway in response to reactive oxygen species sheds new light on the physiology and pathophysiology of neuronal cells and other cell types in the brain. Because there is substantial evidence for the deteriorating role of oxidative stress in neurological and brain dysfunction, manipulating TRPM2 function in neuronal cells may be highly useful in the future for experimental therapies of brain and neurological dysfunctions.
Endogenous ADP-ribose, NAD+ and exogenous H2O2 in neuronal cells have been shown to gate TRPM2 channels although their effects seem to be cell type specific. Similarly, based on studies in cell lines, there appear to be limited indirect TRPM2 channel blockers such as ACA and FFA, and their channel blocking effects seem also to cell type specific. The lack of highly specific and protective TRPM2 blockers unfortunately prevents delineation of the exact relationship between TRPM2 channel activation and neuronal cell death. Hopefully continued research in this important area will produce new and more useful agents leading to a better understanding of the role of TRPM2 channels in physiological and pathological pathways.
Abbreviations
- 2-APB:
-
2-Aminoethoxydiphenyl borate.
- ACA:
-
N-(p-amylcinnomoyl)anthranilic acid
- AD:
-
Alzheimer’s disease
- ADPR:
-
Adenosine diphosphatase ribose
- CHO:
-
Chinese hamster ovary
- CNS:
-
Central nervous system
- DRG:
-
Dorsal rood ganglion
- FFA:
-
Flufenamic acid
- HEK:
-
Human embryonic kidney
- PARG:
-
Poly (ADP-ribose) glycohydrolase
- PARP:
-
Poly(ADP-ribose) polymerase
- PD:
-
Parkinson disease
- PKC:
-
Protein kinase C
- ROS:
-
Reactive oxygen species
- TNF-α:
-
Tumor necrosis factor alpha
- TRP:
-
Transient receptor potential
References
Nazıroğlu M (2007) New molecular mechanisms on the activation of TRPM2 channels by oxidative stress and ADP-ribose. Neurochem Res 32:1990–2001
Halliwell B (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97:1634–1658
Kühn FJ, Heiner I, Luckhoff A (2005) TRPM2: a calcium influx pathway regulated by oxidative stress and the novel second messenger ADP-ribose. Pflugers Arch 451:212–219
Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y (2002) LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 9:163–173
Heiner I, Eisfeld J, Warnstedt M, Radukina N, Jungling E, Luckhoff A (2006) Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem J 398:225–232
Nazıroğlu M, Lückhoff A (2008) A calcium influx pathway regulated separately by oxidative stress and ADP-ribose in TRPM2 channels: Single channel events. Neurochem Res 33:1256–1262
Nazıroğlu M, Lückhoff A (2008) Effects of antioxidants on calcium influx through TRPM2 channels in transfected cells activated by hydrogen peroxide. J Neurol Sci 270:152–158
Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, Knowles HM, Ferraris D, Li W, Zhang J, Stoddard BL, Scharenberg AM (2005) Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem 280:6138–6148
Inamura K, Sano Y, Mochizuki S, Yokoi H, Miyake A, Nozawa K, Kitada C, Matsushime H, Furuichi K (2003) Response to ADP-ribose by activation of TRPM2 in the CRI-G1 insulinoma cell line. J Membr Biol 191:201–207
Hill K, Benham CD, McNulty S, Randall AD (2004) Flufenamic acid is a pH-dependent antagonist of TRPM2 channels. Neuropharmacology 47:450–460
Harteneck C, Frenzel H, Kraft R (2007) N-(p-amylcinnamoyl)anthranilic acid (ACA): a phospholipase A(2) inhibitor and TRP channel blocker. Cardiovasc Drug Rev 25:61–75
Togashi K, Inada H, Tominaga M (2008) Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB). Br J Pharmacol 153:1324–1330
