
Abstract
Deprenyl has been discovered by Knoll and co-workers. The R-enantiomer of deprenyl (selegiline) is a selective and irreversible inhibitor of the B-isoform of monoamine oxidase (MAO-B) enzyme. Due to its dopamine potentiating and possible neuroprotective properties it has an established role in the treatment of parkinsonian patients. By inhibiting MAO-B enzyme, R-deprenyl decreases the formation of hydrogen peroxide, alleviating the oxidative stress also reduced by increased expression of antioxidant enzymes (superoxide dismutases and catalase) reported during chronic treatment. It was shown to prevent the detrimental effects of neurotoxins like MPTP and DSP-4. R-Deprenyl elicits neuroprotective and neuronal rescue activities in concentrations too low to inhibit MAO-B. It is extensively metabolized and some of the metabolites possess pharmacological activities, thus their contribution to neuroprotective properties was also suggested. The recently identified deprenyl-N-oxide is extensively studied in our laboratory. Effects other than neuroprotection, like influencing cell adhesion and proliferation cannot be neglected.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The R-enantiomer of deprenyl (phenyl-isopropyl-methyl-propargylamine; selegiline) is a potent selective, irreversible inhibitor of the B-form of monoamine oxidase (MAO-B) enzyme [37, 48]. The first paper about the pharmacological properties of deprenyl was published in 1965, by Knoll and his co-workers [38]. The primary aim to synthesize MAO inhibitors, in the late fifties and early sixties, was to develop antidepressive agents. The new inhibitors became potent antidepressants, but all of them, except R-deprenyl [39], potentiated the effect of tyramine, and elicited hypertensive crisis, the so called “cheese-effect” [12, 61]. Birkmayer and his co-workers described, that R-deprenyl potentiated the effects of dopamine [8–11] and during the last three decades, from among MAO inhibitors, it became the drug of choice to treat Parkinson’s disease. R-deprenyl in a daily oral dose of 5–10 mg inhibited oxidative deamination of dopamine, as well as the age-dependent increase of MAO-B activity [1, 59, 68]. By reducing the formation of hydrogen peroxide (H2O2), R-deprenyl decreases the oxidative damage caused by the formation reactive oxygen species (ROS). Chronic treatment of rats with R-deprenyl was found to stimulate the expression of antioxidant enzymes, such as superoxide dismutases (SOD1, SOD2) and catalase [13] in the brain, which can neutralize the excessive amount of ROS.
Many papers published that R-deprenyl in a concentration too low to inhibit MAO-B activity, affects some neuronal and extraneuronal functions; such as cellular signaling pathways, antiapoptotic mechanisms, trophic effects, cell to cell adhesion etc. [50, 88, 89]. Many of these are extra-neural and non-central processes. It was published in 2005 by Jenei et al. [31] from our laboratory, that R-deprenyl decreases cell–cell adhesion. It was postulated, that in addition to adhesion, chemotaxis elicited by biologically active ligands, might also be an important component of R-deprenyl’s effects.
During the last decade it was revealed in many laboratories including ours [45], that not only the parent compound, but also its metabolites are responsible for the broad-spectrum of pharmacological activities of R-deprenyl. It seemed reasonable to enroll the metabolites into the pharmacological studies.
Beside R-deprenyl, a new selective inhibitor of MAO-B, named rasagiline has been discovered. Pre-clinical and clinical studies revealed its therapeutic usefulness in the treatment of parkinsonian patients. The effects of rasagiline not related to MAO-B inhibition were compared to that of deprenyl [110, 111].
Pharmacokinetic Characteristics of R-Deprenyl
Deprenyl is an optically active compound, where the enantiomers have different pharmacological activity and/or potency. The R-isomer of deprenyl, also called selegiline, was proved a more potent enzyme inhibitor than its antipode [48].
R-deprenyl is quickly and well absorbed from the gastrointestinal tract, [4, 28, 29, 43, 47, 52, 53, 75], the maximum plasma concentration is reached within 0.5 and 1.5 h after oral administration [29, 47, 75]. R-deprenyl has considerable first pass metabolism [4, 28, 52, 54, 56], the biological availability was found 4% in humans [2]. Concomitant food consumption has increased the absorbed amount of R-deprenyl by about three times, without changing the plasma concentration of its metabolites [5]. Since the parent compound is responsible for the inhibition of MAO-B enzyme, there is a considerable difference in the brain enzyme activity following oral or parenteral drug administration [25]. Recently, orally disintegrating dosage form of R-deprenyl has been developed, avoiding first pass metabolism. This formulation provides higher drug bioavailability, and considerably reduced metabolite concentration [14, 63]. Percutaneous absorption of R-deprenyl is also favorable [6, 7, 23, 69], and as its cutaneous metabolism is insignificant [70], transdermal application provides about 50-fold higher plasma concentration compared to oral drug administration [6]. According to this higher concentration, considerable inhibition of MAO-A activity in the brain is also achieved, while the intestinal MAO-A activity is only slightly affected. This way R-deprenyl does have antidepressant activity without a significant risk of “cheese-effect” [21, 105].
Autoradiographic studies in mice revealed rapid distribution of R-deprenyl. High concentration was detected in lipid-rich tissues, like in the brain, 30 s after i.v. administration. The concentration then dropped rapidly in the brain, while increased in the parenchymal tissues [43]. The main metabolites: methamphetamine (MA) and amphetamine (A) were identified in the brain of the animals after intraperitoneal [62] and subcutaneous [34, 58] administration of R-deprenyl. These metabolites were also detected post mortem in various brain regions of treated parkinsonian patients [66]. In the experiments performed in our laboratory, using dual-labeled R-deprenyl, both 14C-propargyl, indicating the parent compound, and 3H-phenyl, indicating all metabolites were detected in plasma and various brain regions. Both type of radioactivity showed accumulation after repeated drug administration. Steady state concentration of the compounds with or without propargyl group was reached after 4 and 7 days, respectively both in plasma and striatum, the primary site of action [43, 49, 51]. According to the results of positron emission tomography studies with 11C-labeled deprenyl, the enantiomers do not differ in their early phase tissue distribution, as both isomers penetrate quickly to the brain and other tissues. However, the excretion has shown considerable stereoselectivity, as R-deprenyl accumulated significantly in tissues containing high MAO-B activity, such as in the brain, heart and lung. In case of its antipode (S-deprenyl) rapid, exponential decline in the brain, similarly to MA, was observed. The persistent and high radioactivity in the brain can be explained by the irreversible binding of R-deprenyl to MAO-B enzyme [22, 30], confirmed by investigations performed using knock-out mice [19]. The autoradiography studies have shown binding primarily in subcortical parts of the brain, which is rich in MAO-B according to previous studies [17, 40], while the cortex and the white matter was almost lack of radioactivity [32].
Since determination of the pharmacokinetic parameters of R-deprenyl was difficult because of its low plasma concentration due to considerable first pass metabolism and rapid tissue distribution, in early bioequivalence studies plasma concentrations of the main desalkylated metabolites, MA and desmethyldeprenyl (DD) instead of the parent compound were compared in case of the various preparations [57, 60]. In 1994 Mahmood et al. [55] have developed a sensitive fluorimetric method for determination of R-deprenyl concentration in plasma. Since the procedure is based on the inhibition of MAO enzyme, the presence of the metabolites does not disturb the measurement. This method was used for determination of pharmacokinetic parameters of R-deprenyl in dogs after its oral administration. The results have shown rapid absorption (t max = 25 ± 5.8 min), large volume of distribution (6.56 ± 0.56 l/kg), rapid elimination (t 1/2 = 60.24 ± 9.56 min) and low bioavailability (8.51 ± 3.31%) [54]. Similar results have been found in human experiments, maximum plasma concentration was reached within 0.5 and 1.5 h after drug ingestion, the elimination half-life was 70 min. There was no considerable difference in the pharmacokinetic parameters when R-deprenyl was administered orally as a tablet or as a solution [56]. Intensive metabolism in the liver and extrahepatic organs was suggested based on the high oral clearance found in human experiments [53]. Slight accumulation of the parent drug and the metabolites was observed after repeated doses. Elimination half-life of deprenyl and DD was increased by 3–6 times compared to single dose administration, with less variability of the pharmacokinetic parameters [5].
Metabolism and Excretion of R-Deprenyl
Early metabolism studies have revealed the formation of MA and A by desalkylation, and urinary excretion of these metabolites in high amounts in humans [28, 67, 73], and rats [41, 47, 80, 108]. DD was also detected in urine, although in much lower concentration than A derivatives [28, 41, 73, 80].
The amphetamines are pharmacologically active metabolites, and the enantiomers possess considerably different psychostimulant effect; the S-isomers being much stronger inhibitors of noradrenaline and dopamine transporters, than R-amphetamines [93]. The stereoselective effect of deprenyl and its metabolites required studying the stereochemistry of the metabolism. Schachter et al. [71] published first the stereoselective nature of desalkylation. In our laboratory chiral capillary electrophoresis methods were developed for the enantiomeric separation of deprenyl and amphetamine derivatives [79], and we have proved the stereospecific desalkylation of both R- and S-deprenyl in rats. In the urine [80], plasma and tissue samples of animals treated with R-deprenyl [81, 82], only R-A derivatives were identified, while only S-A derivatives were found in specimens from rats treated with S-deprenyl; no racemization or inversion happened during metabolism. The stereospecific nature of the metabolism was also confirmed by Shin analyzing urine samples of healthy volunteers [73]. There is some controversy about the various CYP isoforms desalkylating selegiline. CYP 2D6 [3, 24], CYP 3A4 [18, 85] and CYP 2E1 [103] were found to be involved in the formation of MA and DD. The possible role of flavin-containing-monooxygenase (FMO) enzymes in the formation of A was also suggested [109].
Desalkylated metabolites can be further converted by hydroxylation of the benzene-ring at para-position, or by hydroxylation of the alkyl chain at β-position. High amount of para-hydroxylated amphetamine derivatives were excreted in the urine of rats treated with selegiline [33, 41, 108], and were also identified in human urine, although in somewhat lower concentration [73, 87]. Small amount of para-hydroxy-DD was also found in rat urine [33]. Majority of the para-hydroxylated metabolites were excreted as conjugates [73].
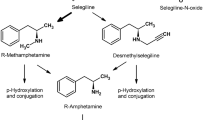
Ephedrine derivatives can also derive by the β-hydroxylation of the alkyl chain. Ephedrine and pseudoephedrine can be formed from MA, while norephedrine and nor-pseudoephedrine from A. These β-hydroxylated metabolites could be identified in the urine of volunteers treated with R-deprenyl, but their total amount was less than 1% of the applied dose [73]. In the pharmacokinetic studies performed in our laboratory, no ephedrine derivatives could be observed in urine of rats treated with R- or S-deprenyl [80]. Beside desalkylation, the possibility of another metabolic pathway was recently suggested by Wu and Ichikawa. In their in vitro experiments, competitive inhibition of FMO-catalyzed metabolism of MPTP by deprenyl and pargylin was observed. Based on their results, these MAO inhibitors are substrates of FMO by themselves, and the formation of N-oxide metabolites from the tertiary-amines was supposed [107]. Previously, highly sensitive and efficient gas-chromatographic methods were used for the wide-spread pharmacokinetic studies of R-deprenyl. However, these were not capable of detecting the N-oxide metabolites, because of the high temperature and sample derivatization needed for the separation, inducing decomposition of the N-oxide. In early thin-layer chromatographic studies of selegiline metabolism, in addition to the desalkylated derivatives, an unidentified metabolite was also observed, but it could not be detected by gas-chromatography [46]. This detected but unidentified metabolite could be deprenyl-N-oxide (DNO). Recently, in vivo formation of N-oxide metabolite from R-deprenyl in humans was shown by HPLC and HPLC–MS methods [35, 36] and its in vitro production by liver microsomal preparations [42, 102] was also demonstrated. The known metabolic pathways of selegiline are shown in Fig. 1.

Metabolic pathway of R-(−)-deprenyl
Oxidation of the tertiary prochiral nitrogen of deprenyl provides quaternary nitrogen, a new chiral center. DNO thus contains two asymmetry centers and its four diastereomers (i.e. two pairs of enantiomers) exist. Each isomer has one enantiomer and two diastereomers among the other isomers (Fig. 2). The metabolic transformation of R-deprenyl can lead to the generation of 1R,NR- and 1R,NS-DNO, while S-deprenyl can be converted to 1S,NR- and 1S,NS-DNO. Since DNO is supposed to contribute to the pharmacological effects of R-deprenyl, the in vivo formation of the isomers was also studied in our laboratory [86].

Isomers of deprenyl-N-oxide
The analysis of the urine samples of rats treated with either R- or S-deprenyl orally revealed that the new chiral centre on the quaternary nitrogen atom was formed stereoselectively, where the configuration of S- over the R- was preferred. 1R,NS- and 1S,NS-isomers were formed in 3–5 fold excess compared to 1R,NR- and 1S,NR-isomers in rats during in vivo metabolism of R- and S-deprenyl, respectively. Based on our in vitro metabolism studies, FMO1 enzyme is responsible for the stereoselective formation of NS-isomers, while FMO3 preferentially forms the NR configuration. Using microsomal preparation, much lower enantioselectivity was observed, indicating, that CYP enzymes do not show stereoselectivity in the formation of the new chiral centre on the quaternary nitrogen [83]. Our results of stereoselective N-oxide formation are in accordance with previous findings [26, 27] with pargylin, a non-selective MAO inhibitor having similar structure to deprenyl, where highly stereoselective and species-dependent N-oxide production was found [26].