Yamamoto S, Takahashi N, Mori Y (2010) Chemical physiology of oxidative stress-activated TRPM2 and TRPC5 channels. Prog Biophys Mol Biol 103:18–27
Clapham DE (2003) TRP channels as cellular sensors. Nature 426:517–524
Watanabe H, Murakami M, Ohba T, Takahashi Y, Ito H (2008) TRP channel and cardiovascular disease. Pharmacol Therap 118:337–351
Nilius B (2007) TRP channels in disease. Biochim Biophys Acta 1772:805–812
Nilius B, Owsianik G, Voets T, Peters JA (2007) Transient receptor potential cation channels in disease. Physiol Rev 87:165–217
Yamamoto S, Wajima T, Hara Y, Nishida M, Mori Y (2007) Transient receptor potential channels in Alzheimer’s disease. Biochim Biophys Acta 1772:958–967
Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, Kaneko S, Lange I, Fleig A, Penner R, Nishi M, Takeshima H, Mori Y (2008) TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med 14:738–747
Hoffmann A, Grimm C, Kraft R, Goldbaum O, Wrede A, Nolte C, Hanisch UK, Richter-Landsberg C, Brück W, Kettenmann H, Harteneck C (2010) TRPM3 is expressed in sphingosine-responsive myelinating oligodendrocytes. J Neurochem 114:654–665
Nagamine K, Kudoh J, Minoshima S, Kawasaki K, Asakawa S, Ito F, Shimizu N (1998) Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics 54:124–131
Hill K, Tigue NJ, Kelsell RE, Benham CD, McNulty S, Schaefer M, Randall AD (2006) Characterization of recombinant rat TRPM2 and a TRPM2-like conductance in cultured rat striatal neurons. Neuropharmacology 50:89–97
Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM (2001) ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 411:595–599
Wehage E, Eisfeld J, Heiner I, Jüngling E, Zitt C, Lückhoff A (2002) Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem 277:23150–23156
Guse AH (2005) Second messenger function and the structure-activity relationship of cyclic adenosine diphosphoribose (cADPR). FEBS J 272:4590–4597
Massullo P, Sumoza-Toledo A, Bhagat H, Partida-Sánchez S (2006) TRPM channels, calcium and redox sensors during innate immune responses. Semin Cell Dev Biol 17:654–666
Virag L, Szabo C (2002) The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev 54:375–429
Fonfria E, Marshall IC, Benham CD, Boyfield I, Brown JD, Hill K, Hughes JP, Skaper SD, McNulty S (2004) TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol 143:186–192
Amina S, Hashii M, Ma WJ, Yokoyama S, Lopatina O, Liu HX, Islam MS, Higashida H (2010) Intracellular calcium elevation induced by extracellular application of cyclic-ADP-ribose or oxytocin is temperature-sensitive in rodent NG108–15 neuronal cells with or without exogenous expression of human oxytocin receptors. J Neuroendocrinol 22:460–466
Kolisek M, Beck A, Fleig A, Penner R (2005) Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell 18:61–69
Nazıroğlu M, Lückhoff A, Jungling E (2007) Antagonist effect of flufenamic acid on TRPM2 cation channels activated by hydrogen peroxide. Cell Biochem Funct 25:383–387
Fonfria E, Marshall IC, Boyfield I, Skaper SD, Hughes JP, Owen DE, Zhang W, Miller BA, Benham CD, McNulty S (2005) Amyloid beta-peptide(1–42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J Neurochem 95:715–723
Gasser A, Glassmeier G, Fliegert R, Langhorst MF, Meinke S, Hein D, Kruger S, Weber K, Heiner I, Oppenheimer N, Schwarz JR, Guse AH (2006) Activation of T cell calcium influx by the second messenger ADP-ribose. J Biol Chem 281:2489–2496