In our in vivo metabolism experiments rats were treated with a single dose of R-deprenyl, and the metabolites excreted in the urine were identified. The amount of DNO, excreted in rat urine collected during 24 h after treatment represented 1–5% of the dose administered. Majority of N-oxide metabolite was found in the fraction of urine collected 0–3 h after single dose drug administration, when its amount was comparable to that of MA and A. After 9 h following treatment, increasing amounts of para-hydroxylated derivatives were detected in the urine, while the excretion of DNO decreased. Considerable amounts of A and MA were found in all urinary fractions. DD and parent compound could be detected only in low amounts in the urine [84]. When repeated dose of R-deprenyl was administered to rats, 55–75% of the dose was recovered in urine 24 h after the last treatment. After repeated drug administration, the amounts of the para-hydroxylated desalkylated metabolites increased in the urine, and became the most abundant metabolites excreted. The total amount of the para-hydroxylated compounds in the urine has reached 50–60% of the dose administered, the majority (50–67%) of which were identified as conjugate. The amount of the parent compound in the urine slightly increased after repeated drug administration (from 0.3 to 2% of the dose), while that of DNO decreased (below 1% of the dose). The amount of DD was very low in all the urine samples (0.01–0.4% of the dose) [84].
When a single dose of diastereomeric mixture (1:1) of deprenyl-N-oxides (5 mg/kg) was administered orally to rats, more than 60% of the dose was excreted unchanged in the urine during 3 h following treatment, and further 10% until 24 h (Fig. 3a). About 10% of the dose was identified as metabolites (R-deprenyl, DD, MA, A, para-hydroxy-MA, para-hydroxy-A, para-hydroxy-DD) in the urine during 24 h after treatment. The urinary excretion of the metabolites formed from DNO within 24 h after its administration is shown in Fig. 3b. The conjugated metabolites were not determined in this experiment. These data indicate that some of the DNO is reduced to deprenyl in rats, and further metabolised by desalkylation and aromatic hydroxylation. This finding also raises the possibility of retro-reduction of DNO metabolite to the parent compound in case of R-deprenyl treatment. Previously, the reduction of N-hydroxylamines by benzamidoxime reductase and human liver microsomes to their parent amines has been demonstrated [15]. This reduction mechanism may also apply in case of N-oxide metabolites formed from tertiary amines.

Urinary excretion of deprenyl metabolites after treatment with deprenyl-N-oxide
The Neuroprotective and Neuronal Rescue Effect of R-Deprenyl and Some of its Metabolites
Starting at 1994 Tatton et al. published that R-deprenyl in a concentration too low to inhibit MAO-B activity (10−9–10−13 M) reduces apoptosis by inducing new protein synthesis. This fact was first made evident by the results of in vitro studies by Tatton et al. [89], who pointed out using in vitro cultured partially NGF-differentiated PC-12 cells of neuroectodermal origin that R-deprenyl prevents apoptosis-inducing effect of serum and nerve growth factor deprivation in such a low dose (10−9 mol/l), which does not exhibit MAO-B inhibitory action. As previous results drew the attention to neuronal apoptosis as likely to be responsible at least in part for cell death in Parkinson’s disease, Huntington’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis, drugs with antiapoptotic, neuroprotective characteristics seem to be promising candidates for future therapy.
R-Deprenyl has been demonstrated to prevent or diminish the effect of various apoptosis-inducing damaging factors both in vitro and in vivo. These factors include serum deprivation, glutathion depletion, poisoning with excitotoxins, okadaic acid, nitric oxide, peroxynitrite, cytosine arabinoside, as well as peripheral nerve crush and axotomy [91].
The anti-apoptotic effect of R-deprenyl has been investigated by our group [44, 45, 50, 51, 76–78] using the human melanoma cell line A2058. Melanocytes are of neuroectodermal origin, like phaeochromocytoma cells used by Tatton et al.; however, our cultures had not been differentiated by NGF, which could account for the discrepancies between the results of the two groups. Serum withdrawal for 5 days resulted in an excessive number of apoptotic cells of the cell cultures, proven by observation of HE-stained cover-slip adherent cultures, TUNEL reaction, flow cytometry and caspase 3 determination [78].Very low doses of R-deprenyl—as given in Tatton’s articles [89, 91] i.e. 10−7–10−13 mol/l—caused a 48 h delay in the onset of apoptosis in case of serum withdrawal, compared to the untreated control (Table 1). However, the same concentrations of S-deprenyl were ineffective. The effect of higher doses of R-deprenyl (10−4–10−2 mol/l) was studied using A2058 melanoma and HT 1080 fibrosarcoma cell lines [77]. The cells were grown in serum supplemented MEM, administration of R-deprenyl was performed 2 h after plating. R-Deprenyl administered at a concentration of 10−2 mol/l resulted in 100% apoptotic cell death of A2058 cells, while 10−3 mol/l R-deprenyl caused apoptosis of 50% of the melanoma cells. Very similar results were published by Tatton et al. [91] after 10−3 mol/l R-deprenyl treatment of PC-3 cells. Administration of 10−4–10−3 mol/l R-deprenyl to HT 1080 fibrosarcoma cell cultures induced apoptosis dose-dependently. However, 10−2 mol/l R-deprenyl caused cytoplasmic, vacuolar, non-lysosomal cell death 24, 48 and 72 h after administration [77].
As mentioned above, 10−7–10−13 mol/l R-deprenyl temporarily prevented apoptosis of A2058 cells after serum deprivation. Simultaneous administration of the non-specific microsomal drug metabolising enzyme inhibitor SKF-525A with R-deprenyl abolished the anti-apoptotic effect [45]. This could mean that R-deprenyl needs metabolic changes in order to exert its anti-apoptotic activity. However, the pro-apoptotic effect of higher doses (10−3–10−4 mol/l) of R-deprenyl could not be influenced by SKF-525A, indicating that R-deprenyl itself and not its metabolites exert pro-apoptotic activity. Nevertheless, it is more reasonable to assess the metabolism of deprenyl directly if in vitro models are used, as it may differ significantly from metabolism in vivo. This has been demonstrated by our group using undifferentiated PC-12 cells, where deprenyl did not undergo any metabolic changes in this culture during the 3 days of experiment (unpublished results). Furthermore, SKF-525A may also possess proapoptotic effects, hindering the interpretation of the results.
Regarding metabolites of R-deprenyl R-DD, MA, A are the best known [36, 73]. DNO became also a possible candidate for being the ultimate effector molecule [75].Our studies are in progress in this respect.
According to Tatton et al. [88] MA and A did not alter the survival of trophically withdrawn PC-12 cells. Similar result has been found by our group using 10−7–10−13 mol/l R-MA- or S-MA-treated serum deprived A2058 melanoma cells. Although R-DD was supposed to be the active metabolite of R-deprenyl [89], our results on serum-deprived A2058 cells showed no anti-apoptotic effect of low doses of DD [77] (Table 1). High doses of DD (10−2–10−4 mol/l) caused death of A2058 melanoma and HT 1080 fibrosarcoma cells. In vivo, 2058 human tumor xenografts, when treated by 0.2–20.0 mg/kg DD, showed dose-dependent growth inhibition [77].
Experiments regarding the anti-apoptotic effect of low doses of R-deprenyl using non-neuronal cells led to different results according to the origin of such cells. Studies on mouse thymocytes [20] showed that R-deprenyl did not exhibit detectable protective effect after dexamethasone treatment which induced apoptosis. On the other hand, it has been proven by our group, that R-deprenyl diminished the apoptosis-inducing effect of ischaemia–reperfusion in experimental rats [101]. The most effective dose was 0.15 mg/kg which equals the human therapeutic dose applied in neurodegenerative disease. Qin and his co-workers [64] reported that R-deprenyl may be a potent inhibitor of non-neuronal apoptosis also in case of cardiac tissue.
The possible mode of apoptosis inhibitory action of R-deprenyl has been widely discussed. R-Deprenyl treatment might protect neurons from oxidative damage mediated by lipid peroxydation of the cell membrane [74], by reducing the production of H2O2 due to inhibition of the normal metabolism of dopamine by MAO-B [16].
As mentioned earlier, R-deprenyl can also increase neuronal survival without inhibiting MAO-B. The mechanism of the established anti-apoptotic property of R-deprenyl is most likely achieved through modulation of gene expression interfering with the apoptotic cascades, preferably the mitochondrial pathway. Regarding this pathway, apoptosis is mediated by loss of mitochondrial membrane potential inducing the opening of the mitochondrial membrane permeability transition pore, leading to a release of pro-apoptotic factors such as cytochrome C and apoptosis-inducing factor (AIF). Activation of the pro-apoptotic protein BAX cause decrease in mitochondrial membrane potential, whereas the maintenance of mitochondrial membrane potential is strongly supported by increased activity of BCL2. Studies of Tatton and others [65, 88, 90, 104] point to the fact, that R-deprenyl alters the expression of these genes in the course of apoptosis in favor of stabilization of mitochondrial membrane potential.
The Effect of R-Deprenyl Outside the Central Nervous System
R-Deprenyl has been shown to affect a number of functions which could explain its beneficial effect in Parkinson’s disease. These functions include its MAO-B inhibitory effect, modulation of cellular signaling pathways, such as pro- and anti-apoptotic pathways, redox signaling or trophic effects of the drug. Although most publications focus on the neuroprotective functions of R-deprenyl, a small but increasing number of publications are discussing its beneficial effects outside the central nervous system. Thyaga Rajan et al. [97–100] in several studies examined the anti-tumor effects of R-deprenyl in rats with either spontaneously growing or carcinogen-induced mammary/pituitary tumors. In old, acyclic female rats treatment with R-deprenyl was shown to temporarily re-establish estrous cycles and significantly reduce the incidence of both mammary and pituitary tumors [98]. R-Deprenyl also reduced the number and size of carcinogen-induced mammary tumors by enhancing splenic IL-2, IFN-γ production, NK cell activity and cathecholaminergic neuronal activities of the central and peripheral nervous system [97, 99, 100]. The effect of R-deprenyl on the neuroendocrine-immune network was discussed in an earlier review [96].
A MAO-B-independent anti-apoptotic effect of R-deprenyl has been shown in a number of tumorigenic cell lines, which further suggests that the effect of the drug cannot be limited to the CNS or cells of neural origin. Interestingly, Seymour and co-workers showed that R-deprenyl was specifically protective in non-malignant human cells (HaCat spontaneously immortalized keratinocytes, normal human urothelial explants) versus tumor cells (H-ras-transfected HaCat cells, HPV16 virus-infected HPV-G keratinocyte cell line, PC-3 prostate adenocarcinoma cell line) against the toxic effects of cobalt-60 gamma ionizing radiation and cisplatin [72]. Although the two studies both used cancer cell lines both the choice of cell lines and the induction of apoptosis was different which could explain why R-deprenyl was protective in certain cancer cell lines against apoptosis but not in others. R-Deprenyl was also shown to protect rat kidney against the apoptosis-inducing effect of ischemia–reperfusion [101], the vascular endothelium from the toxic effects of amyloid-beta peptide [95] and it also reduces myocyte apoptosis in vivo [64].
A novel, also MAO-B-independent effect of R-deprenyl on cell–cell adhesion was described by our research group which could contribute to its protective actions for example in tumour development or neurodegeneration [31]. R-Deprenyl significantly increased cell–cell adhesion of NGF-naïve and NGF-differentiated PC-12 cells (of neuro-ectodermal origin) and also NIH3T3 mouse embryonal fibroblasts (non-neuronal) in a concentration-dependent manner. The effective concentration range which induced cell–cell adhesion in PC-12 cells was slightly higher (10−7 M in NGF-naïve and 10−9 M in NGF-differentiated PC-12 cells) than its anti-apoptotic concentrations (10−11 M in PC-12, 10−13 M in melanoma) [78, 92]. Furthermore the anti-apoptotic actions of R-deprenyl were only limited to NGF-differentiated PC-12 cells [92] which suggest a different mechanism of the two effects. Microsomal metabolism of R-deprenyl is required for its antiapoptotic effects [78, 90] while inhibition of cytochrome P450 had no effect on its cell–cell adhesion increasing function. Interestingly a rarely studied metabolite of R-deprenyl—DNO produced by FMO—was equally effective as the original compound [31].