Lange I, Yamamoto S, Partida-Sanchez S, Mori Y, Fleig A, Penner R (2009) TRPM2 functions as a lysosomal Ca2+-release channel in beta cells. Sci Signal 2:ra23
Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, Schultz G, Harteneck C (2004) Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol 286:C129–C137
Perraud AL, Schmitz C, Scharenberg AM (2003) TRPM2 Ca2+ permeable cation channels: from gene to biological function. Cell Calcium 33:519–531
Olah ME, Jackson MF, Li H, Perez Y, Sun HS, Kiyonaka S, Mori Y, Tymianski M, MacDonald JF (2009) Ca2+-dependent induction of TRPM2 currents in hippocampal neurons. J Physiol 587:965–979
Nazıroğlu M (2007) Molecular mechanisms of vitamin E on intracellular signaling pathways in brain. In: Goth L (ed) Reactive oxygen species and diseases. Research Signpost Press, Kerala, pp 239–256
Ohana L, Newell EW, Stanley EF, Schlichter LC (2009) The Ca2+ release-activated Ca2+ current (ICRAC) mediates store-operated Ca2+ entry in rat microglia. Channels 3:129–139
Morale MC, Serra PA, L’episcopo F, Tirolo C, Caniglia S, Testa N, Gennuso F, Giaquinta G, Rocchitta G, Desole MS, Miele E, Marchetti B (2006) Estrogen, neuroinflammation and neuroprotection in Parkinson’s disease: glia dictates resistance versus vulnerability to neurodegeneration. Neuroscience 138:869–878
Bond CE, Greenfield SA (2007) Multiple cascade effects of oxidative stress on astroglia. Glia 55:1348–1361
Lipski J, Park TI, Li D, Lee SC, Trevarton AJ, Chung KK, Freestone PS, Bai JZ (2006) Involvement of TRP-like channels in the acute ischemic response of hippocampal CA1 neurons in brain slices. Brain Res 1077:187–199
Bai JZ, Lipski J (2010) Differential expression of TRPM2 and TRPV4 channels and their potential role in oxidative stress-induced cell death in organotypic hippocampal culture. Neurotoxicology 31:204–214
Staaf S, Franck MC, Marmigère F, Mattsson JP, Ernfors P (2010) Dynamic expression of the TRPM subgroup of ion channels in developing mouse sensory neurons. Gene Expr Patterns 10:65–74
Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, Takada Y, Kume T, Katsuki H, Mori Y, Akaike A (2006) A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J Pharmacol Sci 101:66–76
Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, Mori Y (2006) Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol 2:596–607
Hermosura MC, Cui AM, Go RC, Davenport B, Shetler CM, Heizer JW, Schmitz C, Mocz G, Garruto RM, Perraud AL (2008) Altered functional properties of a TRPM2 variant in Guamanian ALS and PD. Proc Natl Acad Sci USA 105:18029–18034
Yase Y (1972) The pathogenesis of amyotrophic lateral sclerosis. Lancet 2:292–296
Spencer PS, Nunn PB, Hugon J, Ludolph AC, Ross SM, Roy DN, Robertson RC (1987) Guam amyotrophic lateral sclerosis-parkinsonism-dementia linked to a plant excitant neurotoxin. Science 237:517–522
Yanagihara R, Garruto RM, Gajdusek DC, Tomita A, Uchikawa T, Konagaya Y, Chen KM, Sobue I, Plato CC, Gibbs CJ Jr (1984) Calcium and vitamin D metabolism in Guamanian Chamorros with amyotrophic lateral sclerosis and parkinsonism-dementia. Ann Neurol 15:42–48
Dreses-Werringloer U, Lambert JC, Vingtdeux V, Zhao H, Vais H, Siebert A, Jain A, Koppel J, Rovelet-Lecrux A, Hannequin D, Pasquier F, Galimberti D, Scarpini E, Mann D, Lendon C, Campion D, Amouyel P, Davies P, Foskett JK, Campagne F, Marambaud P (2008) A polymorphism in CALHM1 influences Ca2+ homeostasis, Aβ levels, and Alzheimer’s disease risk. Cell 133:1149–1161