Very recently two studies reported the inhibitory effect of R-deprenyl on hyperpermeability of vascular endothelial cells caused by burn or hemorrhagic shock [94, 106]. The authors attribute the protective effects of the drug to its antioxidant actions, maintenance of mitochondrial membrane potential, inhibition of cytochrome C release from the mitochondria and inhibition of caspase 3 activation. Interestingly, the authors describe reduced intercellular adhesion as part of the mechanism of hyperpermeabilty. Furthermore, they also mention that apoptosis might contribute to these loosening intercellular junctions and cell–matrix adhesions, therefore leading to the detachment of endothelial cells from the underlying basement membrane. Both studies hypothesize that R-deprenyl acts by antagonizing apoptosis in these cells, therefore preventing disruption of intercellular junctions caused by apoptosis. Unfortunately, the effect of R-deprenyl on endothelial cell–cell adhesion was not studied in these papers [94, 106]. Their hypothesis, however, is very well supported by our finding that R-deprenyl actively enhances cell–cell adhesion [31]. The direct effect of R-deprenyl on cell–cell adhesion might have contributed to its inhibitory effects on hyperpermeability in vascular endothelial cells. The concentrations used in the aforementioned papers in vitro were (10−6–10−5 M) certainly within the concentration range of our cell aggregation experiments where we observed significant increase of cell–cell adhesion in PC-12 or NIH3T3 cells (10−9–10−5 M or 10−11–10−5 M, respectively) [31]. As the authors in these papers did not test the effects of R-deprenyl metabolites on vascular hyperpermeability it is difficult to speculate on any further similarities or differences between the two mechanisms.
Although more investigation is required to clarify the effects of R-deprenyl outside the central nervous system the above results suggest that R-deprenyl might be more than an agent against Parkinson’s disease.
Conclusions
R-Deprenyl is an original Hungarian drug, which has been discovered by Knoll and his co-workers and during the past three decades it was used to treat the disabling Parkinson’s disease. R-Deprenyl (selegiline) has been the only selective, irreversible MAO-B inhibitor, which in an oral dose of 10 mg/day delays the need for levodopa administration. It proved to be effective even in monotherapy, when it was administered at the early stage of the disease, and its effectiveness lasts till functioning neurons exist in the CNS. Recently, a new MAO-B inhibitor, rasagiline was introduced to therapy. The question arises, whether there is a need for two MAO inhibitors in the treatment of Parkinson’s disease. According to our knowledge, the answer is yes, but not because of their different MAO inhibitory effects, but due to the other versatile pharmacological properties of the molecules. Both selegiline and rasagiline in a concentration too low to inhibit the enzyme activity distinctly modify the regulation of apoptotic cascades. The metabolism of selegiline is rather intensive. It has high capacity “first pass” metabolism and the microsomal enzymes convert it to amphetamine, methamphetamine, desmethyldeprenyl, para-hydroxy metabolites and the conjugates of the formers. In addition, the flavin-containing monooxygenase synthesizes deprenyl-N-oxide, which has two chiral centers, thus exists in the forms of four enantiomers. Additionally, deprenyl-N-oxide has a quaternary character, which alters membrane penetration. As N-oxides, it can be reduced back to the parent compound. Both selegiline and its metabolites can elicit diverse pharmacological activities, which play a role in the complex effects of selegiline. R-Deprenyl significantly increased cell–cell adhesion of NGF-naive and NGF-differentiated PC-12 cells of neuro-ectodermal origin. Parenteral administration increases the bioavailability of selegiline and the same dose, which is used for oral treatment, effectively inhibits the brain MAO-A enzyme; by overcoming “first pass” metabolism, antidepressive activity can be elicited. Deprenyl-N-oxide, the metabolite of selegiline possessing propargyl group also modifies the function of apoptotic cascades. The effect of deprenyl-N-oxide is under further studies in our laboratory, and it was found to increase mitotic activity. The mechanisms of the pleiotropic effects of MAO-B inhibitors are thus worth for further studies to understand their complex pharmacological effects.
References
Adolfsson R, Gottfries CG, Oreland L et al (1980) Increased activity of brain and platelet monoamine oxidase in dementia of Alzheimer type. Life Sci 27:1029–1034
Azzaro AJ, Ziemniak J, Kemper E et al (2007) Pharmacokinetics and absolute bioavailability of selegiline following treatment of healthy subjects with the selegiline transdermal system (6 mg/24 h): a comparison with oral selegiline capsules. J Clin Pharmacol 47:1256–1267
Bach MV, Coutts RT, Baker GB (2000) Metabolism of N,N-dialkylated amphetamines, including deprenyl, by CYP2D6 expressed in a human cell line. Xenobiotica 30:297–306
Barrett JS, Szego P, Rohatagi S et al (1996) Absorption and presystemic metabolism of selegiline hydrochloride at different regions in the gastrointestinal tract in healthy males. Pharm Res 13:1535–1540
Barrett JS, Rohatagi S, DeWitt KE et al (1996) The effect of dosing regimen and food on the bioavailability of the extensively metabolized, highly variable drug Eldepryl(R) (selegiline hydrochloride). Am J Ther 3:298–313
Barrett JS, Hochadel TJ, Morales RJ et al (1996) Pharmacokinetics and safety of a selegiline transdermal system relative to single-dose oral administration in the elderly. Am J Ther 3:688–698
Barrett JS, DiSanto AR, Thomford PJ et al (1997) Toxicokinetic evaluation of a selegiline transdermal system in the dog. Biopharm Drug Dispos 18:165–184
Birkmayer W, Riederer P, Youdim MB et al (1975) The potentiation of the anti akinetic effect after L-dopa treatment by an inhibitor of MAO-B, Deprenil. J Neural Transm 36:303–326
Birkmayer W, Riederer P, Ambrozi L et al (1977) Implications of combined treatment with ‘Madopar’ and L-deprenil in Parkinson’s disease. A long-term study. Lancet 1:439–443
Birkmayer W, Knoll J, Riederer P et al (1983) (−)-Deprenyl leads to prolongation of L-dopa efficacy in Parkinson’s disease. Mod Probl Pharmacopsychiatry 19:170–176
Birkmayer W, Knoll J, Riederer P et al (1985) Increased life expectancy resulting from addition of L-deprenyl to Madopar treatment in Parkinson’s disease: a longterm study. J. Neural Transm 64:113–127
Blackwell B, Marley E, Price J et al (1967) Hypertensive interactions between monoamine oxidase inhibitors and foodstuffs. Br J Psychiatry 113:349–365
Carrillo MC, Kanai S, Nokubo M et al (1991) (−)-Deprenyl induces activities of both superoxide dismutase and catalase but not of glutathione peroxidase in the striatum of young male rats. Life Sci 48:517–521
Clarke A, Brewer F, Johnson ES et al (2003) A new formulation of selegiline: improved bioavailability and selectivity for MAO-B inhibition. J Neural Transm 110:1241–1255
Clement B, Behrens D, Moller W et al (2000) Reduction of amphetamine hydroxylamine and other aliphatic hydroxylamines by benzamidoxime reductase and human liver microsomes. Chem Res Toxicol 13:1037–1045
Cohen G, Spina MB (1989) Deprenyl supresses the oxidant stress associated with increased dopamine turnover. Ann Neurol 26:689–690
Denney RM, Fritz RR, Patel NT et al (1983) Use of a monoclonal antibody for comparative studies of monoamine oxidase B in mitochondrial extracts of human brain and peripheral tissues. Mol Pharmacol 24:60–68
Dragoni S, Bellik L, Frosini M et al (2003) L-Deprenyl metabolism by the cytochrome P450 system in monkey (Cercopithecus aethiops) liver microsomes. Xenobiotica 33:181–195
Ekblom J, Oreland L, Chen K et al (1998) Is there a “non-MAO” macromolecular target for L-deprenyl?: studies on MAOB mutant mice. Life Sci 63:PL181–PL186
Fang J, Zuo DM, Yu PH (1995) Lack of protective effect of R(−)-deprenyl on programmed cell death of mouse thymocytes induced by dexamethasone. Life Sci 57:15–22
Feiger AD, Rickels K, Rynn MA et al (2006) Selegiline transdermal system for the treatment of major depressive disorder: an 8 week, double-blind, placebo-controlled, flexible-dose titration trial. J Clin Psychiatry 67:1354–1361
Fowler JS, MacGregor RR, Wolf AP et al (1987) Mapping human brain monoamine oxidase A and B with 11C-labeled suicide inactivators and PET. Science 235:481–485
Gaál J, Szebeni G, Székács G et al (2000) Transdermal formulations of deprenyl: guinea pig and pig models. Neurobiology (Bp) 8:143–166
Grace JM, Kinter MT, Macdonald TL (1994) Atypical metabolism of deprenyl and its enantiomer, (S)-(+)-N, alpha-dimethyl-N-propynylphenethylamine, by cytochrome P450 2D6. Chem Res Toxicol 7:286–290
Haberle D, Szökő É, Magyar K (2002) The influence of metabolism on the MAO-B inhibitory potency of selegiline. Curr Med Chem 9:47–51
Hadley MR, Svajdlenka E, Damani LA et al (1994) Species variability in the stereoselective N-oxidation of pargyline. Chirality 6:91–97
Hadley MR, Gabriac SD, Hutt AJ (1999) Resolution of enantiomeric N-oxides by capillary electrophoresis using cyclodextrins as chiral selectors. Chirality 11:409–415
Heinonen EH, Myllyla V, Sotaniemi K et al (1989) Pharmacokinetics and metabolism of selegiline. Acta Neurol Scand Suppl 126:93–99
Heinonen EH, Anttila MI, Lammintausta RA (1994) Pharmacokinetic aspects of l-deprenyl (selegiline) and its metabolites. Clin Pharmacol Ther 56:742–749
Inoue O, Axelsson S, Lundqvist H et al (1990) Effect of reserpine on the brain uptake of carbon 11 methamphetamine and its N-propagyl derivative, deprenyl. Eur J Nucl Med 17:121–126
Jenei V, Zor K, Magyar K et al (2005) Increased cell-cell adhesion, a novel effect of R-(−)-deprenyl. J Neural Transm 112:1433–1445
Jossan SS, Dargy R, Gillberg PG et al (1989) Localization of monoamine oxidase-B in human-brain by autoradiographical use of C-11-labelled L-deprenyl. J Neural Transm 77:55–64
Kalász H, Kerecsen L, Knoll J et al (1990) Chromatographic studies on the binding, action and metabolism of (−)-deprenyl. J Chromatogr 499:589–599
Kalász H, Bartók T, Szökő É et al (1999) Analysis of deprenyl metabolites in the rat brain using HPLC-ES-MS. Curr Med Chem 6:271–278
Katagi M, Tatsuno M, Miki A et al (2001) Simultaneous determination of selegiline-N-oxide, a new indicator for selegiline administration, and other metabolites in urine by high-performance liquid chromatography-electrospray ionization mass spectrometry. J Chromatogr B Biomed Sci Appl 759:125–133
Katagi M, Tatsuno M, Tsutsumi H et al (2002) Urinary excretion of selegiline N-oxide, a new indicator for selegiline administration in man. Xenobiotica 32:823–831
Knoll J, Magyar K (1972) Some puzzling pharmacological effects of monoamine oxidase inhibitors. Adv Biochem Psychopharmacol 5:393–408
Knoll J, Ecseri Z, Kelemen K et al (1965) Phenylisopropylmethylpropinylamine (E-250), a new spectrum psychic energizer. Arch Int Pharmacodyn Ther 155:154–164
Knoll J, Vizi ES, Somogyi G (1968) Phenylisopropylpropanylamine (E-250), a monoamine oxidase inhibitoe antagonizing the effects of tyramine. Arzneimittelforschung 18:109–112
Konradi C, Svoma E, Jellinger K et al (1988) Topographic immunocytochemical mapping of monoamine oxidase-A, monoamine oxidase-B and tyrosine hydroxylase in human post mortem brain stem. Neuroscience 26:791–802
Lengyel J, Magyar K, Hollósi I et al (1997) Urinary excretion of deprenyl metabolites. J Chromatogr A 762:321–326
Lévai F, Fejer E, Szeleczky G et al (2004) In vitro formation of selegiline-N-oxide as a metabolite of selegiline in human, hamster, mouse, rat, guinea-pig, rabbit and dog. Eur J Drug Metab Pharmacokinet 29:169–178
Magyar K (1994) Behaviour of (−)-deprenyl and its analogues. J Neural Transm Suppl 41:167–175
Magyar K, Szende B (2000) The neuroprotective and neuronal rescue effect of (−)-deprenyl. In: Cameron RG, Feuer G (eds) Handbook Exp. Pharm. Springer, Heidelberg, pp 457–472
Magyar K, Szende B (2004) (−)-Deprenyl, a selective MAO-B inhibitor, with apoptotic and anti-apoptotic properties. Neurotoxicology 25:233–242