Cicchetti F, Drouin-Ouellet J, Gross RE (2009) Environmental toxins and Parkinson’s disease: what have we learned from pesticide-induced animal models? Trends Pharmacol Sci 30:475–483
Bove J, Prou D, Perier C, Przedborski S (2005) Toxin-induced models of Parkinson’s disease. NeuroRx 2:484–494
Freestone PS, Chung KK, Guatteo E, Mercuri NB, Nicholson LF, Lipski J (2009) Acute action of rotenone on nigral dopaminergic neurons–involvement of reactive oxygen species and disruption of Ca2+ homeostasis. Eur J Neurosci 30:1849–1859
Kahlert S, Zündorf G, Reiser G (2005) Glutamate-mediated influx of extracellular Ca2+ is coupled with reactive oxygen species generation in cultured hippocampal neurons but not in astrocytes. J Neurosci Res 79:262–271
Bähler M, Rhoads A (2002) Calmodulin signaling via the IQ motif. FEBS Lett 513:107–113
Li SS (2005) Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Biochem J 390:641–653
Yuan JP, Kiselyov K, Shin DM, Chen J, Shcheynikov N, Kang SH, Dehoff MH, Schwarz MK, Seeburg PH, Muallem S, Worley PF (2003) Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell 114:777–789
McQuillin A, Bass NJ, Kalsi G, Lawrence J, Puri V, Choudhury K, Detera-Wadleigh SD, Curtis D, Gurling HM (2006) Fine mapping of a susceptibility locus for bipolar and genetically related unipolar affective disorders, to a region containing the C21ORF29 and TRPM2 genes on chromosome 21q22.3. Mol Psychiatry 11:134–142
Xu C, Macciardi F, Li PP, Yoon IS, Cooke RG, Hughes B, Parikh SV, McIntyre RS, Kennedy JL, Warsh JJ (2006) Association of the putative susceptibility gene, transient receptor potential protein melastatin type 2, with bipolar disorder. Am J Med Genet B Neuropsychiatr Genet 141:36–43
Kühn FJP, Kühn C, Nazıroğlu M, Lückhoff A (2009) Role of an N-terminal splice segment in the activation of the cation channel TRPM2 by ADP-Ribose and hydrogen peroxide. Neurochem Res 34:227–233
Pedersen MØ, Larsen A, Stoltenberg M, Penkowa M (2009) Cell death in the injured brain: roles of metallothioneins. Prog Histochem Cytochem 44:1–27
Cook NL, Vink R, Helps SC, Manavis J, van den Heuvel C (2010) Transient receptor potential melastatin 2 expression is increased following experimental traumatic brain injury in rats. J Mol Neurosci 42:192–199
Hill K, McNulty S, Randall AD (2004) Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn Schmiedebergs Arch Pharmacol 370:227–237
Smith MA, Herson PS, Lee K, Pinnock RD, Ashford ML (2003) Hydrogen-peroxide-induced toxicity of rat striatal neurons involves activation of a nonselective cation channel. J Physiol 547:417–425
Fonfria E, Mattei C, Hill K, Brown JT, Randall A, Benham CD, Skaper SD, Campbell CA, Crook B, Murdock PR, Wilson JM, Maurio FP, Owen DE, Tilling PL, McNulty S (2006) TRPM2 is elevated in the tMCAO stroke model, transcriptionally regulated and functionally expressed in C13 microglia. J Recept Signal Transduct Res 26:179–198
Nazıroğlu M (2009) Role of selenium on calcium signaling and oxidative stress-induced molecular pathways in epilepsy. Neurochem Res 34:2181–2191
Acknowledgments
The author wishes thanks to Prof. Dr. James W. Putney (Calcium Regulation Section, NIEHS, USA) and Ercan Sözbir (Department of Biophysics, SDU, Isparta, Turkey) for polishing English of the paper and preparation of Figures, respectively.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nazıroğlu, M. TRPM2 Cation Channels, Oxidative Stress and Neurological Diseases: Where Are We Now?. Neurochem Res 36, 355–366 (2011). https://doi.org/10.1007/s11064-010-0347-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-010-0347-4