Magyar K, Szűts T (1982) The fate of (−)-deprenyl in the body—Preclinical studies. In: Szebeni R (eds) Proceedings of the international symposium on (−)-deprenyl, Jumex. Chinoin, Budapest, pp 25–31
Magyar K, Tóthfalusi L (1984) Pharmacokinetic aspects of deprenyl effects. Pol J Pharmacol Pharm 36:373–384
Magyar K, Vizi ES, Ecseri Z et al (1967) Comparative pharmacological analysis of the optical isomers of phenyl-isopropyl-methyl-propinylamine (E-250). Acta Physiol Acad Sci Hung 32:377–387
Magyar K, Lengyel J, Szatmári I et al (1995) The distribution of orally administered (−)-deprenyl-propynyl-14C and (−)-deprenyl-phenyl-3H in rat brain. Prog Brain Res 106:143–153
Magyar K, Szende B, Lengyel J et al (1996) The pharmacology of B-type selective monoamine oxidase inhibitors; milestones in (−)-deprenyl research. J Neural Transm Suppl 48:29–43
Magyar K, Szende B, Lengyel J et al (1998) The neuroprotective and neuronal rescue effects of (−)-deprenyl. J Neural Transm Suppl 52:109–123
Magyar K, Pálfi M, Tábi T et al (2004) Pharmacological aspects of (−)-deprenyl. Curr Med Chem 11:2017–2031
Mahmood I (1997) Clinical pharmacokinetics and pharmacodynamics of selegiline. An update. Clin Pharmacokinet 33:91–102
Mahmood I, Peters DK, Mason WD (1994) The pharmacokinetics and absolute bioavailability of selegiline in the dog. Biopharm Drug Dispos 15:653–664
Mahmood I, Neau SH, Mason WD (1994) An enzymatic assay for the MAO-B inhibitor selegiline in plasma. J Pharm Biomed Anal 12:895–899
Mahmood I, Marinac JS, Willsie S et al (1995) Pharmacokinetics and relative bioavailability of selegiline in healthy volunteers. Biopharm Drug Dispos 16:535–545
Mascher HJ, Kikuta C, Millendorfer A et al (1997) Pharmacokinetics and bioequivalence of the main metabolites of selegiline: desmethylselegiline, methamphetamine and amphetamine after oral administration of selegiline. Int J Clin Pharmacol Ther 35:9–13
Melega WP, Cho AK, Schmitz D et al (1999) L-methamphetamine pharmacokinetics and pharmacodynamics for assessment of in vivo deprenyl-derived l-methamphetamine. J Pharmacol Exp Ther 288:752–758
Milgram NW, Racine RJ, Nellis P et al (1990) Maintenance on L-deprenyl prolongs life in aged male rats. Life Sci 47:415–420
Müller FO, Schall R, Hundt HK et al (1996) Bioavailability of two selegiline hydrochloride tablet products. Arzneimittelforschung 46:1037–1040
Natoff IL (1964) Cheese and monoamine oxidase inhibitors. Interactions in anaesthetised cats. Lancet 1:532–533
Philips SR (1981) Amphetamine, p-hydroxyamphetamine and beta-phenylethylamine in mouse-brain and urine after (−)- and (+)-deprenyl administration. J Pharm Pharmacol 33:739–741
Poston KL, Waters C (2007) Zydis selegiline in the management of Parkinson’s disease. Expert Opin Pharmacother 8:2615–2624
Qin F, Shite J, Mao W et al (2003) Selegiline attenuates cardiac oxidative stress and apoptosis in heart failure: association with improvement of cardiac function. Eur J Pharmacol 461:149–158
Reed JC (1996) Mechanisms of Bcl-2 family protein function and dysfunction in health and disease. Behring Inst Mitt Oct(97):72–100
Reynolds GP, Riederer P, Sandler M et al (1978) Amphetamine and 2-phenylethylamine in post-mortem Parkinsonian brain after (−)deprenyl administration. J Neural Transm 43:271–277
Reynolds GP, Elsworth JD, Blau K et al (1978) Deprenyl is metabolized to methamphetamine and amphetamine in man. Br J Clin Pharmacol 6:542–544
Riederer P, Youdim MB (1986) Monoamine oxidase activity and monoamine metabolism in brains of parkinsonian patients treated with L-deprenyl. J Neurochem 46:1359–1365
Rohatagi S, Barrett JS, DeWitt KE et al (1997) Integrated pharmacokinetic and metabolic modeling of selegiline and metabolites after transdermal administration. Biopharm Drug Dispos 18:567–584
Rohatagi S, Barrett JS, McDonald LJ et al (1997) Selegiline percutaneous absorption in various species and metabolism by human skin. Pharm Res 14:50–55
Schachter M, Marsden CD, Parkes JD et al (1980) Deprenyl in the management of response fluctuations in patients with Parkinson’s disease on levodopa. J Neurol Neurosurg Psychiatry 43:1016–1021
Seymour CB, Mothersill C, Mooney R et al (2003) Monoamine oxidase inhibitors l-deprenyl and clorgyline protect nonmalignant human cells from ionising radiation and chemotherapy toxicity. Br J Cancer 89:1979–1986
Shin HS (1997) Metabolism of selegiline in humans. Identification, excretion, and stereochemistry of urine metabolites. Drug Metab Dispos 25:657–662
Simonian NA, Coyle JT (1996) Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol 36:83–106
Szebeni G, Lengyel J, Székács G et al (1995) Gas chromatographic procedure for simultaneous determination of selegiline metabolites, amphetamine, methamphetamine and demethyl-deprenyl in pig plasma. Acta Physiol Hung 83:135–141
Szende B (2004) Cell proliferation and cell death in relation to dose of various chemical substances. In: Török T, Klebovich I (eds) Monoamine oxidase inhibitors and their role in neurotransmission. Medicina, Budapest, pp 133–140
Szende B, Magyar K, Szegedi Z (2000) Apoptotic and antiapoptotic effect of (−)-deprenyl and (−)-desmethyl-deprenyl on human cell lines. Neurobiology (Bp) 8:249–255
Szende B, Bokonyi G, Bocsi J et al (2001) Anti-apoptotic and apoptotic action of (−)-deprenyl and its metabolites. J Neural Transm 108:25–33
Szökő É, Magyar K (1995) Chiral separation of deprenyl and its major metabolites using cyclodextrine-modified capillary zone electrophoresis. J Chromatogr A 709:157–162
Szökő É, Magyar K (1996) Enantiomer identification of major metabolites of (−)-deprenyl in rat urine by capillary electrophoresis. Int J Pharm Advances 1:320–328
Szökő É, Kalász H, Magyar K (1999) Biotransformation of deprenyl enantiomers. Eur J Drug Metab Pharmacokinet 24:315–319
Szökő É, Kalász H, Magyar K (1999) Metabolic transformation of deprenyl enantiomers in rats. Neurobiology (Bp) 7:247–254
Szökő É, Tábi T, Borbás T et al (2004) Assessment of the N-oxidation of deprenyl, methamphetamine, and amphetamine enantiomers by chiral capillary electrophoresis: an in vitro metabolism study. Electrophoresis 25:2866–2875
Szökő É, Tábi T, Halász AS et al (2004) Chiral characterization and quantification of deprenyl-N-oxide and other deprenyl metabolites in rat urine by capillary electrophoresis. Chromatographia 60:S245–S251
Taavitsainen P, Anttila M, Nyman L et al (2000) Selegiline metabolism and cytochrome P450 enzymes: in vitro study in human liver microsomes. Pharmacol Toxicol 86:215–221
Tábi T, Magyar K, Szökő É (2003) Chiral characterization of deprenyl-N-oxide and other deprenyl metabolites by capillary electrophoresis using a dual cyclodextrin system in rat urine. Electrophoresis 24:2665–2673
Tarjányi Z, Kalász H, Szebeni G et al (1998) Gas-chromatographic study on the stereoselectivity of deprenyl metabolism. J Pharm Biomed Anal 17:725–731
Tatton WG, Chalmers-Redman RM (1996) Modulation of gene expression rather than monoamine oxidase inhibition: (−)-deprenyl-related compounds in controlling neurodegeneration. Neurology 47:S171–S183
Tatton WG, Ju WY, Holland DP et al (1994) (−)-Deprenyl reduces PC12 cell apoptosis by inducing new protein synthesis. J Neurochem 63:1572–1575
Tatton WG, Wadia JS, Ju WY et al (1996) (−)-Deprenyl reduces neuronal apoptosis and facilitates neuronal outgrowth by altering protein synthesis without inhibiting monoamine oxidase. J Neural Transm Suppl 48:45–59
Tatton WG, Chalmers-Redman RM, Elstner M et al (2000) Glyceraldehyde-3-phosphate dehydrogenase in neurodegeneration and apoptosis signaling. J Neural Transm Suppl:77–100
Tatton WG, Chalmers-Redman RM, Ju WJ et al (2002) Propargylamines induce antiapoptotic new protein synthesis in serum-and nerve growth factor (NGF)-withdrawn, NGF-differentiated PC-12 cells. J Pharmacol Exp Ther 301:753–764
Tekes K, Tóthfalusi L, Gaál J et al (1988) Effect of MAO inhibitors on the uptake and metabolism of dopamine in rat and human brain. Pol J Pharmacol Pharm 40:653–658
Tharakan B, Whaley JG, Hunter FA et al (2010) (−)-Deprenyl inhibits vascular hyperpermeability after hemorrhagic shock. Shock 33:56–63
Thomas T, McLendon C, Thomas G (1998) L-deprenyl: nitric oxide production and dilation of cerebral blood vessels. Neuroreport 9:2595–2600
Thyaga Rajan S, Felten DL (2002) Modulation of neuroendocrine-immune signaling by L-deprenyl and L-desmethyldeprenyl in aging and mammary cancer. Mech Ageing Dev 123:1065–1079
Thyaga Rajan S, Quadri SK (1999) L-deprenyl inhibits tumor growth, reduces serum prolactin, and suppresses brain monoamine metabolism in rats with carcinogen-induced mammary tumors. Endocrine 10:225–232
Thyaga Rajan S, Meites J, Quadri SK (1995) Deprenyl reinitiates estrous cycles, reduces serum prolactin, and decreases the incidence of mammary and pituitary tumors in old acyclic rats. Endocrinology 136:1103–1110
Thyaga Rajan S, Madden KS, Stevens SY et al (1999) Inhibition of tumor growth by L-deprenyl involves neural-immune interactions in rats with spontaneously developing mammary tumors. Anticancer Res 19:5023–5028
Thyaga Rajan S, Madden KS, Stevens SY et al (2000) Anti-tumor effect of L-deprenyl is associated with enhanced central and peripheral neurotransmission and immune reactivity in rats with carcinogen-induced mammary tumors. J Neuroimmunol 109:95–104
Toronyi E, Hamar J, Magyar K et al (2002) Antiapoptotic effect of (−)-deprenyl in rat kidney after ischemia-reperfusion. Med Sci Monit 8:BR65–BR68
Tsutsumi H, Katagi M, Nishikawa M et al (2004) In vitro confirmation of selegiline N-oxidation by flavin-containing monooxygenase in rat microsome using LC-ESI MS. Biol Pharm Bull 27:1572–1575
Valoti M, Fusi F, Frosini M et al (2000) Cytochrome P450-dependent N-dealkylation of L-deprenyl in C57BL mouse liver microsomes: effects of in vivo pretreatment with ethanol, phenobarbital, beta-naphthoflavone and L-deprenyl. Eur J Pharmacol 391:199–206
Wadia JS, Chalmers-Redman RM, Ju WJ et al (1998) Mitochondrial membrane potential and nuclear changes in apoptosis caused by serum and nerve growth factor withdrawal: time course and modification by (−)-deprenyl. J Neurosci 18:932–947
Wecker L, James S, Copeland N et al (2003) Transdermal selegiline: targeted effects on monoamine oxidases in the brain. Biol Psychiatry 54:1099–1104
Whaley JG, Tharakan B, Smith B et al (2009) (−)-Deprenyl inhibits thermal injury-induced apoptotic signaling and hyperpermeability in microvascular endothelial cells. J Burn Care Res 30:1018–1027
Wu RF, Ichikawa Y (1995) Inhibition of 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine metabolic activity of porcine FAD-containing monooxygenase by selective monoamine oxidase-B inhibitors. FEBS Lett 358:145–148
Yoshida T, Yamada Y, Yamamoto T et al (1986) Metabolism of deprenyl, a selective monoamine oxidase (MAO) B inhibitor in rat: relationship of metabolism to MAO-B inhibitory potency. Xenobiotica 16:129–136
Yoshida T, Oguro T, Kuroiwa Y (1987) Hepatic and extrahepatic metabolism of deprenyl, a selective monoamine oxidase (MAO) B inhibitor, of amphetamines in rats: sex and strain differences. Xenobiotica 17:957–963
Youdim MB, Weinstock M (2002) Novel neuroprotective anti-Alzheimer drugs with anti-depressant activity derived from the anti-Parkinson drug, rasagiline. Mech Ageing Dev 123:1081–1086
Youdim MB, Wadia A, Tatton W et al (2001) The anti-Parkinson drug rasagiline and its cholinesterase inhibitor derivatives exert neuroprotection unrelated to MAO inhibition in cell culture and in vivo. Ann N Y Acad Sci 939:450–458
Acknowledgments
We would like to join those scientists who want to express their sincere thanks to Professor Abel Lajtha for serving as an editor of the journal of Neurochemical Research for the last 35 years. Dr. Lajtha was born in Hungary. We sentence our review of deprenyl research to him, the drug which is an original Hungarian product, used for the treatment of Parkinson’s disease due to its selective irreversible inhibition on MAO-B. We would like to thank Dr. Lajtha for his personal help and keeping his eyes on the progression of deprenyl studies carried out during the last decades.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Magyar, K., Szende, B., Jenei, V. et al. R-Deprenyl: Pharmacological Spectrum of its Activity. Neurochem Res 35, 1922–1932 (2010). https://doi.org/10.1007/s11064-010-0238-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-010-0238-8